Introduction
Gastric cancer is often diagnosed at an advanced
stage and it commonly recurs even after complete resection. Thus,
chemotherapy is required for the treatment of unresectable cases
and prevention of cancer recurrence. However, consistent exposure
to chemotherapeutics often results in drug resistance, which is a
serious impediment to cancer treatment. Hence, research has been
conducted to reveal the mechanism of cancer chemoresistance to
overcome this problem.
The development of drug resistance is a complex
process due to various mechanisms. These include overexpression of
efflux pumps, including ABCC, upregulation of survival pathways,
elevation of the antioxidant system, defects in apoptotic
signaling, and alterations in the dynamics of cytoskeletons
(1–4).
The cytoskeleton is one of the main targets for cancer treatment,
since the cytoskeletal network plays pivotal roles in cytokinesis,
mitosis, membrane transport, and many other processes essential for
rapid growth and metastasis of cancer cells. The roles of actin and
microtubule cytoskeletons are relatively well investigated in
cancer biology, and many efforts have been made to develop
therapeutic strategies targeting them in carcinomas (5–7). Recently,
it was found that certain keratin molecules interact with multiple
factors that are involved in cellular survival processes and are
associated with cancer cell malignancy and resistance to apoptosis.
Several of them are recognized as markers for tumor prognosis as
well as for tumor diagnosis.
The keratin filament is a 10-nm thick intermediate
filament that belongs to the three major cytoplasmic filament
networks, together with microfilaments and microtubules. Keratins
are mainly found in the outer layer of the skin, hair, and nails,
and they provide mechanical strength and protection against various
external stimuli, including heat, UV, septic infection, and
physical stress (8,9). In contrast to microtubules and
microfilaments that are uniform, there are diverse types of keratin
filaments composed of different isoforms of keratin proteins. The
keratin gene family consists of 54 genes in humans, which are
either found as the acidic type I or the basic or neutral type II
keratin genes (28 type I genes, 26 type II genes). Complete keratin
filaments are assembled as heteropolymers by non-covalent
interaction between type I and type II keratin chains, and
expression of keratin proteins is also paired with acidic and basic
keratins in a tissue- and differentiation-specific fashion
(10–13). Since different keratin pairs are
present in different regions or different developmental stages of
epithelia, the regulation of their cell type- and stage-specific
expression pattern may be strictly associated with their own unique
function. Indeed, recently identified results indicate that keratin
filaments are involved in the regulation of various cellular
functions including cell survival, differentiation and
transformation, by influencing intracellular signaling. For
instance, K8 expression correlates with c-FLIP level and ERK1/2
signaling in epithelial cells and protects cells from death
ligand-induced apoptosis (14).
K8/K18 influences insulin receptor signaling via modulation of
phosphoinositide-dependent Akt and Rab5 signaling in hepatocytes
(15), and is also implicated in
colonic epithelial cell differentiation through regulation of the
Notch1 signaling (16). K5/K14 is
involved in the mediation of Tap63 and Notch-1 signaling during
cell transformation (17).
Furthermore, elevated expression of certain keratins, such as K8,
K17, K18 and K20 is shown to be linked to malignant transformation
and unfavorable prognosis of various types of cancer (18). In addition, primary breast cancer
exhibits an altered expression profile of keratin during metastatic
progression, and K81, known as hair keratin, which is not found in
normal epithelial cells, is expressed in breast cancer cells, where
it was found to be involved in metastasis. In this study, it was
demonstrated that keratin 6 (K6) is extremely overexpressed in
cisplatin-resistant variants of human gastric cancer SNU601 cells,
and we investigated the role of K6 in drug responsiveness.
Materials and methods
Cell culture and drug treatment
Human gastric cancer SNU-601 cells were obtained
from the Korean Cell Line Bank (Seoul, Korea), and the
cisplatin-resistant sublines (SNU601-cis2 and SNU601-cis10),
selected by gradually increasing cisplatin concentrations from 2 to
10 µg/ml, were a gift from Professor C. H. Choi of the Department
of Pharmacology, Chosun University, Korea (19). The cells were cultured in RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented
with 10% (v/v) fetal bovine serum and 1% penicillin-streptomycin at
37°C in an atmosphere containing 5% CO2. Oxaliplatin
(L-OHP) was obtained from Boryung Pharmaceutical (Seoul, Korea),
and cisplatin (CDDP) was purchased from Sigma-Aldrich; Merck KGaA.
Unless specified otherwise, drugs were purchased from Calbiochem;
EMD/Merck KGaA.
