Introduction
Breast cancer is the most common cancer among women
and a global public health crisis (1). This cancer readily metastasizes, which
limits further treatment (2).
Therefore, the molecular mechanisms underlying breast cancer must
be better understood. Epigenetic alterations are fundamental for
the formation and progression of cancer, and histone modifications,
such as histone methylation, are associated with breast cancer
prognosis (3). Histone methylation
commonly occurs at histone 3 (H3) and H4 arginine or lysine
residues, and is catalyzed by an enzyme family with a conserved
catalytic domain or su(var)3-9, enhancer-of-zeste and trithorax
(SET) domain (4).
The SET domain bifurcated 1 (SETDB1) gene is located
on human chromosome 1q21 and encodes a histone H3 lysine 9
methyltransferase, which is also known as ESET or KMT1E (5). SETDB1 can be targeted to the
KRAB-associated protein-1 (KAP-1) corepressor and enhance binding
of heterochromatin protein 1 proteins that contribute to gene
expression silencing (6,7). SETDB1 is also involved in
transcriptional repression and heterochromatin formation with
methyl-CpG-binding domain protein 1 and the human homologue of
mouse ATFa-associated modulator (8–10).
Studies suggest that SETDB1 is essential for early embryonic
development and maintenance of embryonic stem cells (11–15).
SETDB1 was previously identified as an oncogene in melanoma
(16). Subsequently, SETDB1 was
demonstrated to be overexpressed and required for cancer cell
proliferation and metastasis in various solid tumors, including
glioma, and prostate, lung and colorectal cancer (17–21).
Furthermore, the promoter activity of SETDB1 was inhibited by p53
protein, leading to paclitaxel induced-cell death in human lung
cancer cells (22). These findings
suggest that SETDB1 has an important role in cancer cell growth and
metastasis, and in drug resistance. Thus, as a H3K9 special histone
methyltransferase, SETDB1 may be a promising epigenetic target for
clinical treatment (23,24).
In the present study, SETDB1 overexpression was
demonstrated to increase breast cancer cell proliferation,
migration and invasion, and depletion of SETDB1 decreased
metastasis in vivo. Furthermore, SETDB1 upregulation induced
epithelial-mesenchymal transition (EMT) in MCF7 cells. Finally,
SETDB1 was identified as an EMT inducer that binds directly to the
promoter of the Snail transcription factor.
Materials and methods
Cell culture
The cell lines MCF7, T47D, BT549 and MDA-MB-231
breast cancer were obtained from the Type Culture Collection of
Chinese Academy of Sciences (Shanghai, China). MCF7 and M231 cells
were maintained in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). T47D and BT549
cells were maintained in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.). All media were supplemented with 10% fetal
bovine serum (FBS; Biowest, SAS, Nuaille, France) and cell lines
were cultured at 37°C with 5% CO2.
Western blot analysis
Total protein was extracted from cells using a
radioimmunoprecipitation lysis buffer (cat. no. sc-24948; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA). A bicinchoninic acid
protein Assay (Thermo Fisher Scientific, Inc.) was used to quantify
total protein concentration. Equal amounts of proteins (50 µg/well)
were separated by SDS-PAGE on 10% gels and electrophoretically
transferred onto polyvinylidene fluoride membranes (cat. no.
IPFL00010; Merck KGaA, Darmstadt, Germany). The membranes were
blocked with 5% milk in Tris-HCl buffered solution for 2 h at room
temperature, and then incubated overnight at 4°C with primary
antibodies and secondary antibody (1:2,000, cat. no. 7074; Cell
Signaling Technology, Inc., Danvers, MA, USA) at 37°C for 1 h. The
signals were visualized using an enhanced chemiluminescent
substrate (Thermo Fisher Scientific, Inc.) and detected by a
FluorChem Q imaging system (ProteinSimple, San Jose, CA, USA).
Images from western blot analysis were quantified using Quantity
One® software 4.6.6 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The expression level was normalized with
respect to an internal control. Primary antibody against SETDB1 was
obtained from ProteinTech Group, Inc. (Chicago, IL, USA; cat. no.
11231-1-AP), and primary antibodies against E-cadherin (cat. no.
3195), β-catenin (cat. no.8480), vimentin (cat. no. 5741), Snail
(cat. no. 3879) and Slug (cat. no. 9585) were obtained from Cell
Signaling Technology, Inc. (EMT Antibody Sampler Kit; cat. no.
9782). β-actin rabbit monoclonal antibody (mAb) internal control
was obtained from Cell Signaling Technology, Inc. (cat. no.
8457).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was obtained using RNAiso plus (cat. no.
9108; Takara Biotechnology Co., Ltd., Dalian, China), and 2 µg
total RNA was reversed transcribed to cDNA using PrimeScript RT
Master Mix (cat. no. DRRO36A; Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. Primers used to
detect SETDB1 (forward, 5′-GATGAGGAACTGGAGAAGATG-3′ and reverse,
5′-ATTAGTCACTGCCCTGGATG-3′); transforming growth factor 1 (TGF1
forward, 5′-ACCTGAACCCGTGTTGCTCT-3′ and reverse,
5′-GAACCCGTTGATGTCCACTT-3′); zinc finger E-box binding homeobox 1
(ZEB1 forward, 5′-GAAAATCCAGTCGCTACAA-3′ and reverse,
5′-CACACAGAAGACAAGTGCTA-3′); Snail (forward,
5′-CCCAATCGGAAGCCTAACT-3′ and reverse, 5′-GCTGCTGGAAGGTAAACTCT-3′);
Slug (forward, 5′-CTCAGAAAGCCCCATTAGT-3′; reverse,
5′-GCCCAGAAAAAGTTGAATAG-3′); tight junction protein 1 (ZO-1
forward, 5′-GCAGCAAGAGATGGCAATA-3′ and reverse,
5′-CAGGGACATTCAATAGCGTA); and GAPDH (internal control forward,
5′-CTGACTTCAACAGCGACACC-3′ and reverse,
5′-TGCTGTAGCCAAATTCGTTGT-3′); transcripts were obtained from BioTNT
(Shanghai, China). qPCR was performed using SYBR Premix Ex Taq
(cat. no. RR420A; Takara Biotechnology Co., Ltd.) on a StepOnePlus
Real-Time System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermal cycle conditions consisted of one cycle at 95°C
for 10 min, followed by 40 cycles of amplification at 95°C for 15
sec, and then 60°C for 1 min. The expression level of mRNA was
obtained by calculating the 2−∆∆Cq values (25).
Lentiviral-vector infections
pLVX-SETDB1-wt-IRES-Puro and
pLKD-EF1a-EGFP-P2A-LUC-F2A-Puro-U6 shSETDB1 vectors were generated
by Obio Technology (Shanghai) Co., Ltd. (Shanghai, China). The
empty vector acts as control group. The three shRNA sequences were
as follows: S1, 5′-GGGCAGTGACTAATTGTGA-3′; S2,
5′-GCATGCGAATTCTGGGCAAGA-3′; S3, 5′-GGGAGGACATAGAAGACATCT-3′. The
negative control shRNA (shNC) sequences were as follows: Y009
forward,
5′-CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTG-3′;
Y009 reverse,
5′-AATTCAAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAA-3′.
