Introduction
Hepatocellular carcinoma (HCC) is a common malignant
tumor with high incidence and mortality (1,2).
Surgical resection and liver transplantation are the most effective
treatments for HCC, but the survival rate of HCC patients remains
at only 30–40% (3). The diagnostic
methods for HCC include biopsy, computed tomography (CT), and
magnetic resonance imaging (MRI) (4,5);
however, most patients are diagnosed at advanced stages without
curative approaches (6). Moreover,
the current molecular pathogenesis of HCC is poorly understood
(7). Therefore, elucidation of key
mechanism underlying HCC is still essential for the development of
effective therapeutic strategies for HCC therapy.
Hepatitis B virus X protein (HBx) plays a key role
in the replication of viral genomes and the development of chronic
hepatitis B (CHB) to HCC (8,9). HBx
has been found to be involved in HCC development via regulating the
Notch signaling pathway (10–12).
The Notch signaling pathway has been described to promote cell
survival, and angiogenesis in a variety of human cancers and may
serve as a promising therapeutic strategy for cancer therapy
(13–15). Accumulating evidence has confirmed
the role of Notch1 in regulating cell invasion and metastasis in
prostate (16), breast (17) and colorectal cancer (18). In addition, Notch1 was found to
participate in the pathogenesis of liver cancer by various
functions and pathways. Sun et al (19) confirmed that Notch1 could regulate
the Wnt signaling pathway, and further affect the proliferation of
hepatocellular carcinoma cells. Importantly, constitutively active
Notch1 alone failed to transform immortalized L02 cells in
vivo, yet it synergized with the Ras pathway to promote hepatic
cell transformation (20). However,
there is a discrepancy concerning the role of Notch1 in cancer
development due to the high context dependency of the Notch cascade
(21). In addition to the limited
and inconsistent data regarding Notch1 involvement in HCC
considering anti-tumoral effects following its inhibition (22,23),
there is still an urgent need for exploring the effects of Notch1
on the tumorigenesis and progression of HCC.
In the present study, we knocked down the expression
of Notch1, and then detected the regulatory relationship between
Notch1 and the Notch signaling pathway. We then investigated the
effect of Notch1 knockdown on the proliferation, cell cycle and
apoptosis of L02/HBx cells in vitro and the tumor formation
ability of L02/HBx cells in vivo.
Materials and methods
Cell culture
The L02/HBx cells were obtained from Institute of
Biochemistry and Cell Biology, Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences (Shanghai, China) and
cultured in Gibco™ Dulbecco's modified Eagle's medium (DMEM)
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal
bovine serum (FBS) and 250 µg/ml G418 in a 37°C incubator with 5%
CO2.
Cell transfection
The short hairpin RNAs specially targeting Notch1
(Notch1-shRNAs) were purchased from Shanghai GenePharma Co., Ltd.
(Shanghai, China). The sequences of Notch1-shRNAs were as follows:
shRNA1,
5′-CCGGGATGCCAAATGCCTGCCAGAACTCGAGTTCTGGCAGGCATTTGGCATCTTTTT-3′;
shRNA2,
5′-CCGGCAAAGACATGACCAGTGGCTACTCGAGTAGCCACTGGTCATGTCTTTGTTTTT-3′;
shRNA3,
5′-CCGGCTTTGTTTCAGGTTCAGTATTCTCGAGAATACTGAACCTGAAACAAAGTTTTT-3′;
and shNC,
5′-CCGGGCCGAACCAATACAACCCTCTCTCGAGAGAGGGTTGTATTGGTTCGGCTTTTT-3′.
For the in vitro group, L02/HBx cells were divided into 5
groups by transfection with Notch1-shRNAs, Notch2-shRNAs,
Notch3-shRNAs, sh-NC or without any treatment using Invitrogen™
Oligofectamine (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The shRNA, which induced the lowest
relative mRNA of Notch1, was used for subsequent experiments.
Quantitative reverse-transcription PCR
(qRT-PCR)
When the cell reached 80–90% confluence, cell were
harvested and total RNA was extract by TRIzol RNA kit (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The concentration and purity of total RNA were
assessed by a ultraviolet spectrophotometer. Reverse transcription
into cDNA was then conducted by a reverse cDNA transcription Kit
(Takara Bio Inc., Otsu, Japan). Quantitative SYBR ExScript qRT-PCR
Kit (Takara Bio Inc.) was used to detect the expression of target
genes and the primers used in this study are showed in Table I. The reactions were performed at
95°C for 10 min followed by 40 cycles of 95°C, 15 sec and 60°C for
1 min. Finally, the expression levels of target genes were
normalized to GAPDH and then calculated using the 2−ΔΔCq
method (24).
 | Table I.Primer sequences for the
amplification of target genes. |
Table I.
Primer sequences for the
amplification of target genes.
| Gene | Sequences | PCR product
(bp) |
|---|
| Notch1 |
5′-CCGCAGTTGTGCTCCTGAA-3′ | 109 |
|
|
5′-ACCTTGGCGGTCTCGTAGCT-3′ |
|
| Hes1 |
5′-GCTAAGGTGTTTGGAGGCT-3′ | 122 |
|
|
5′-CCGCTGTTGCTGGTGTA-3′ |
|
| NICD |
5′-GATGTCACCAGGTCTTACTAC-3′ | 132 |
|
|
5′-GTATATCTTCAGCAGAAATGG-3′ |
|
| Cyclin D1 |
5′-GGCTGAAGTCACCTCTTGGTTACAG-3′ | 177 |
|
|
5′-TAGCGTATCGTAGGAGTGGGACA-3′ |
|
| CDK4 |
5′-TGGTGTCGGTGCCTATGG-3′ | 128 |
|
|
5′-GAACTGTGCTGATGGGAAGGG-3′ |
|
| E2F1 |
5′-CCAGGAAAAGGTGTGAAATC-3′ | 74 |
|
|
5′-AAGCGCTTGGTGGTCAGATT-3′ |
|
| P21 |
5′-TGATTAGCAGCGGAACAAGGAG-3′ | 254 |
|
|
5′-GGAGAAACGGGAACCAGGACA-3′ |
|
| Rb |
5′-TCAAGGGTCATTATGGGTTAGGC-3′ | 115 |
|
|
5′-CTTTAGGTGTAGGGGAGGGGAG-3′ |
|
| Cyclin E1 |
5′-CCTGGATGTTGACTGCCTTGA-3′ | 116 |
|
|
5′-CTATGTCGCACCACTGATACCCT-3′ |
|
| Caspase-3 |
5′-GGTTCATCCAGTCGCTTTG-3′ | 99 |
|
|
5′-ATTCTGTTGCCACCTTTCG-3′ |
|
| Caspase-9 |
5′-GCGAACTAACAGGCAAGCA-3′ | 149 |
|
|
5′-CATCACCAAATCCTCCAGAAC-3′ |
|
| Caspase-8 |
5′-AGAGTCTGTGCCCAAATCAAC-3′ | 78 |
|
|
5′-GCTGCTTCTCTCTTTGCTGAA-3′ |
|
| Actin |
5′-GTTGCGTTACACCCTTTCTTG-3′ | 157 |
|
|
5′-GACTGCTGTCACCTTCACCGT-3′ |
|
Western blot analysis
Total protein was extracted using
radioimmunoprecipitation assay (RIPA) (Sangon Biotech, Shanghai,
China). The protein samples were run by 10% SDS-PAGE gel
electrophoresis, transferred to the polyvinylidene fluoride (PVDF)
membranes for 2 h, blocked using skimmed milk powder antigen for 30
min, and incubated with primary rat antibodies against Notch1 (cat.