Analysis of apoptosis
Treated cells were stained with 1 µg/ml Hoechst
33342 (HO) for 15 min, and both suspended and attached cells were
collected and centrifuged. The pooled cell pellets were washed and
fixed in 3.7% formaldehyde, and an aliquot of the suspension was
centrifuged at 600 × g for 10 min in a Cytospin centrifuge
(Shandon, Thermo Fisher Scientific, Inc.). Slides were prepared,
air-dried, mounted in anti-fade solution, and observed under a
fluorescence microscope (DM5000, Leica, Germany) using excitation
and emission wavelengths of 340 and 425 nm, respectively. Condensed
or fragmented nuclei were considered indicative of apoptosis. A
total of 500 cells distributed across random microscope fields of
view were counted, and the number of apoptotic and non-apoptotic
cells was expressed as a percentage of the total.
Immunoblotting
Treated cells were lysed in a lysis buffer (50 mM
HEPES, 150 mM NaCl, 1% Triton X-100, 5 mM EGTA, and protease
inhibitor cocktail) and equal amounts of protein extracts were
electrophoretically separated using 10–12% SDS-PAGE and transferred
to a nitrocellulose membrane using a standard technique. Antibodies
were used to probe for K6 (dilution 1:1,000; cat. no. sc-22479;
Santa Cruz Biotechnology), K16 (dilution 1:1,000; cat. no.
sc-53255; Santa Cruz Biotechnology), K17 (dilution 1:500; cat. no.
sc-393002; Santa Cruz Biotechnology), pan-keratin (dilution
1:1,000; cat. no. sc-8018; Santa Cruz Biotechnology), cytochrome
c (dilution 1:500; cat. no. sc-13156; Santa Cruz
Biotechnology), pro-caspase-8 (dilution 1:500; cat. no. sc-73526;
Santa Cruz Biotechnology), caveolin-1 (dilution 1:500; cat. no.
sc-894; Santa Cruz Biotechnology), Fas associated protein with
death domain (FADD) (dilution 1:500; cat. no. sc-13156; Santa Cruz
Biotechnology), β-actin (dilution 1:1,000; cat. no. sc-8432; Santa
Cruz Biotechnology), cleaved caspase-3 (cysteinyl
aspartate-specific protease-3; dilution 1:500; cat. no. 9661; Cell
Signaling Technology) and DR5 (TRAIL-R2; dilution 1:500; cat. no.
2019; ProSci). As second antibodies, anti-rabbit HRP (dilution
1:3,000; cat. no. sc-2030; Santa Cruz Biotechnology), anti-mouse
HRP (dilution 1:3,000; cat. no. sc-2031; Santa Cruz Biotechnology)
and anti-goat HRP (dilution 1:2,000; cat. no. sc-2020; Santa Cruz
Biotechnology) were used. Anti-a-tubulin (dilution 1:2,000; cat.
no. B-5-1-2; Invitrogen; Thermo Fisher Scientific, Inc.) was used
as a loading control. Signals were acquired using an Image Station
4000MM image analyzer (Kodak).
RNA interference (RNAi)
For the RNAi experiment, the small interfering RNA
(siRNA) of K6 #1 was purchased from Bioneer (Daejeon, Korea) and
the siRNA of K6 #2 was purchased from Ambion (Thermo Fisher
Scientific, Inc.). For the control siRNA,
5′-CCUACGCCACCAAUUUCGU(dtdt)-3′ (sense) and
5′-ACGAAAUUGGUGGCGUAGG(dtdt)-3′ (antisense) were used. Cells were
individually transfected with siRNA oligonucleotides using an Amaxa
Transfection System™ (Basel, Switzerland) and grown for 24 h prior
to drug treatment.
Real-time reverse
transcription-polymerase chain reaction (PCR)
Real-time PCR was performed with the Light Cycler
2.0 (Roche, Switzerland) using the Fast Start DNA Master SYBR-Green
I Kit (Roche). For verification of the correct amplification
product, PCR products were analyzed on a 2% agarose gel stained
with ethidium bromide. The sequences of the primers were as
follows: For β-actin, 5′-GACTATGACTTAGTTGCGTTA-3′ and
5′-GCCTTCATACATCTCAAGTTG-3′; for K6A,
5′-TTCAGAACAACTTCCACTTACTTTCC-3′ and
5′-GTCACTTGTGCTTTCATGGATACTG-3′; for K6B, 5′-GTAAAACGACGGCCAGT-3′
and 5′-TAATACGACTCACTATAGG-3′; for K6C, 5′-GTAAAACGACGGCCAGT-3′ and
5′-TAATACGACTCACTATAGG-3′; for K16, 5′-AAGGAGGAGCTGGCCTA-3′ and
5′-CTCTGCCATCTGCTCGTA-3′; and for K17, 5′-CATGCAGGCCTTGGAGATAGA-3′
and 5′-CACGCAGTAGCGGTTCTCTGT-3′. PCR was conducted at 95°C for 10
min, followed by 45 cycles of 95°C for 15 sec, 60°C for 20 sec, and
72°C for 21 sec. Melting curve analysis was performed to confirm
production of a single product. Negative controls without template
were included for each run. Data were analyzed using Light Cycler
software version 4.0 (Roche). Relative gene expression was
calculated by the 2−ΔΔCq method (20).