Cells (5×104) were seeded onto 24-well plates.
Lentiviral vectors were introduced into breast cancer cells at
different multiplicity of infection (MOI) values (MDA-MB-231 and
BT549, MOI 10; MCF7 and T47D, MOI 40). Different final
concentrations (2, 4, 6, 8 and 10 µg/ml) of puromycin were applied
to the breast cancer cells for 24 h, and the concentration of 6
µg/ml was selected to kill non-transfected cells for 2 weeks to
obtain stably expressed cells. Transfection efficiency was
validated with RT-PCR and western blot analysis.
Cell proliferation assays
A total of 2×103 cells/well were cultured
in 96-multiwell plates for 96 h or 7 days. Cells were incubated
with CCK-8 reagent (10 µl/well; cat. no. CK04-11; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) for 1 h at 37°C. Absorbance at
450 nm was read using a Multiscan Go-1510 microplate reader (Thermo
Fisher Scientific, Inc.).
Migration and invasion assay
Cells (~5×104) were resuspended in 100 µl
serum-free medium and plated in the upper chamber of a Transwell
assay system (cat. no. 3422; Corning Incorporated, Corning, NY,
USA). Then, 10% FBS-containing medium was added to the lower
chamber. For the invasion assay, the Transwell membrane was
precoated with Matrigel (cat. no. 354234; BD Biosciences, Franklin
Lakes, NJ, USA) and incubated at 37°C for 4 h. After cells were
cultured for 24 h, the upper chambers were fixed with 100% methanol
for 10 min at room temperature, and stained with 0.1% crystal
violet for 20 min at room temperature. Cells were counted under a
light microscope in five predetermined fields and averaged.
Wound healing assay
Cells (~5×105) were seeded onto 6-well
plates and incubated overnight. A 1,000 µl sterile pipette tip was
used to scratch the cell monolayer (point of zero migration). After
cell incubation for 24 and 48 h, phase-contrast images of the
scratched area were captured using computer-assisted light
microscopy to assess gap closure.
Animal experiments
Female nude mice aged 4–6 weeks (n=12) were obtained
from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China).
Cells (~1×105 cells in 100 µl) were resuspended in
serum-medium and injected into the tail vein of mice. The animals
were monitored twice weekly. Body condition score was used to
assessment of overall health of the animal from 1
(emaciated/wasted) to 5 (obese) (25). After 2 months of observation, the
maximum body weight loss was 5% of original weight. When the body
condition score was 1/5, all mice were euthanized and organs were
dissected. Lungs were fixed with 10% buffered formalin for 24 h at
room temperature, and embedded in paraffin. Sections (4 µm-thick)
were dewaxed in xylene, rehydrated through decreasing
concentrations of ethanol (100, 95 and 80%) and washed in PBS. Then
stained with hematoxylin for 10 min and eosin for 30 sec at room
temperature. Following staining, sections were dehydrated through
increasing concentrations of ethanol (80, 95 and 100%) and xylene.
Animal experiments were approved by the Ethics Committee of Fudan
University (Shanghai, China).
Chromatin immunoprecipitation (CHIP)
assay
CHIP assay was performed with a SimpleCHIP Enzymatic
Chromatin IP kit (cat. no. 9003; Cell Signaling Technology, Inc.)
according the manufacturer's instructions. Briefly,
~4×106 MCF7 cells overexpressing SETDB1 for each
immunoprecipitation (IP) were crosslinked with 1% formaldehyde at
room temperature for 10 min. Then, 0.5 µl micrococcal nuclease was
added/IP preparation to digest DNA to 150–900 bp. IP antibodies
histone H3 (D2B12) XP Rabbit mAb (10 µl; positive control; cat. no.
4620), normal rabbit IgG (2 µl; negative control; cat. no. 2729)
and SETDB1 rabbit polyclonal antibody (2 µg; cat. no. 11231–1-AP;
Proteintech Group, Inc.) were added to 500 µl 1X CHIP buffer
containing 10 µg fragmented chromatin DNA at 4°C with rotation
overnight. Then, 30 µl CHIP-Grade Protein G Magnetic Beads (cat.
no. 9006; Cell Signaling Technology, Inc.) were used to pull-down
the antibody/chromatin complex at 4°C with rotation for 2 h.
Chromatin was eluted from the antibody/protein G magnetic beads at
65°C for 30 min and reversed cross-linked with 6 µl 5 M NaCl and 2
µl proteinase K (cat. no. 10012) at 65°C for 2 h. DNA was purified
using spin columns (centrifuged at 18,500 × g in a microcentrifuge
for 30 sec.) and quantified by PCR. A master reaction mix was
prepared including 12.5 µl nuclease-free H2O, 2.0 µl 10X
PCR buffer, 1.0 µl 4 mM dNTP Mix, 2.0 µl 5 µM primers, 0.5 µl Taq
DNA Polymerase (cat. no. EP0404; Thermo Fisher Scientific, Inc.).
The following PCR reaction program was used: Initial denaturation,
95°C 5 min; denaturation, 95°C 30 sec; annealing, 62°C 30 sec;
extension, 72°C 30 sec; denaturation, annealing and extension were
repeated for a total of 34 cycles; final extension of 72°C 5 min.
PCR products (10 µl) were resolved on 2% agarose gels and ethidium
bromide was used to visualize DNA. Six pairs of Snail primers were
used (P2 forward, 5′-GGAAGCCAGCGTGAAAGATC-3′ and reverse,
5′-TCCTCTCCTCAGCCAACTCG-3′; P3 forward, 5′-GAGAGGACTTTGGCTTTTAC-3′
and reverse, 5′-TCATTAAGCGGAATACTCCC-3′; P4 forward,
5′-GTATTCCGCTTAATGACTGC-3′ and reverse, 5′-AAAACCTATAAGCACCCCAC-3′;
P5 forward, 5′-GTGGTGTGGGGTGCTTATAG-3′ and reverse,
5′-CTGTAACACGGCTCCATAGG-3′; P6 forward, 5′-AGCCGTGTTACAGCCTTTAG-3′
and reverse, 5′-CGTAGGAGTTTGGACTTTGC-3′; P7 forward,
5′-AAAGTCCAAACTCCTACGAG-3′ and reverse,
5′-ATTATCAAGGGAAAAGGCCC-3′).