no. ab44986), Hes1 (cat. no. ab108937), NICD (cat. no. ab8925),
Ki-67 (cat. no. ab15580), proliferating cell nuclear antigen (PCNA)
(cat. no. ab18197), Bcl-2 (cat. no. ab182858), cyclin D1 (cat. no.
ab16663), CDK4 (cat. no. ab199728), E2F1 (cat. no. ab4070), p21
(cat. no. ab109199), retinoblastoma gene (Rb) (cat. no. ab181616),
cyclin E1 (cat. no. ab133266), caspase-3 (cat. no. ab13585),
caspase-9 (cat. no. ab32539), caspase-8 (cat. no. ab25901) and
actin (cat. no. ab8226) (Abcam, Cambridge, MA, USA; 1:1,000
dilution) overnight at 4°C, respectively. Actin was used as the
internal control. Then the samples were incubated with secondary
rabbit anti-rat antibody (cat. no. sc-516132; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) (1:5,000 dilution) at room
temperature for 2 h, followed by luminescence development using an
enhanced chemiluminescence (ECL) analysis system (Santa Cruz
Biotechnology).
CCK-8 assay
Cells were seeded in 96-well plates at a density of
5×104 cells/well. After transfection for 24, 48, 72 and
96 h, the cells in the corresponding well were replaced with
primary culture medium (no red phenol), and WST-8 solution was
added to each well at 37°C for 4 h. The absorbance of each well at
450 nm was detected by a microplate spectrophotometer (BioTek
Instruments, Winooski, VT, USA).
Colony formation assay
Cells were digested by 0.25% trypsin and filtered
using a 40-µm nylon mesh. Then, cells were seeded in 24-well plates
and maintained in a 37°C incubator with 5% CO2 for 14
days. Thereafter, the cells were fixed with 4% methanol for 15 min,
followed by incubation with Giemasa (Kaiji Biotechnology, Shanghai,
China) for 30 min. The colonies containing >30 cells were
counted under a light microscope (Olympus IX83; Olympus Corp.,
Tokyo, Japan).
Cell cycle assay
Cells were digested by 0.25% trypsin. Cells
(1×106 cells/ml) were collected and then fixed with 70%
volume ethanol overnight at 4°C. After discarding the supernatant
by centrifugation (in 5 min × 1,500 g), 50 µl RNase A was added to
the cell precipitation for 30 min at 37°C and then 200 µl propidium
iodide (PI) (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was
added for staining. Cell cycle was then analyzed by flow cytometry
(Sysmex Partec, Munster, Germany).
Cell apoptosis
Cells were digested by 0.25% trypsin. Cells
(1×106 cells/ml) were collected and then suspended in 1X
binding buffer. Thereafter, the cells were stained with Annexin
V-PE and 7-amino-actinomycin (7-AAD) at room temperature for 15
min. Cell apoptosis was then detected by flow cytometry within 1
h.
In vivo tumor xenograft test
A total of 12 male nude mice (4–6 weeks old, 18–20 g
in weight; Cyagen, Shanghai, China) were housed for one week and
then divided into 2 groups: Notch1-shRNA and sh-NC groups (n=6 each
group). The mice were housed in temperature-controlled cages with a
12-h light/dark cycle and given free access to water and normal
chows. The maximum allowable tumor size was diameter of 2.0 cm and
volume of 4.2 cm3. Then Notch1-shRNA and sh-NC
transfected cells at logarithmic growth phase were digested and
suspended in DMEM respectively, at a cell density of
2×106. Next, 0.2 ml cells were injected subcutaneously
into the neck of each nude mouse, respectively. The nude mice were
sacrificed by ether anesthesia after 25 days. The tumors were
removed, and the tumor volume was calculated according to the
formula: V = (LxW2)/2 (V, volume, L, length and W,
width). Then, the tissues were fixed in 10% formalin for
haematoxylin and eosin (H&E) staining and immunohistochemistry
(IHC) straining. Non-retrospective ethical approval was obtained
for the animal experiments conducted in the study and tumor burden
did not exceed the recommended dimensions. The animal experiments
in this study were approved by the Animal Care and Research
Committee of Chengdu Military General Hospital. All experiments
were performed in compliance with relevant laws and guidelines. In
addition, all experiments were conducted following the
institutional guidelines of Chengdu Military General Hospital
(Chengdu, Sichuan, China).
H&E staining
To assess the pathological changes, the fixed
samples of the lungs in 10% formaldehyde were embedded in paraffin.
Tissue sections cut from paraffin-embedded samples were then
stained with H&E to detect inflammatory cell infiltration in
the lung tissue.
IHC staining
Paraffin sections (3–5 µm in thickness) on slides
with suitable tissue adhesive were processed for deparaffinization
and hydration. Endogenous peroxidase was inactivated by incubation
with 3% hydrogen peroxidase for 15–20 min. Antigen retrieval was
conducted with a microwave oven heating for 30 min with citrate
buffer (0.01 M, pH 6.0). After blocked with 5% bovine serum albumin
for 1 h to reduce nonspecific reactions, the sections were
incubated with the appropriate monoclonal antibodies, including
Notch1 (cat. no. ab44986), Hes1 (cat. no. ab108937), NICD (cat. no.
ab8925), Ki-67 (cat. no. ab15580), cyclin D1 (cat. no. ab16663),
retinoblastoma gene (Rb) (cat. no. ab181616), caspase-3 (cat. no.
ab13585) and actin (cat. no. ab8226) (Abcam, Cambridge, MA, USA;
1:1,000 dilution) overnight at 4°C, followed by incubation with the
secondary rabbit anti-rat antibody (cat. no. sc-516132; Santa Cruz
Biotechnology) (1:5,000 dilution). The reaction was visualized
using DAB (3,3′-diaminobenzidine), and nuclei were counterstained
with hematoxylin.
Statistical analysis
Data were derived from at least three independent
experiments. The data were analyzed using SPSS Software v10.0 (IBM
Corp., Armonk, NY, USA). Data are presented as mean ± standard
deviation and experiments were repeated at least three times. Group
statistical comparisons were assessed by one-way analysis of
variance (ANOVA) followed by Bonferroni's post hoc test. A value of
P<0.05 indicated a statistically significant difference.