Confocal microscopy
To detect keratins, cells grown on coverslips were
fixed with 4% paraformaldehyde for 10 min, permeabilized with 2%
Triton X-100 for 10 min, and blocked with 5% BSA/PBS for 30 min.
Then, the coverslips were incubated with primary antibodies and
reacted with fluorescein isothiocyanate (FITC)-conjugated secondary
antibody [anti-mouse FITC (dilution 1:100; cat. no. sc-2010; Santa
Cruz Biotechnology) and anti-goat FITC (dilution 1:100; cat. no.
sc-2024; Santa Cruz Biotechnology)]. To visualize lipid rafts,
cells grown on coverslips were fixed with 1.5% paraformaldehyde for
5 min at −20°C and stained with 10 µg/ml FITC-labeled cholera toxin
B (CTxB). After washing, drying, and mounting, the prepared slides
were observed under a laser-scanning confocal microscope (×600
magnification; FV1000; Olympus Optical Co.). Using an argon laser,
FITC was excited at 488 nm, and the evoked emission was filtered
with a 515 nm band-pass filter.
Lipid raft isolation
Lipid rafts were isolated using sucrose
density-gradient centrifugation. A total of 108 cells
were lysed for 30 min in 1 ml lysis buffer (1% Brij35 in HEPES
buffer; 25 mM HEPES, 1 mM EDTA, and 150 mM NaCl, pH 6.5)
supplemented with a protease inhibitor cocktail and homogenized
with a glass Dounce homogenizer. The homogenates were mixed with 1
ml of 80% sucrose in HEPES buffer and placed at the bottom of a
centrifuge tube. The samples were then overlaid with 6.5 ml of 30%
sucrose and 3 ml of 5% sucrose, and centrifuged at 188,000 x g for
18 h at 4°C. Fractions (1 ml) were collected from the bottom to the
top of the gradient, and rafts were determined by measurement of
the total cholesterol level. Fractions 3 through 5 of the sucrose
gradients were pooled as the raft fraction; the rest was used as
the non-raft fraction.
Stable gene expression
pCMV6-Entry, pCMV6-K6A, and pCMV6-K6B vectors were
purchased from OriGene. These vectors were transfected individually
into SNU601 cells using Amaxa Transfection System™. Stably
transfected cells were selected and maintained in 200 µg/ml G418
containing growth media.
Statistical analysis
All numerical data are presented as the mean ± SE.
All data represent the results of at least three independent
experiments. Student's t-test was used for simple comparison, and
one-way ANOVA with Tukey's test was applied to multiple comparisons
to analyze differences in gene silencing or gene overexpression. A
significant difference was assumed at P<0.05.
Results
K6, K16, and K17 expression is
elevated in the cisplatin- resistant SNU601 cell sublines
It has been reported that human cancer cells that
have developed resistance to cisplatin exhibit cross-resistance to
various anticancer agents (21,22).
Consistent with this result, the cisplatin-resistant variants,
SNU601-cis2 and SNU601-cis10, which were previously developed by
culturing SNU601 human gastric cancer cells in 2 and 10 µM
cisplatin, respectively, demonstrate a strong resistance to a wide
range of cytotoxic agents apart from cisplatin (19). This is probably due to changes in the
expression of a variety of genes involved in multiple pathways
related to cell survival during the process of acquiring
chemoresistance by cancer cells. It has been known that
cytoskeletal protein plays an important role in the survival and
protection of cancer cells against apoptotic stimuli, and has been
used as a target for anticancer treatment for a long time. Thus, we
investigated whether there was a change in the expression profile
of intracellular cytoskeletal filaments in the
chemoresistance-acquired cells. As shown in Fig. 1A, there was no change in the
expression level of tubulin or actin; however, notably, the
expression levels of K6, K16, and K17 in the keratin, constituting
the intermediated filament, were found to be highly increased in
both SNU601-cis2 and SNU601-cis10 resistant cells as detected by
immunoblot assay. However, there was no difference in the detection
of total Ks as recognized by the antibody against pan-K, suggesting
that there was no significant difference in the overall level of
keratin expression. This result was also confirmed by
immunofluorescence assay using confocal microscopy (Fig. 1B). Subsequently, the mRNA levels of
K6A (KRT6A), K6B (KRT6B), K6C (KRT6C), K16 (KRT16)
and K17 (KRT17) and genes were determined by real-time
quantitative PCR. Consistently, mRNA levels of these genes were
highly increased in the SNU601-cis2 and SNU601-cis10 resistant
cells (Fig. 2) indicating that
expression of these proteins is upregulated at the transcriptional
level.