Knockout of Snail by clustered
regularly interspaced short palindromic repeats (Crispr)/Cas9
Single guide RNA (sgRNA) was designed using an
online Optimized Crispr Design Tool (genome-engineering.org). The Snail gene sequence was
downloaded from Ensemble Genome Browser (ensembl.org). The 25 nucleotide sequences followed by
protospacer adjacent motif sequence were designed as sgRNA (sense,
5′-CACCGTGTAGTTAGGCTTCCGATTG-3′ and antisense,
5′-AAACCAATCGGAAGCCTAACTACAC-3′). The sequence of NC sgRNA was as
follows: 5′-GTATTACTGATATTGGTGGG-3′. Following phosphorylation and
annealing (37°C for 30 min, 95°C for 5 min, and then ramping down
to 25°C at 5°C/min), the sgRNA oligos were cloned into a
BbsI-digested pX330 vector in a thermocycler (37°C for 5
min, 23°C for 5 min for a total of six cycles and then held at 4°C
hold until analysis). Then, 1–2 µl of the final product was
transformed into DH5α competent cells. Colonies were selected and
verified by sequencing performed by Sangon Biotech Co., Ltd.
(Shanghai, China). Cells (5×104) were seeded into 24
well plates. DNA (1 µg) and 2 µl Lipofectamine® 3000
(cat. no. L3000001; Thermo Fisher Scientific, Inc.) were added to
cells for 24 h.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA).
Three independent experiments were performed. The data of western
blot analyses were quantified using Quantity One®
software 4.6.6 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Student's t-test was used to compare data the difference between
two groups. One-way analysis of variance multiple comparison with
Tukey's post hoc test was used for multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
SETDB1 promotes breast cancer cell
growth in vitro
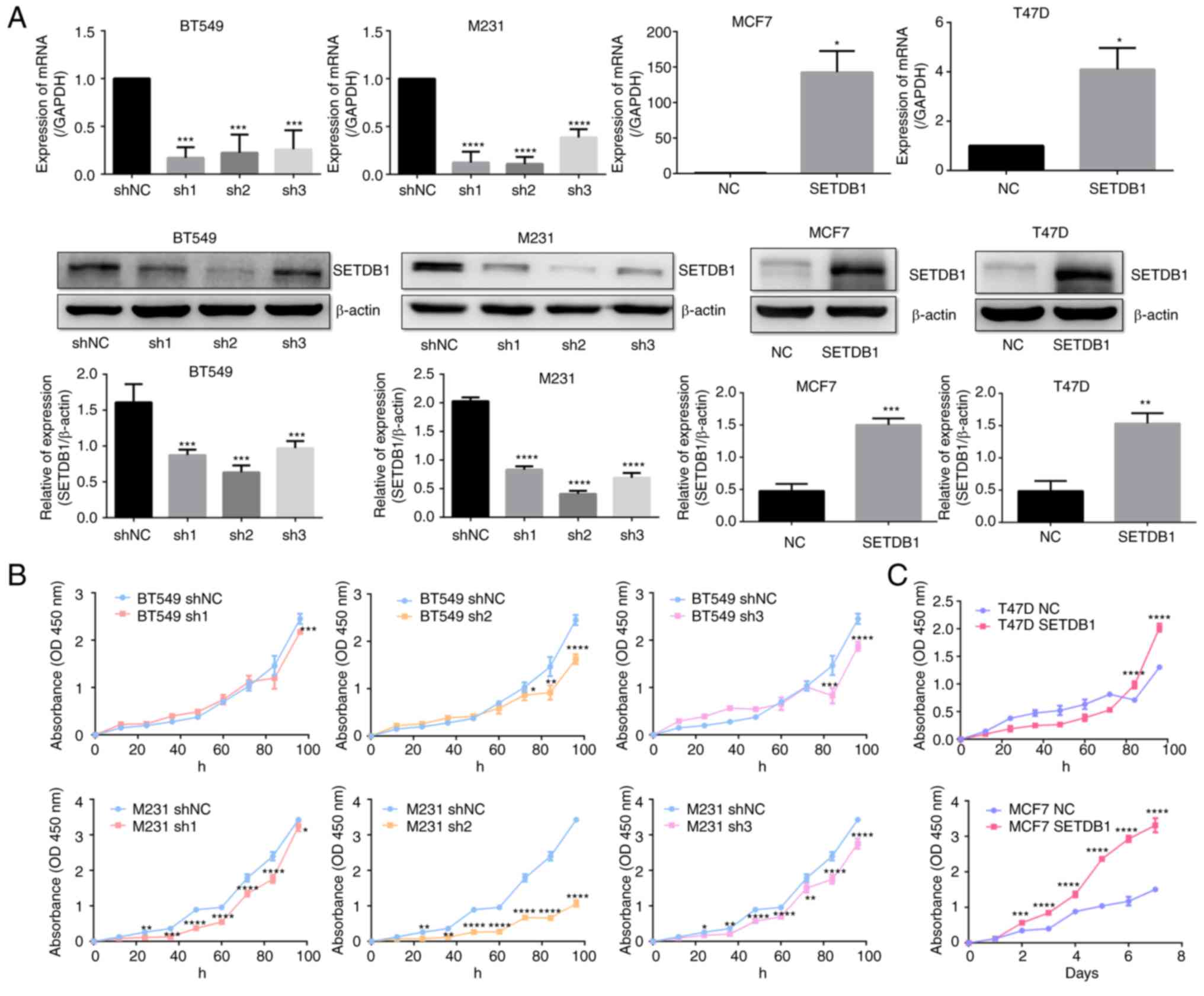
SETDB1 was overexpressed in the MCF7 and T47D cells,
and knocked down in BT549 and MDA-MB-231 cells via
lentiviral-vector infections. Transfection efficiency was validated
by RT-qPCR and western blot analysis (Fig. 1A). Proliferation assays revealed
that knocking down SETDB1 expression resulted in a decrease in the
proliferation of BT549 cells at 84 h and at 24 h in MDA-MB-231
cells (Fig. 1B). By contrast,
overexpression of SETDB1 increased proliferation of T47D cells
after 84 h and MCF7 cells after 2 days (Fig. 1C).