Results
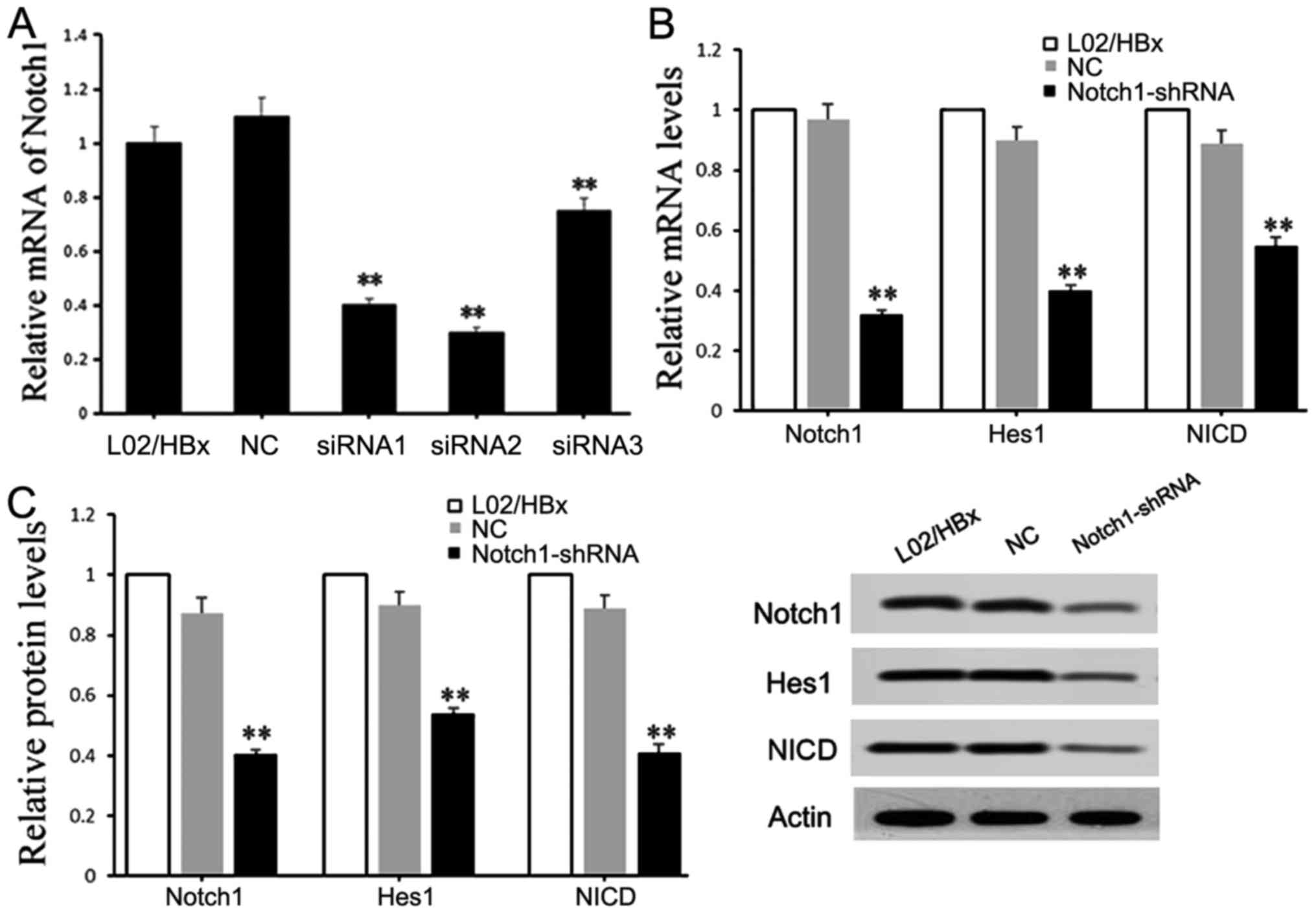
Knockdown of Notch1 inhibits the
activation of the Notch1 signaling pathway
Three pairs of chemically synthesized shRNAs
(shRNA1, 2, 3) targeting Notch1 and negative controls (sh-NC group)
were transfected into L02/HBx cells, respectively. The results of
qRT-PCR showed that compared with the blank control, the expression
of Notch1 was obviously inhibited by all siRNAs (P<0.01), and
the inhibition efficiencies were 64.2% (shRNA1), 72.9% (shRNA2) and
22.4% (shRNA3), respectively (Fig.
1A). The L02/HBx cells were transfected with shRNA2 for
subsequent experiments. Moreover, the effects of recombinant
plasmid Notch1-shRNA on Notch1 signaling pathway were detected. The
results showed that the mRNA and protein expression levels of
Notch1 signaling pathway-related molecules, including Notch1, Hes1
and NICD, were significantly decreased in the Notch1-shRNA
transfected cells compared with those in the blank control
(P<0.01) (Fig. 1B and C),
indicating that Notch1-shRNA could inhibit the activation of the
Notch1 signaling pathway.
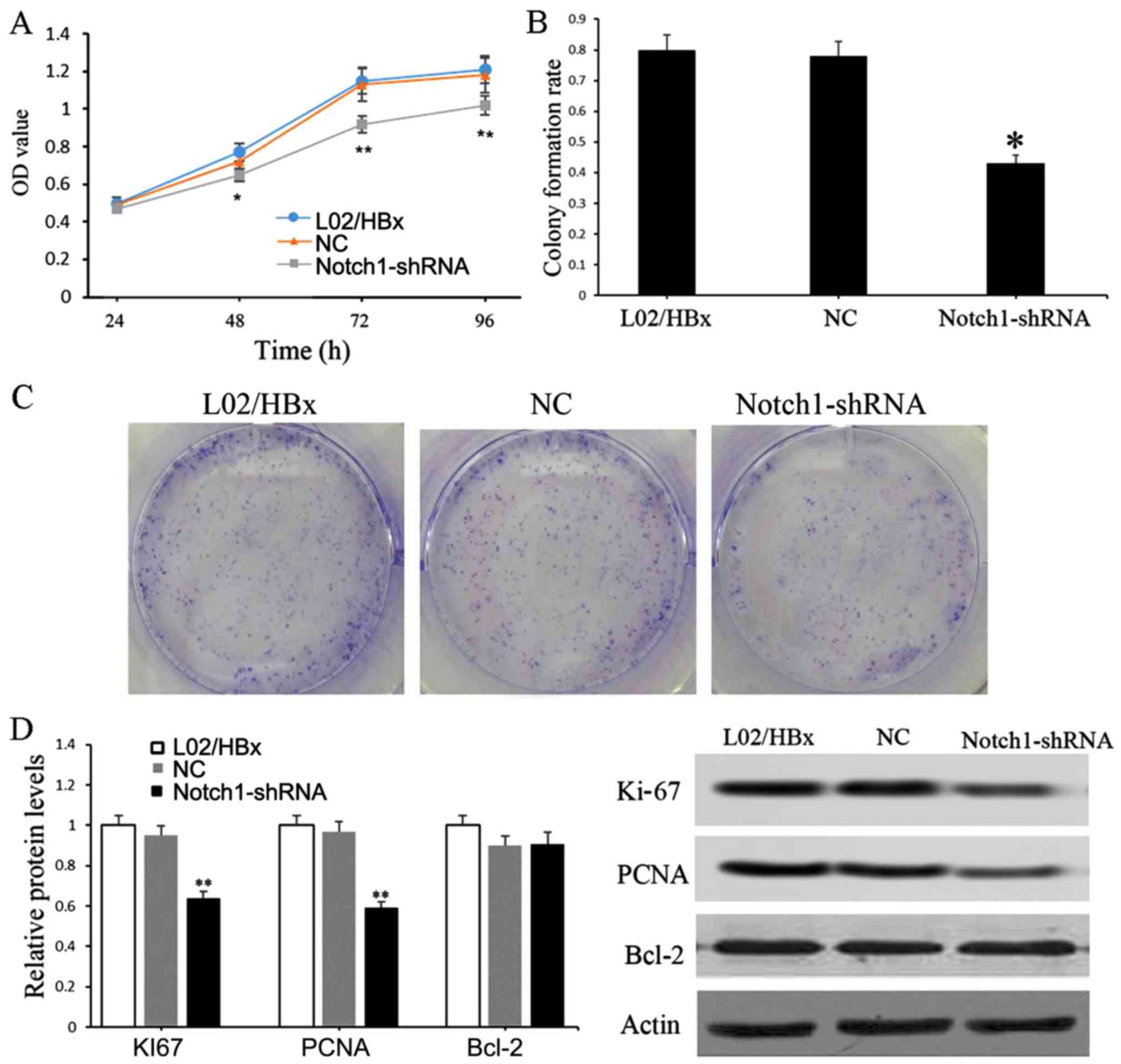
Knockdown of Notch1 impairs the
proliferation of L02/HBx cells
The effect of Notch1 on the proliferation of L02/HBx
cells was detected by CCK-8 and colony formation assays. Compared
with the blank control, the cell viability of the Notch1-shRNA
group was significantly decreased after 48 h of transfection
(P<0.05) (Fig. 2A). Consistent