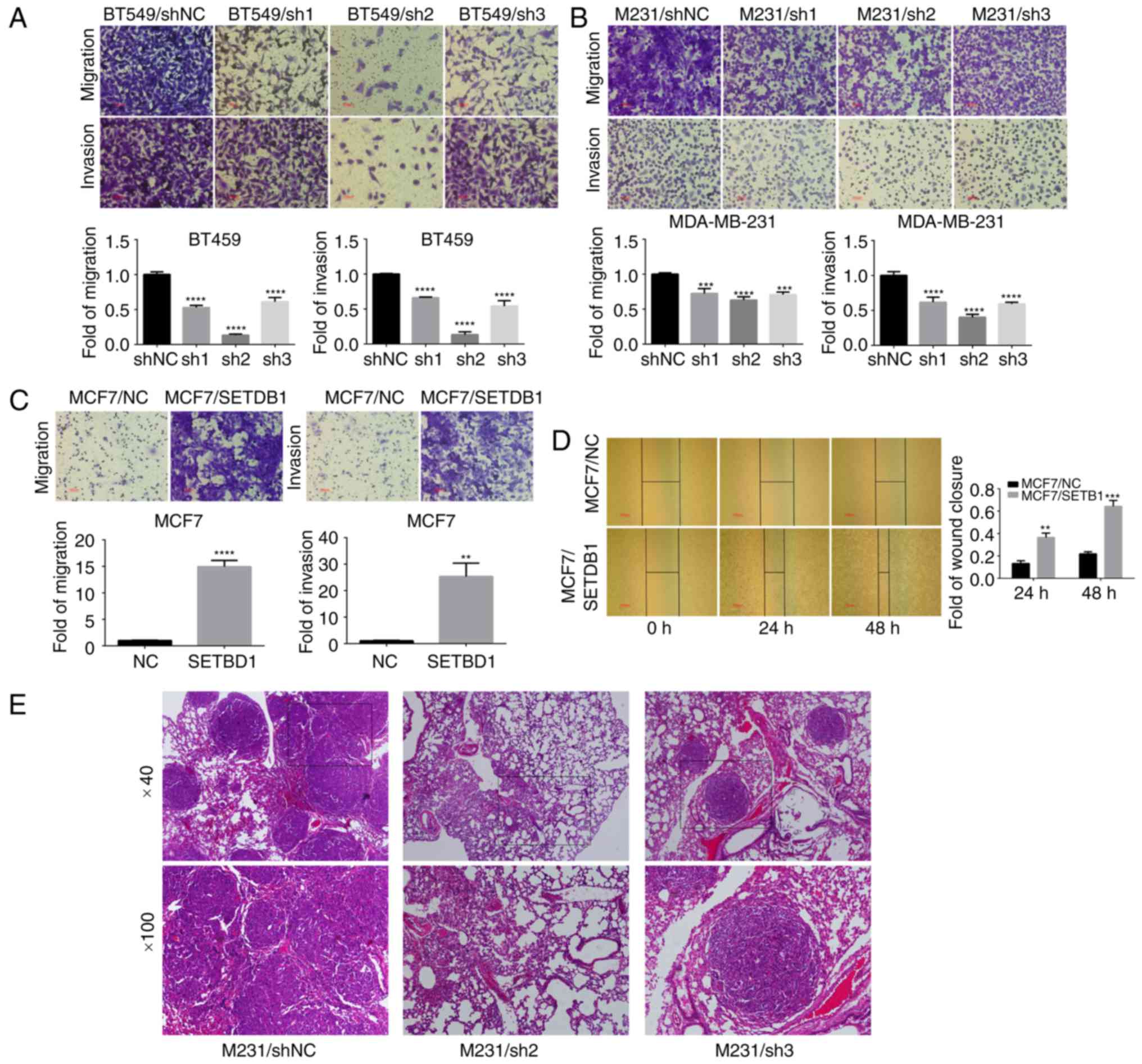
SETDB1 downregulation inhibited breast
cancer cell migration and invasion in vitro as well as metastasis
in vivo
Transwell assay results revealed that downregulation
of SETDB1 in BT549 (Fig. 2A) and
MDA-MB-231 (Fig. 2B) cells
significantly decreased migration and invasiveness, particularly in
breast cancer cells transfected with lentiviral containing the sh2
sequence compared with the negative control. Ectopic expression of
SETDB1 facilitated migration and invasiveness in MCF7 cells
(Fig. 2C). A wound-healing assay
revealed that SETDB1 overexpression in MCF7 cells significantly
increased cell migration compared with controls (Fig. 2D). In MDA-MB-231-treated
immunodeficient mice, H&E staining revealed that multiple tumor
nodules were observed in lungs of MDA-MB-231/NC animals (4/4 in
M231 shNC group), and few or no tumors were found in mice injected
with SETDB1-deficient cells (0/4 in M231 sh2 group; 1/4 in M231 sh3
group) (Fig. 2E). No tumor nodules
were observed in other tissues in shNC and shSETDB1 groups.
SETDB1 acts as an EMT inducer by
activating Snail expression
MDA-MB-231 cell morphology changed from long spindle
shapes to round shapes following knockdown of SETDB1 expression
(Fig. 3A). Thus, SETDB1 may be
involved EMT in breast cancer. In SETDB1-overexpressing MCF7 cells,
there was no difference between MCF7/SETDB1 and MCF7/NC cells after
puromycin treatment for 2 weeks. However, most MCF7/SETDB1 cells
changed from epithelial to mesenchymal phenotypes on the 26th day
(Fig. 3A). Immunoblotting data
revealed that E-cadherin and β-catenin protein was significantly
decreased in MCF7/SEDB1 cells on the 26th day after infection and
selection, with no difference was observed on the 14th day. By
contrast, expression of vimentin was significantly increased in
MCF7/SETDB1 cells on the 26th and 14th days (Fig. 3B). Thus, SETDB1 overexpression
induced EMT in breast cancer cells. SETDB1 RNA and protein levels
decreased after EMT formation on the 26th day compared with the
14th day in the MCF/SETDB1 cells (Fig.
3C). To further address the mechanism of how SETDB1 induces EMT
in breast cancer, RT-qPCR to detect the RNA level of EMT markers,
including TGF1, ZEB1, Snail, Slug and ZO-1. Slug expression was
significantly increased following SETDB1 overexpression in MCF7
cells (Fig. 3D). Western blot
analysis revealed no significant difference in Slug protein
expression on the 26 and 14th days, whereas Snail protein was
significantly increased in MCF7/SETDB1 cells on the 26th day
(Fig. 3E).