with the results of the CCK-8 assay, the number of clones formed by
Notch1-shRNA transfected cells was significantly reduced in
comparison with the blank control or sh-NC groups (P<0.05)
(Fig. 2B and C). Furthermore, the
protein expression levels of Ki-67, PCNA and Bcl-2 were detected.
The results showed that the protein expression levels of Ki-67 and
PCNA were significantly downregulated in the Notch1-shRNA
transfected cells relative to those in blank control (P<0.01)
(Fig. 2D).
Knockdown of Notch1 leads to L02/HBx
cell cycle arrest at the G0/G1 phase
The effect of Notch1 on L02/HBx cell cycle was
detected by flow cytometry. The result showed that the proportion
of cells at the G0/G1 phase in the
Notch1-shRNA group was increased significantly compared with the
blank control group (P<0.01), and the proportion of cells at S
phase was decreased significantly (P<0.01); no obvious change
was detected in the proportion of cells at G2/M (Fig. 3A and B). These data indicated that
inhibition of Notch1 significantly arrested the cell cycle at the
G0/G1 phase. Moreover, we determined the
expression changes in cell cycle-related molecules. As shown in
Fig. 3C and D, the mRNA and protein
expression levels of Cyclin D1, CDK4 and E2F1 were significantly
decreased in the Notch1-shRNA group (P<0.01), while the mRNA and
protein levels of P21 and Rb were significantly increased compared
with the blank control group, (P<0.01). These results indicate
that knockdown of Notch1 led to L02/HBx cell cycle arrest at
G0/G1 phase.
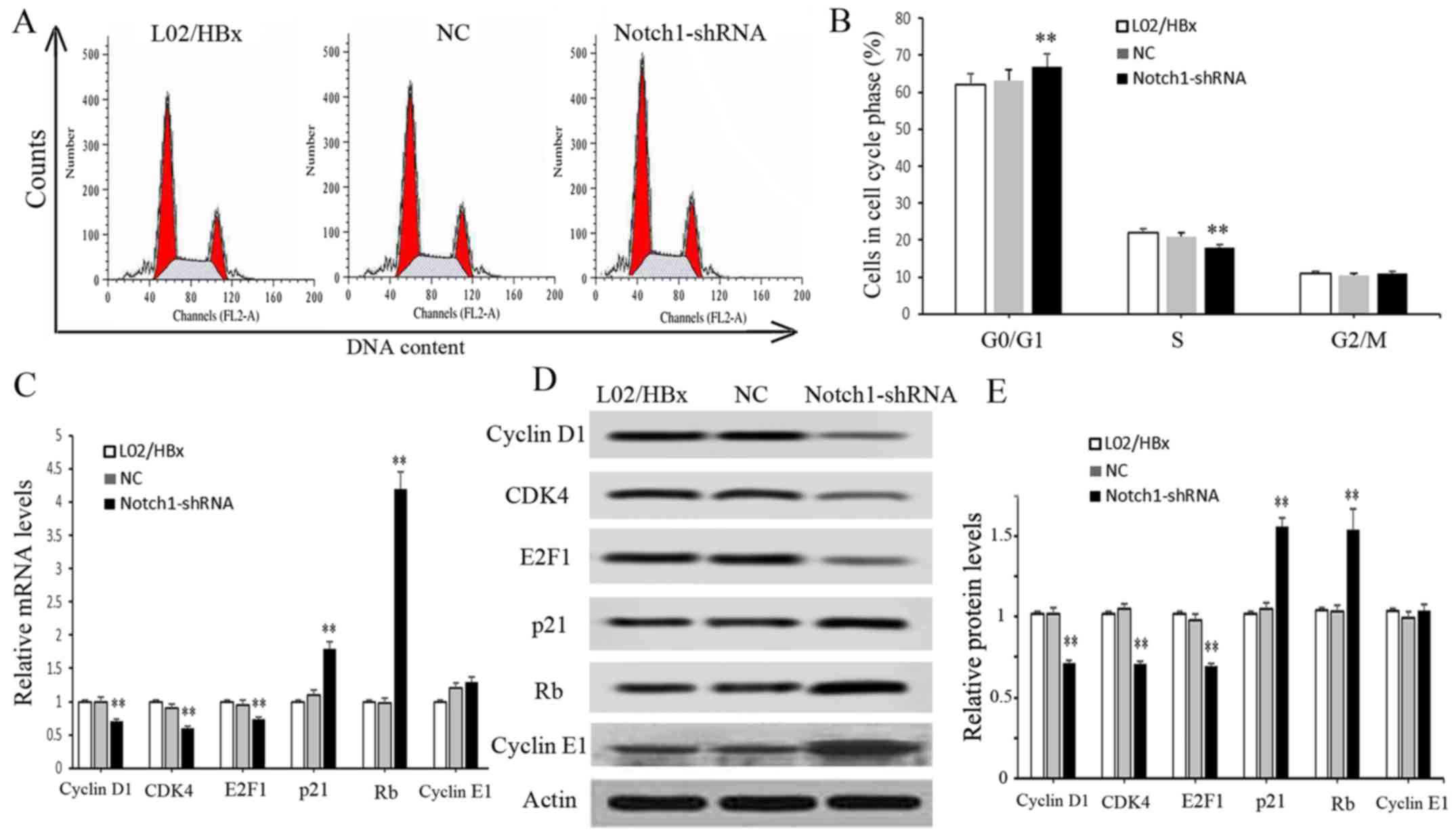 | Figure 3.Notch1 knockdown arrests L02/HBx cell
cycle in vitro. L02/HBx cells were transfected with
Notch1-shRNAs, sh-NC or without any treatment. (A and B) The cell
cycle phase was detected by flow cytometry. (C) The mRNA levels of
cell cycle-related molecules were detected by RT-PCR, including
cyclin D1, CDK4, E2F1, P21 and Rb. (D) The protein levels of cell
cycle-related molecules including cyclin D1, CDK4, E2F1, p21, Rb,
cyclin E1 and actin, were detected by western blot analysis. (E)
Relative protein levels of cell cycle-related molecules were
analyzed by bar graph. Data are represented as the mean ± SD of at
least three independent experiments. **P<0.01 vs. LO2/HBX
group. |
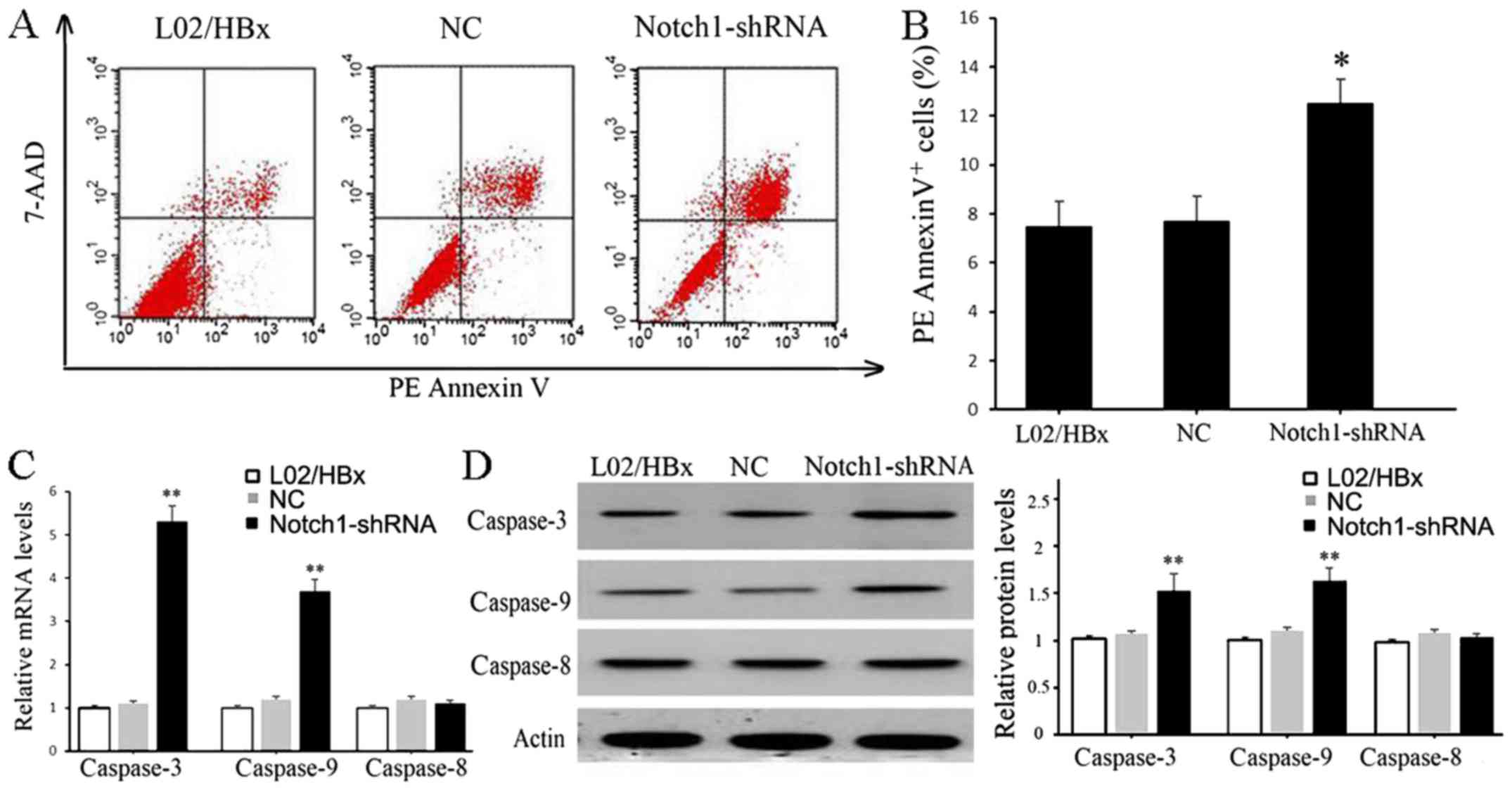
Knockdown of Notch1 promoted apoptosis
of L02/HBx cells
The effect of Notch1 on L02/HBx cell apoptosis was
also assessed by flow cytometry. As shown in Fig. 4A and B, the percentage of apoptotic
cells in the Notch1-shRNA transfected group was significantly
increased compared with the blank control or sh-NC groups
(P<0.05). Furthermore, the mRNA and protein expression levels of
caspase-3 and caspase-9 were significantly increased in the
Notch1-shRNA group compared with the blank control (P<0.01), but
the expression of caspase-8 exhibited no significant difference
(Fig. 4C and D).