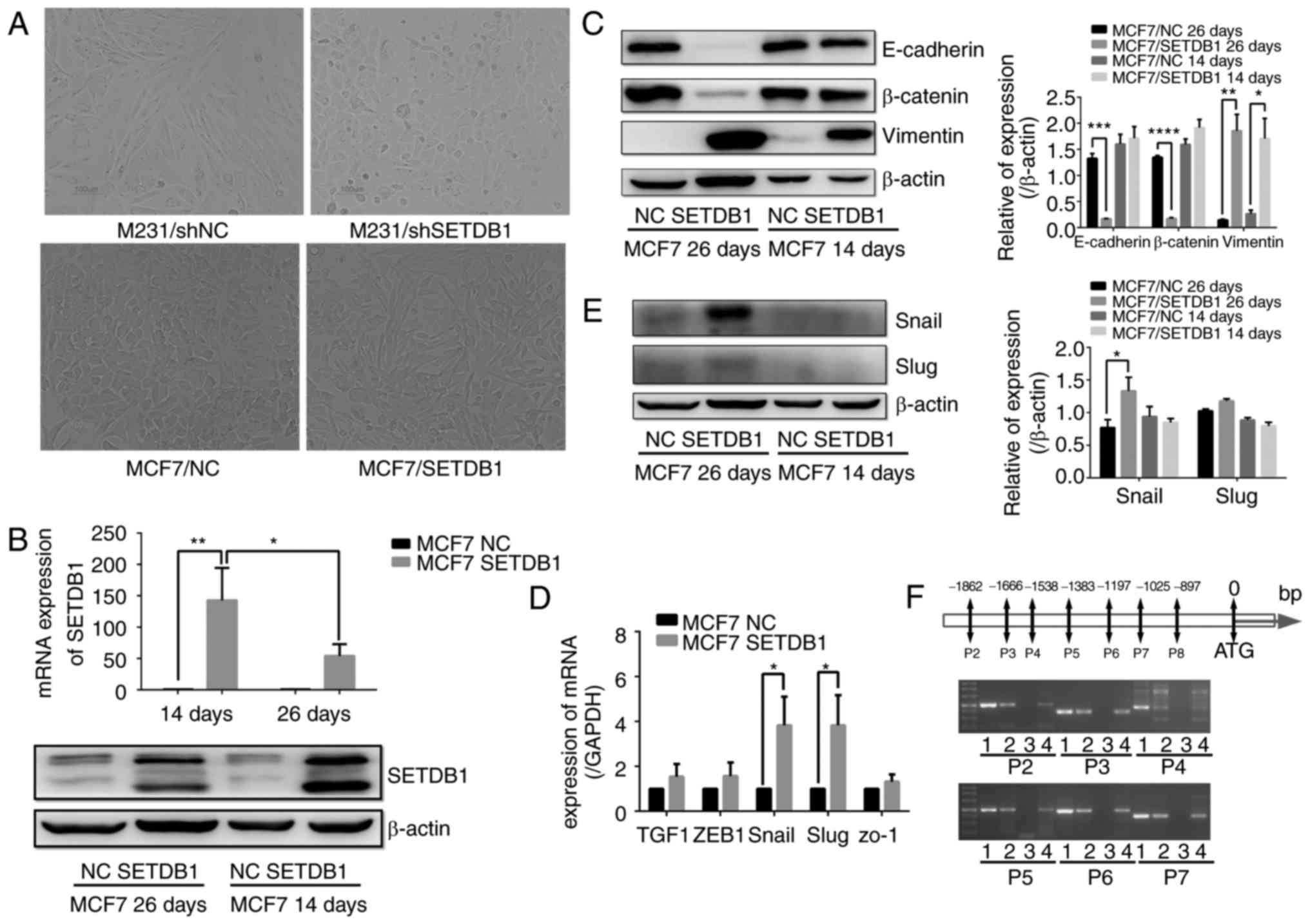 | Figure 3.SETDB1 induces EMT by increasing
Snail expression. (A) Alterations in the morphology of the
MDA-MB-231 at day 14 and MCF7 cells day 26 after puromycin
selection were observed following knockdown and overexpression of
SETDB1; magnification, ×200. (B) Immunoblot of decreased E-cadherin
and β-catenin protein in SETDB1-overexpressing MCF7 cells after 26
days of puromycin selection, but no difference at 14 days. Vimentin
expression was significantly increased in MCF7/SETDB1 cells at 26
and 14 days after puromycin selection. (C) SETDB1 RNA (upper) and
protein levels (lower) decreased at 26 days compared to 14 days.
(D) Reverse transcription-quantitative polymerase chain reaction
validation expression of genes associated with EMT including TCF1,
ZEB1, Snail, Slug and ZO-1 after SETDB1 upregulation in MCF7 cells;
Slug expression was increased in MCF7/SETDB1 cells. (E) Snail
protein was significantly increased in MCF7/SETDB1 cells at day 26
but not day 14. Slug protein showed no difference on either day.
Data are presented as the mean ± standard deviation and represent
three independent experiments. (F) An enrichment of fragments
hybridized with the CHIP-grade SETDB1 antibody was observed in the
Snail promoter regions of P2, P3, P4, P5, P6 and P7 in groups 1
(input group), 2 (H3 group), 3 (IgG group) and 4 (SETDB1 group).
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. NC.
EMT, epithelial-mesenchymal transition; sh, short hairpin RNA; NC,
negative control; SETDB1, SET domain bifurcated 1; TGF1,
transforming growth factor 1; ZEB1, zinc finger E-box binding
homeobox 1; ZO-1, tight junction protein 1. |
Subsequently, CHIP was performed to investigate
whether SETDB1 regulated the Snail gene by directly binding to its
promoter region. The nucleotide segments from 0–2,000 bp downstream
from the initiation start site (ATG) were divided into 13 regions.
An enrichment of fragments that hybridized with the CHIP-grade
SETDB1 antibody was observed in the promoter regions of P2, P3, P4,
P5, P6 and P7 (Fig. 3F). Thus,
SETDB1 may be involved in EMT by upregulating the expression of
Snail transcription factor.
Knockout of Snail with Crispr-Cas9
partly reversed SETDB1-induced EMT
Following Crispr-Cas9 knockout of Snail expression
in SETDB1-overexpressing MCF7 cells, MCF7/SETDB1 cell morphology
changed to an epithelial phenotype following Snail knockout,
whereas this did not occur in the Crispr-NC group (Fig. 4A). Thus, depletion of Snail may
reverse EMT in SETDB1-overexpressing MCF7 cells. Cell migration and
invasiveness was significantly decreased in MCF7/SETDB1 +
Crispr-Snail cells compared with MCF7/SETDB1 and MCF7/SETDB1 +
Crispr-NC cells (Fig. 4B).
Immunoblotting results revealed that E-cadherin and β-catenin
protein were significantly increased following knockout of Snail
expression, and vimentin was decreased in MCF7/SETDB1 +
Crispr-Snail cells (Fig. 4C). These
results highlight a critical role of Snail in SETDB1-induced EMT of
breast cancer cells. However, compared to MCF7/NC group cells, the
expression of E-cadherin and β-catenin was still significantly
decreased and vimentin was increased in MCF7/SETDB1 + Crispr-Snail
cells (Fig. 4C).
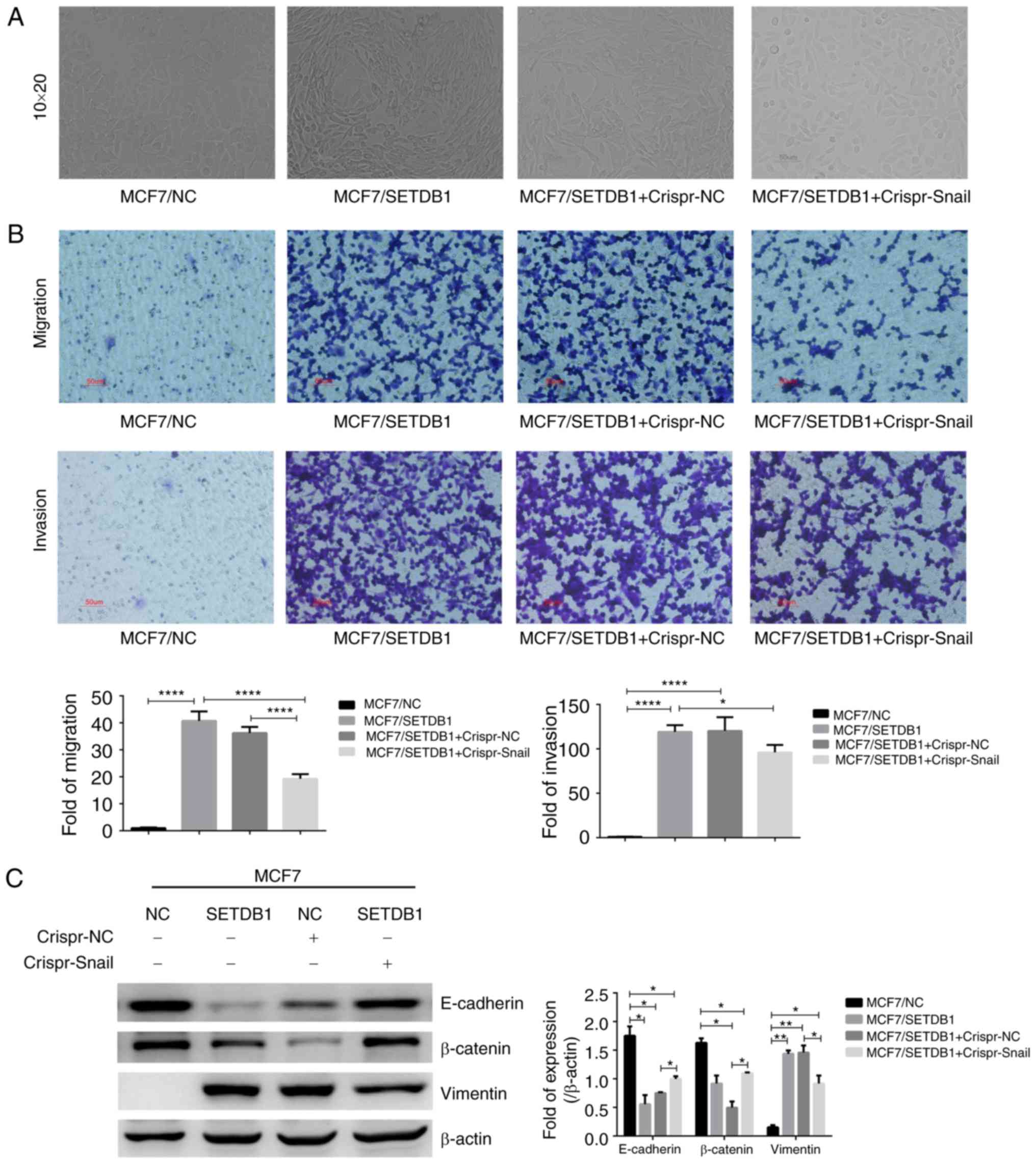 | Figure 4.Knockout of Snail via Crispr-Cas9
partially reversed SETDB1-induced EMT. (A) Morphology of
MCF7/SETDB1 cells changed from mesenchymal to epithelial phenotypes
following Snail knockdown; ×200. (B) Compared with the MCF7/NC
group, the migration and invasion significantly increased in the
MCF7/SETDB1 group (****P<0.0001). However, depletion of Snail by
Crispr-Cas9 decreased migration (****P<0.0001) and invasion
(*P<0.05) of MCF7/SETDB1 cells. The red scale bar shows 50 µm.
Magnification, ×200. (C) Immunoblot of EMT markers E-cadherin,
β-catenin and vimentin protein in SETDB1-overexpressing and
Snail-Crispr knockdown MCF7 cells. Data presented as the mean ±
standard deviation and represent three independent experiments.
*P<0.05, **P<0.01, ****P<0.0001. EMT,
epithelial-mesenchymal transition; NC, negative control; SETDB1,
SET domain bifurcated 1; Crispr, clustered regularly interspaced
short palindromic repeats. |
Discussion
The SETDB1 gene is located at human chromosome 1q21,
and many recurrent translocations occur in this region (5). The gene encodes SETDB1 protein, which
contains an unusual bifurcated SET domain, suggesting that SET
domains may have two distinct functional domains (5). SETDB1, which acts as a histone lysine
methyltransferase (HMT), is involved in transcriptional repression
and embryonic development (8–15).
SETDB1 was demonstrated to be an important oncogene in a zebrafish
melanoma model (16). Other studies
suggest that SETDB1 overexpression increases cell growth and
migration in gliomagenesis and prostate cancer (17,18).
Notably, studies have indicated that SETDB1 was amplified in lung
cancer, and enhanced cell growth via the WNT signaling pathway and
downregulated p53 expression (19,26).
However, SETDB1 has been reported to interact with the mothers
against decapentaplegic homolog 2/3 (Smad2/3) repressor complex on
the Annexin A2 promoter to suppress lung cancer metastasis
(27). Thus, further investigation
is required to uncover the function and mechanism underlying the
role of SETDB1 in cancer.
According to a meta-analysis, five HMTs, including
SETDB1, were amplified in >10% of breast cancer samples,
particularly for triple-negative breast cancer (28). In the present study, SETDB1
expression was detected in breast cancer cell lines, and
demonstrated that SETDB1 protein and mRNA were overexpressed in the
MCF7 and T47D cells compared to the immortalized mammalian
epithelial cell line MCF10A (data not shown). However, compared to
its upregulation in MCF7 and T47D, which have low invasion
capability, the high level of invasiveness of the breast cancer
cells showed lower levels of SETDB1 expression (data not shown),
which suggests that SETDB1 may play different roles in the
development of breast cancer via multiple mechanisms.
Downregulation of SETDB1 in the BT549 and MDA-MB-231 cells reduced
cell proliferation, whereas SETDB1 upregulation in MCF7 and T47D
cells enhanced proliferation. Recently, Regina et al
(29) reported that SETDB1 is
overexpressed in breast cancer cell lines and enhanced tumor cell
growth, which is consistent with the findings of the current study.
Additionally, depletion of SETDB1 suppressed cell migration and
invasion in vitro and reduced lung metastasis in
vivo. By contrast, SETDB1 overexpression enhanced cell
migration and invasiveness. In a nude mice model, SETDB1
downregulation significantly suppressed the distant metastasis
ability of breast cancer cell in lungs. Thus, SETDB1 has an
oncogenic role in breast cancer, but how it causes metastasis is
unclear. A previous study reported that microRNA-7 directly
inhibits SETDB1 expression, which leads to a reduction in STAT3
expression and partially reverses EMT in breast cancer stem cells
(30). Additionally, SETDB1 is a
novel interactor of ΔNp63 and contributes to stable p63 protein in
breast cancer (29).