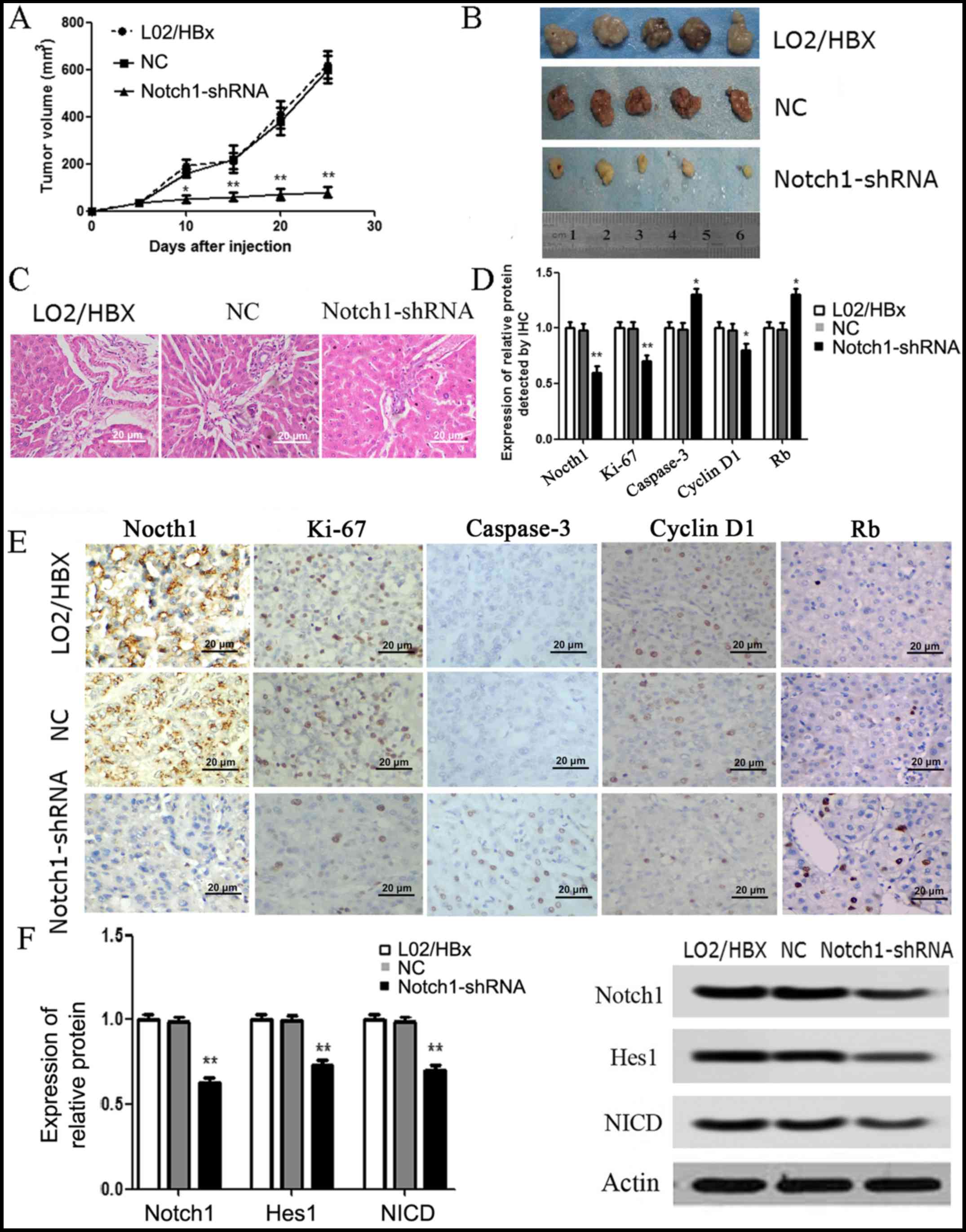
Knockdown of Notch1 inhibits the
tumorigenicity of L02/HBx cells in vivo
The in vivo tumor xenograft model was
established to further explore the effects of Notch1. The results
showed that the tumor volume was significantly inhibited in the
Notch1-shRNA group compared with that noted in the L02/HBx group
(P<0.05) (Fig. 5A and B). A
statistically difference was detected at 10 days after inoculation
(Fig. 5A). In addition, the results
of H&E staining revealed that the tumor tissues were
hepatocellular carcinoma, characterized by a cord like arrangement,
with round nuclei, large nuclear shelves and frequent nuclear
division (Fig. 5C). The results of
IHC staining showed that the expression levels of Notch1, Ki-67 and
cyclin D1 in the Notch1-shRNA group were significantly decreased
compared with these levels in the L02/HBx group, while the
expression levels of Rb and caspase-3 were significantly increased
(P<0.05) (Fig. 5D and E).
Moreover, the results of western blot analysis showed that the
expression levels of Notch1 pathway-related proteins, including
Notch1, Hes1 and NICD were all significantly decreased in the
Notch1-shRNA tumor group in comparison with the L02/HBx group
(P<0.01) (Fig. 5F). There was no
significant difference in the above indices between the NC and
blank groups.
Discussion
HCC is the leading cause of cancer-related death
around the world with limited effective treatments. In addition,
the key mechanism underlying HCC is largely unknown. HBx functions
by interfering with cellular functions, causing aberration in
cellular behavior and transformation, while Notch signaling is
involved in cellular differentiation, cell survival and cell death
processes in various types of cells (12). Notably, Notch1 has been found to
regulate cell invasion and metastasis in prostate (16), breast (17) and colorectal cancer (18). In 2017, Niloofar et al
(25) used bioinformatic methods to
predict targets for Notch1 and HBX genes in chronic hepatitis
B-induced HCC. However, reports concerning the specific role of
Notch1 in the tumorigenesis and progression of HCC are limited. The
present study firstly demonstrated a regulatory relationship
between Notch1 and the Notch signaling pathway, and investigated
the inhibitory effect of Notch1 knockdown on the proliferation,
cell cycle and apoptosis of L02/HBx cells in vitro and the
tumor formation ability of L02/HBx cells in vivo.
One important finding of this study was that the
viability and colony formation ability of L02/HBx cells were
inhibited and the protein expression levels of Ki-67 and PCNA were
markedly downregulated after knockdown of Notch1. Ki-67 is
considered a key marker to assess cell proliferation in breast
cancer (26). Li et al
confirmed that Ki-67 expression is associated with tumor cell
proliferation and growth, and may be a promising target in the
diagnosis and treatment of cancer (27). In addition, PCNA is also regarded as
a marker of cell proliferation in cancer (28). Upregulation of PCNA can mediate the
role of long non-coding RNA CCHE1 in the proliferation of cervical
cancer (29). Given the key role of
Ki-67 and PCNA in proliferation, we speculated that knockdown of
Notch1 may inhibit the proliferation of L02/HBx cells by
suppressing the expression levels of Ki-67 and PCNA.
Cell apoptosis is a key step in cancer development.
Caspase-9 is the major molecule of the endogenous pathway, and
caspase-8 is the major molecule of the exogenous pathway. These two
pathways eventually cause caspase-3 activation. Some studies have
found that HBx does not directly induce apoptosis, but increases
the sensitivity of these cells to apoptotic factors (30–33).
Overall, it seems that the effects of HBx on cell apoptosis are
related to the cell type and duration of expression of caspases
(34). In addition, knockdown of
Notch-1 is found to induce apoptosis of prostate cancer cells
(35). Thus, we aimed to ascertain
whether the Notch1 pathway could regulate the apoptosis of L02/HBx
cells, and to assess the expression of caspase-8, caspase-9 and
caspase-3 apoptotic proteins. Our results showed that the apoptosis
of L02/HBx cells was increased after Notch1 knockdown, together
with increased expression of caspase-3 and capase-9. The expression
of caspase-8 did not exhibit significant changes after Notch1
knockdown. Therefore, we speculate that Notch1 may cause
dysregulation of cell apoptosis via regulating caspase-9 and
caspase-3, an endogenous apoptosis pathway.