SETDB1 was identified as an EMT inducer in breast
cancer cells. EMT is a process by which cells acquire molecular
alterations that facilitate dysfunctional cell-cell adhesive
interactions and junctions, and a more spindle-shaped morphology
(31). SETDB1 knockdown lead to
MDA-MB-231 cell morphological changes from long spindle to round
shapes. Furthermore, the morphology of SETDB1-overexpression MCF7
cells changed from epithelial to a mesenchymal phenotype following
lentiviral-vector infection for 26 days. Additionally, Transwell
assay and wound healing assay were performed to detect cell
migration and invasion, and protein and RNA were extracted to
detect the expression of EMT-associated markers. Immunoblotting
revealed that epithelial markers, E-cadherin and β-catenin, were
significantly decreased in MCF7/SEDB1 cells, and mesenchymal
marker, vimentin, was significantly increased. Notably, vimentin
expression was significantly increased following EMT (day 26), and
also at the beginning of the transition (day 14). It is speculated
that SETDB1 increased the abilities of migration and invasion
mainly via increasing vimentin expression in MCF7 cells. Taken
together, these findings demonstrate that SETDB1 is involved in the
EMT process in breast cancer. However, changes in the SETDB1
expression in MCF7 cells were not detected until the 26th day after
infection with vectors. This finding suggests that the effects of
SETDB1 overexpression on inducement of EMT in breast cancer involve
a long process. EMT is induced by various signaling pathway such as
TGF-β (32), bone morphogenetic
protein (33), Hedgehog,
WNT-β-catenin and Notch signaling (34). These signaling pathways induce
multiple transcription factors during EMT. For instance, ZEB1,
ZEB2, and Snail, are induced by TGF-β-signaling and are critical
for TGF-β-induced EMT (35).
Expression of Snail was increased following SETDB1 overexpression
in MCF7 cells in the current study. Snail family proteins are zinc
finger transcription regulators and bind to the E-box motif via
conserved C2H2 domains (36).
Additionally, Snail is a strong transcriptional repressor of the
E-cadherin gene and induces epithelial-mesenchymal conversion in
epithelial cells (37). A previous
study reported that zinc finger protein 274, which contains five
C2H2 domain binding sites, co-localizes with SETDB1, KAP1 and
H3K9me3 at the 3′-ends of zinc finger genes (38). To investigate the interaction
between SETDB1 and Snail, CHIP was performed and demonstrated that
SETDB1 binds to the promoter of Snail. Furthermore, knocking down
Snail expression with Crispr-Cas9 partially reversed SETDB1-induced
EMT. Unexpectedly, the expression of E-cadherin and β-catenin were
different between the MCF7/SETDB1 and MCF7/SETDB1-Crispr-NC groups.
It was speculated that Cripsr vector infection had a marginal
effect on the state of cells, which may contributes to the
differences. However, compared with the MCF7/NC group, E-cadherin
and β-catenin protein levels were still significantly decreased. As
SETDB1 was stably expressed in MCF7 cells, there were no off target
effects caused by the Crispr control vector. Notably, the
expression of EMT-associated markers remained significantly
different in MCF7/SETDB1 + Crispr-Snail cells compared with the
MCF7/NC group cells. These findings indicated that SETDB1 induces
EMT via multiple pathways, not only by regulating Snail. Recently,
Du et al (39) reported that
SETDB1 is downregulated in a TGF-β-induced EMT model and its
silencing enhances EMT by repressing Snail. Notably, SETDB1 was
detected expression in the MCF7/SETDB1 cells on the 26th and 14th
days after infection with overexpression vectors, SETDB1 expression
was also decreased following EMT (day 26 compared with day 14).
Thus, when the phenotype of epithelial cells changes to
mesenchymal, SETDB1 is potentially negatively regulated by another
mechanism that leads to reduced SETDB1 expression, and differs from
that observed during early and aggressive breast cancer.
Alternatively, SETDB1 may have different roles in various stages of
breast cancer, which may be associated its unusual bifurcated SET
domain. In summary, SETDB1 is required for breast cancer cell
proliferation and invasion. Additional investigations on the
functional and pathological roles of SETDB1 in breast cancer are
required.
Acknowledgements
Not applicable.
Funding
The present study was fund by The National Natural
Science Foundation of China (grant nos. 81772796 and 81470857) and
PhD Research Foundation Affiliated Hospital of Jining Medical
University (grant no. 2017-BS-022).
Availability of data and materials
The dataset generated or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY conceived and designed the study, analysed and
interpreted the data and drafted the manuscript. YS and CH
performed cell culture and lentiviral infection, and proliferation,
migration, invasion and wound healing assay. LC performed western
blot analyses and RT-PCR. DZ performed CHIP assay and standard PCR.
KR contributed to Crispr-Cas9 construction and western blot
analysis. ZZ contributed to animal experiment and H&E staining.
RZ and XL reviewed the manuscript, supervised the study and were
also involved in the conception of the study. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Animal experiments were approved by the Ethics
Committee of Fudan University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benson JR and Jatoi I: The global breast
cancer burden. Future Oncol. 8:697–702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elsheikh SE, Green AR, Rakha EA, Powe DG,
Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, et
al: Global histone modifications in breast cancer correlate with
tumor phenotypes, prognostic factors, and patient outcome. Cancer
Res. 69:3802–3809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Albert M and Helin K: Histone
methyltransferases in cancer. Semin Cell Dev Biol. 21:209–220.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harte PJ, Wu W, Carrasquillo MM and Matera
AG: Assignment of a novel bifurcated SET domain gene, SETDB1, to
human chromosome band 1q21 by in situ hybridization and radiation
hybrids. Cytogenet Cell Genet. 84:83–86. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schultz DC, Ayyanathan K, Negorev D, Maul
GG and Rauscher FJ III: SETDB1: A novel KAP-1-associated histone
H3, lysine 9-specific methyltransferase that contributes to
HP1-mediated silencing of euchromatic genes by KRAB zinc-finger
proteins. Genes Dev. 16:919–932. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verschure PJ, van der Kraan I, de Leeuw W,
van der Vlag J, Carpenter AE, Belmont AS and van Driel R: In vivo
HP1 targeting causes large-scale chromatin condensation and
enhanced histone lysine methylation. Mol Cell Biol. 25:4552–4564.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang H, An W, Cao R, Xia L,
Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG and Zhang Y:
mAM facilitates conversion by ESET of dimethyl to trimethyl lysine
9 of histone H3 to cause transcriptional repression. Mol Cell.
12:475–487. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarraf SA and Stancheva I: Methyl-CpG
binding protein MBD1 couples histone H3 methylation at lysine 9 by
SETDB1 to DNA replication and chromatin assembly. Mol Cell.
15:595–605. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ichimura T, Watanabe S, Sakamoto Y, Aoto
T, Fujita N and Nakao M: Transcriptional repression and
heterochromatin formation by MBD1 and MCAF/AM family proteins. J
Biol Chem. 280:13928–13935. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bilodeau S, Kagey MH, Frampton GM, Rahl PB
and Young RA: SetDB1 contributes to repression of genes encoding
developmental regulators and maintenance of ES cell state. Genes
Dev. 23:2484–2489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu S, Brind'Amour J, Karimi MM, Shirane
K, Bogutz A, Lefebvre L, Sasaki H, Shinkai Y and Lorincz MC:
Setdb1 is required for germline development and silencing of
H3K9me3-marked endogenous retroviruses in primordial germ cells.