A growing body of evidence suggests that
downregulation of cell cycle progression is one of the significant
reasons for the early stage of liver cancer (36). p21 is a major target gene for
p53-induced cell cycle arrest through DNA damage (37,38),
and DNA damage repair and activation of p53 can both activate p21,
thereby regulating the expression of pRb (39). In addition, Rb protein was found to
play an important role in the initiation and maintenance phase of
cell cycle arrest (39). Kunter
et al confirmed that inhibition of PI3K/Akt signaling could
arrest HCC cells at the G0/G1 phase by
regulating Rb/E2F1 (40). Chen
et al indicated that silencing of cyclin D1 could arrest HCC
cells in the G0/G1 phase (41). In addition, CDK4 was found to be
enzymatically activated by D-type cyclins to inactivate
retinoblastoma, and thus arrest G1/S transition (42). In addition, a previous study showed
that the activation of the Notch1 signaling pathway led to tumor
cell growth and proliferation in HepG2 and SMMC7721 cells (43). Qi et al demonstrated that
Notch1 signaling could induce cell cycle arrest and apoptosis, thus
inhibiting the growth of human HCC (44). In this study, we investigated the
effect of Notch1 inhibition on the cell cycle and the expression of
critical cycle regulators. The results showed that inhibition of
Notch1 led to l02/HBx cell cycle arrest at the
G0/G1 phase. Moreover, the expression levels
of cyclin D1, E2F1 and CDK4 were decreased and the expression
levels of p21 and Rb were increased. Therefore, we hypothesized
that Notch1 may cause cell cycle disturbances in L02/HBx cells via
the cyclin D1/CDK4 pathway. The Notch pathway is a major member of
the network that is involved in the pathogenesis of HBx-induced
HCC. Activated Notch1 was observed in HBx-induced L02 cell
malignant transformation, while inhibition of the Notch1 pathway
decreased cell proliferation (19).
In addition, Villanueva et al suggested that Notch signaling
is activated in human HCC samples and promotes formation of liver
tumors in mice. These reports indicate that Notch signaling
inhibition contributes to the suppression of HBx-induced HCC
(45). Consistent with this
research, knockdown of Notch1 in this study was found to inhibit
tumor growth and induce cell cycle arrest in L02/HBx cells in
vitro and in vivo. These findings confirmed that
knockdown of Notch1 inhibited the tumorigenicity of L02/HBx
cells.
In conclusion, our findings revealed that knockdown
of Notch1 can inhibit the activation of the Notch1 signaling
pathway, and thus suppress the development of HBx-induced HCC.
Knockdown of Notch1 may inhibit the proliferation of L02/HBx cells
by regulating the expression levels of Ki-67 and PCNA, and induce
cell cycle arrest at the G0/G1 phase by
decreasing the expression of cyclin D1, CDK4, E2F1 and increasing
the expression of p21 and retinoblastoma gene (Rb). Moreover,
knockdown of Notch1 may promote apoptosis of L02/HBx cells by
activation of caspase-3 and caspase-9. In vivo experiments
also confirmed that knockdown of Notch1 inhibited the
tumorigenicity of L02/HBx cells. Thus, Notch1 may serve as a
promising therapeutic target for HCC.
Acknowledgements
The authors would like to thank the members of
Cangzhou Central Hospital, for providing technical support
concerning the present study.
Funding
The present study was funded by the Applied basic
research project of sichuan science and technology department (no.
16PJ010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YP interpreted the main data regarding the cell
transfection and cell function analysis. DZ and YY were involved in
the in vivo tumor xenograft test. CK and ZJ were responsible
for immunohistochemistry. ZZ and XW interpreted the western blot
analysis and conducted the statistical analysis. JX was responsible
for the design and drafting of the manuscript. All authors read and
approved the final manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
The animal experiments in this study were approved
by the Animal Care and Research Committee of Chengdu Military
General Hospital. All experiments were performed in compliance with
relevant laws and guidelines. In addition, all experiments were
conducted following the institutional guidelines of Chengdu
Military General Hospital (Chengdu, Sichuan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maida M, Orlando E, Cammà C and Cabibbo G:
Staging systems of hepatocellular carcinoma: A review of
literature. World J Gastroenterol. 20:4141–4150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li CP, Cai MY, Jiang LJ, Mai SJ, Chen JW,
Wang FW, Liao YJ, Chen WH, Jin XH, Pei XQ, et al: CLDN14 is
epigenetically silenced by EZH2-mediated H3K27ME3 and is a novel
prognostic biomarker in hepatocellular carcinoma. Carcinogenesis.
37:557–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raimondo G, Allain JP, Brunetto MR,
Buendia MA, Chen DS, Colombo M, Craxì A, Donato F, Ferrari C, Gaeta
GB, et al: Statements from the Taormina expert meeting on occult
hepatitis B virus infection. J Hepatol. 49:652–657. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kabel AM, Al-Joaid AA, Al-Ghamdi AA,
Al-Joaid AA and Al-Zaidi WS: Hepatocellular carcinoma: A focus on
the new lines of management. J Cancer Res Treat. 4:55–60. 2016.
|
|
5
|
El-Serag HB, Marrero JA, Rudolph L and
Reddy KR: Diagnosis and treatment of hepatocellular carcinoma.
Gastroenterology. 134:1752–1763. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Villanueva A, Lachenmayer A and
Finn RS: Advances in targeted therapies for hepatocellular
carcinoma in the genomic era. Nat Rev Clin Oncol. 12:408–424. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim SU, Kim DY, Ahn SH, Kim HM, Lee JM,
Chon CY, Park YN, Han KH and Park JY: The impact of steatosis on
liver stiffness measurement in patients with chronic hepatitis B.
Hepatogastroenterology. 57:832–838. 2010.PubMed/NCBI
|
|
8
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cirrhosis: Incidence and risk
factors. Gastroenterology. 127 (5 Suppl 1):S35–S50. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wen J, Liu Y, Liu J, Liu L, Song C, Han J,
Zhu L, Wang C, Chen J, Zhai X, et al: Expression quantitative trait
loci in long non-coding RNA ZNRD1-AS1 influence both HBV infection
and hepatocellular carcinoma development. Mol Carcinog.
54:1275–1282. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Papatheodoridis GV, Lampertico P,
Manolakopoulos S and Lok A: Incidence of hepatocellular carcinoma
in chronic hepatitis B patients receiving nucleos(t)ide therapy: A
systematic review. J Hepatol. 53:348–356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
You X, Liu F, Zhang T, Lv N, Liu Q, Shan
C, Du Y, Kong G, Wang T, Ye L and Zhang X: Hepatitis B virus X
protein upregulates Lin28A/Lin28B through Sp-1/c-Myc to enhance the
proliferation of hepatoma cells. Oncogene. 33:449–460. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kongkavitoon P, Tangkijvanich P, Hirankarn
N and Palaga T: Hepatitis B virus HBx activates Notch signaling via
delta-like 4/Notch1 in hepatocellular carcinoma. PLoS One.
11:e01466962016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Purow B: Notch inhibition as a promising
new approach to cancer therapy. Adv Exp Med Biol. 727:305–319.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Braune EB and Lendahl U: Notch-a
goldilocks signaling pathway in disease and cancer therapy. Discov
Med. 21:189–196. 2016.PubMed/NCBI
|
|
15
|
Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S,
Wu GS and Wu K: Notch signaling: An emerging therapeutic target for
cancer treatment. Cancer Lett. 369:20–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lefort K, Ostano P, Mello-Grand M, Calpini
V, Scatolini M, Farsetti A, Dotto GP and Chiorino G: Dual tumor
suppressing and promoting function of Notch1 signaling in human
prostate cancer. Oncotarget. 7:48011–48026. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shao S and Zhao X, Zhang X, Luo M, Zuo X,
Huang S, Wang Y, Gu S and Zhao X: Notch1 signaling regulates the
epithelial-mesenchymal transition and invasion of breast cancer in
a Slug-dependent manner. Mol Cancer. 14:282015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fender AW, Nutter JM, Fitzgerald TL,
Bertrand FE and Sigounas G: Notch-1 promotes stemness and
epithelial to mesenchymal transition in colorectal cancer. J Cell
Biochem. 116:2517–2527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Q, Wang R, Luo J, Wang P, Xiong S, Liu
M and Cheng B: Notch1 promotes hepatitis B virus X protein-induced
hepatocarcinogenesis via Wnt/β-catenin pathway. Int J Oncol.
45:1638–1648. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fan R, Chen P, Zhao D, Tong JL, Li J and
Liu F: Cooperation of deregulated Notch signaling and Ras pathway
in human hepatocarcinogenesis. J Mol Histol. 42:473–481. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ranganathan P, Weaver KL and Capobianco
AJ: Notch signalling in solid tumours: A little bit of everything
but not all the time. Nat Rev Cancer. 11:338–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu
X, Sun J, Yang S, Poon RT and Fan ST: Notch1-Snail1-E-cadherin
pathway in metastatic hepatocellular carcinoma. Int J Cancer.
131:E163–E172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim SO, Kim HS, Quan X, Ahn SM, Kim H,
Hsieh D, Seong JK and Jung G: Notch1 binds and induces degradation
of Snail in hepatocellular carcinoma. BMC Biol. 9:832011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Niloofar M, Paryan M, Khansarinejad B,
Rafiei M and Mondanizadeh M: Bioinformatic prediction of miRNAs
targeting Notch1 and HBx genes in chronic hepatitis B-induced
hepatocellular carcinoma. Majallah-i dānishgāh-i ulūm-i pizishkī-i
Arāk. 19:89–101. 2017.
|
|
26
|
Pathmanathan N and Balleine RL: Ki67 and
proliferation in breast cancer. J Clin Pathol. 66:512–516. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li LT, Jiang G, Chen Q and Zheng JN: Ki67
is a promising molecular target in the diagnosis of cancer
(review). Mol Med Rep. 11:1566–1572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bologna-Molina R, Mosqueda-Taylor A,
Molina-Frechero N, Mori-Estevez AD and Sánchez-Acuña G: Comparison
of the value of PCNA and Ki-67 as markers of cell proliferation in
ameloblastic tumor. Med Oral Patol Oral Cir Bucal. 18:e174–e179.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang M, Zhai X, Xia B, Wang Y and Lou G:
Long noncoding RNA CCHE1 promotes cervical cancer cell
proliferation via upregulating PCNA. Tumor Biol. 36:7615–7622.
2015. View Article : Google Scholar
|
|
30
|
Huang Y, Tai AW, Tong S and Lok AS: HBV
core promoter mutations promote cellular proliferation through
E2F1-mediated upregulation of S-phase kinase-associated protein 2
transcription. J Hepatol. 58:1068–1073. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sanz-Cameno P, Martín-Vílchez S,
Lara-Pezzi E, Borque MJ, Salmerón J, Muñoz de Rueda P, Solís JA,
López-Cabrera M and Moreno-Otero R: Hepatitis B virus promotes
angiopoietin-2 expression in liver tissue: Role of HBV X protein.
Am J Pathol. 169:1215–1222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang TW, Yevsa T, Woller N, Hoenicke L,
Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova
A, et al: Senescence surveillance of pre-malignant hepatocytes
limits liver cancer development. Nature. 479:547–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chung TW, Lee YC and Kim CH: Hepatitis B
viral HBx induces matrix metalloproteinase-9 gene expression
through activation of ERK and PI-3K/AKT pathways: Involvement of
invasive potential. FASEB J. 18:1123–1125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang F, Xia X, Wang J, Sun Q, Luo J and
Cheng B: Notch1 signaling contributes to the oncogenic effect of
HBx on human hepatic cells. Biotechnol Lett. 35:29–37. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ye QF, Zhang YC, Peng XQ, Long Z, Ming YZ
and He LY: Silencing Notch-1 induces apoptosis and increases the
chemosensitivity of prostate cancer cells to docetaxel through
Bcl-2 and Bax. Oncol Lett. 3:879–884. 2012.PubMed/NCBI
|
|
36
|
Park IY, Sohn BH, Yu E, Suh DJ, Chung YH,
Lee JH, Surzycki SJ and Lee YI: Aberrant epigenetic modifications
in hepatocarcinogenesis induced by hepatitis B virus X protein.
Gastroenterology. 132:1476–1494. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lv WP, Zhou KL, Gao B, Chen YL, Xiang X,
Su M and Dong JH: Hepatitis B virus X protein induces malignant
transformation tendency of Chang liver cells involving p16(INK4a)
hypermethylation. Beijing Da Xue Xue Bao Yi Xue Ban. 44:932–936.
2012.(In Chinese). PubMed/NCBI
|
|
39
|
Wei X, Tan C, Tang C, Ren G, Xiang T, Qiu
Z, Liu R and Wu Z: Epigenetic repression of miR-132 expression by
the hepatitis B virus × protein in hepatitis B virus-related
hepatocellular carcinoma. Cell Signal. 25:1037–1043. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kunter I, Atabey N and Erdal E: Inhibition
of the PI3K/Akt signalling caused G0/G1 arrest through the
mechanism of Rb/E2F1 on the HCC cell lines. Int Cong Mol Med.
61:2009.
|
|
41
|
Chen J, Li X, Cheng Q, Ning D, Ma J, Zhang
ZP, Chen XP and Jiang L: Effects of cyclin D1 gene silencing on
cell proliferation, cell cycle, and apoptosis of hepatocellular
carcinoma cells. J Cell Biochem. 119:2368–2380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Calvisi D and Eferl R: CDK4/6 inhibition
and sorafenib: A ménage à deux in HCC therapy? Gut. 66:1179–1180.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gao J, Dong Y, Zhang B, Xiong Y, Xu W,
Cheng Y, Dai M, Yu Z, Xu H and Zheng G: Notch1 activation
contributes to tumor cell growth and proliferation in human
hepatocellular carcinoma HepG2 and SMMC7721 cells. Int J Oncol.
41:1773–1781. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qi R, An H, Yu Y, Zhang M, Liu S, Xu H,
Guo Z, Cheng T and Cao X: Notch1 signaling inhibits growth of human
hepatocellular carcinoma through induction of cell cycle arrest and
apoptosis. Cancer Res. 63:8323–8329. 2003.PubMed/NCBI
|
|
45
|
Villanueva A, Alsinet C, Yanger K, Hoshida
Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Solé M, Thung S,
Stanger BZ and Llovet JM: Notch signaling is activated in human
hepatocellular carcinoma and induces tumor formation in mice.
Gastroenterology. 143:1660–1669.e7. 2012. View Article : Google Scholar : PubMed/NCBI
|