Genes Dev. 29:1082015.
|
|
13
|
Koide S, Oshima M, Takubo K, Yamazaki S,
Nitta E, Saraya A, Aoyama K, Kato Y, Miyagi S, Nakajima-Takagi Y,
et al: Setdb1 maintains hematopoietic stem and progenitor cells by
restricting the ectopic activation of nonhematopoietic genes.
Blood. 128:638–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thompson PJ, Dulberg V, Moon KM, Foster
LJ, Chen C, Karimi MM and Lorincz MC: hnRNP K coordinates
transcriptional silencing by SETDB1 in embryonic stem cells. PLoS
Genet. 11:e10049332015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fei Q, Yang X, Jiang H, Wang Q, Yu Y, Yu
Y, Yi W, Zhou S, Chen T, Lu C, et al: SETDB1 modulates PRC2
activity at developmental genes independently of H3K9
trimethylation in mouse ES cells. Genome Res. 25:1325–1335. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ceol CJ, Houvras Y, Jane-Valbuena J,
Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ,
Ferré F, et al: The histone methyltransferase SETDB1 is recurrently
amplified in melanoma and accelerates its onset. Nature.
471:513–517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spyropoulou A, Gargalionis A, Dalagiorgou
G, Adamopoulos C, Papavassiliou KA, Lea RW, Piperi C and
Papavassiliou AG: Role of histone lysine methyltransferases SUV39H1
and SETDB1 in gliomagenesis: Modulation of cell proliferation,
migration, and colony formation. Neuromolecular Med. 16:70–82.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun Y, Wei M, Ren SC, Chen R, Xu WD, Wang
FB, Lu J, Shen J, Yu YW, Hou JG, et al: Histone methyltransferase
SETDB1 is required for prostate cancer cell proliferation,
migration and invasion. Asian J Androl. 16:319–324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rodriguez-Paredes M, Martinez de Paz A,
Simó-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A,
Vázquez-Cedeira M, Lazo PA, Carneiro F, et al: Gene amplification
of the histone methyltransferase SETDB1 contributes to human lung
tumorigenesis. Oncogene. 33:2807–2813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen K, Zhang F, Ding J, Liang Y, Zhan Z,
Zhan Y, Chen LH and Ding Y: Histone methyltransferase SETDB1
promotes the progression of colorectal cancer by inhibiting the
expression of TP53. J Cancer. 8:3318–3330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ho Y, Lin YM, Huang YC, Chang J, Yeh KT,
Lin LI, Gong Z, Tzeng TY and Lu JW: Significance of histone
methyltransferase SETDB1 expression in colon adenocarcinoma. APMIS.
125:985–995. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Noh HJ, Kim KA and Kim KC: p53
Down-regulates SETDB1 gene expression during paclitaxel
induced-cell death. Biochem Biophys Res Commun. 446:43–48. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cicchini C, Battistelli C and Tripodi M:
SETDB1 is a new promising target in HCC therapy. Chin Clin Oncol.
5:732016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karanth AV, Maniswami RR, Prashanth S,
Govindaraj H, Padmavathy R, Jegatheesan SK, Mullangi R and
Rajagopal S: Emerging role of SETDB1 as a therapeutic target.
Expert Opin Ther Targets. 21:319–331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang W, Wang JG, Xu J, Zhou D, Ren K, Hou
C, Chen L and Liu X: HCRP1 inhibits TGF-β induced
epithelial-mesenchymal transition in hepatocellular carcinoma. Int
J Oncol. Mar 8–2017.(Epub ahead of print). doi:
10.3892/ijo.2017.3903. View Article : Google Scholar
|
|
26
|
Sun QY, Ding LW, Xiao JF, Chien W, Lim SL,
Hattori N, Goodglick L, Chia D, Mah V, Alavi M, et al: SETDB1
accelerates tumourigenesis by regulating the WNT signalling
pathway. J Pathol. 235:559–570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu PC, Lu JW, Yang JY, Lin IH, Ou DL, Lin
YH, Chou KH, Huang WF, Wang WP, Huang YL, et al: H3K9 histone
methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3
pathway to suppress lung cancer metastasis. Cancer Res.
74:7333–7343. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu L, Kimball S, Liu H, Holowatyj A and
Yang ZQ: Genetic alterations of histone lysine methyltransferases
and their significance in breast cancer. Oncotarget. 6:2466–2482.
2015.PubMed/NCBI
|
|
29
|
Regina C, Compagnone M, Peschiaroli A,
Lena A, Annicchiarico-Petruzzelli M, Piro MC, Melino G and Candi E:
Setdb1, a novel interactor of ΔNp63, is involved in breast
tumorigenesis. Oncotarget. 7:28836–28848. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Cai K, Wang J, Wang X, Cheng K,
Shi F, Jiang L, Zhang Y and Dou J: MiR-7, inhibited indirectly by
lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of
breast cancer stem cells by downregulating the STAT3 pathway. Stem
Cells. 32:2858–2868. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Creighton CJ, Chang JC and Rosen JM:
Epithelial-mesenchymal transition (EMT) in tumor-initiating cells
and its clinical implications in breast cancer. J Mammary Gland
Biol Neoplasia. 15:253–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kahata K, Dadras MS and Moustakas A: TGF-β
family signaling in epithelial differentiation and
epithelial-mesenchymal transition. Cold Spring Harb Perspect Biol.
10:a0221942018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McCormack N and O'Dea S: Regulation of
epithelial to mesenchymal transition by bone morphogenetic
proteins. Cell Signal. 25:2856–2862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zardawi SJ, O'Toole SA, Sutherland RL and
Musgrove EA: Dysregulation of Hedgehog, Wnt and Notch signalling
pathways in breast cancer. Histol Histopathol. 24:385–398.
2009.PubMed/NCBI
|
|
35
|
Miyazono K: Transforming growth
factor-beta signaling in epithelial-mesenchymal transition and
progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci.
85:314–323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kataoka H, Murayama T, Yokode M, Mori S,
Sano H, Ozaki H, Yokota Y, Nishikawa S and Kita T: A novel
snail-related transcription factor Smuc regulates basic
helix-loop-helix transcription factor activities via specific E-box
motifs. Nucleic Acids Res. 28:626–633. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frietze S, O'Geen H, Blahnik KR, Jin VX
and Farnham PJ: ZNF274 recruits the histone methyltransferase
SETDB1 to the 3′ ends of ZNF genes. PLoS One. 5:e150822010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Du D, Katsuno Y, Meyer D, Budi EH, Chen
SH, Koeppen H, Wang H, Akhurst RJ and Derynck R: Smad3-mediated
recruitment of the methyltransferase SETDB1/ESET controls Snail1
expression and epithelial-mesenchymal transition. EMBO Rep.
19:135–155. 2018. View Article : Google Scholar : PubMed/NCBI
|