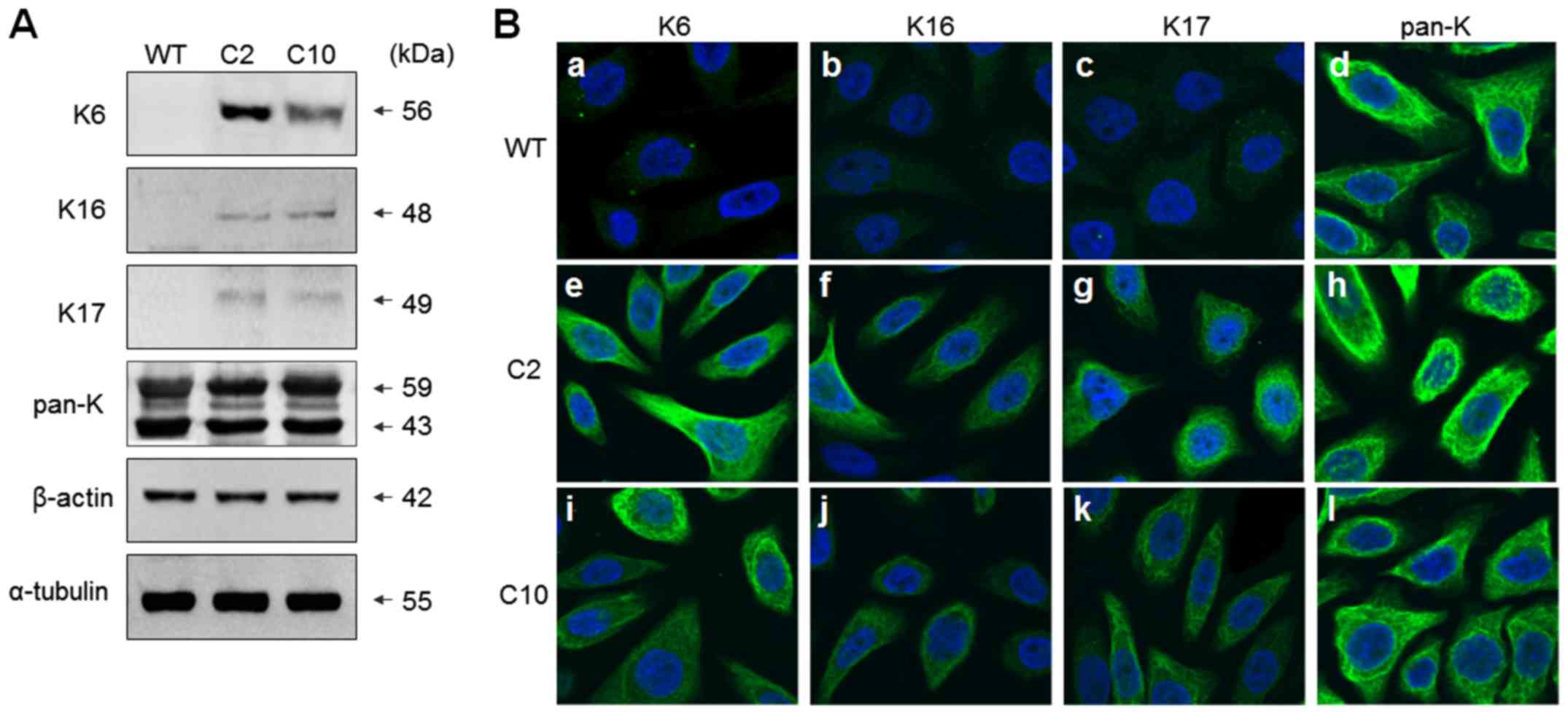 | Figure 1.Expression of K6, K16 and K17 is
increased in the cisplatin-resistant sublines of SNU601 cells. (A)
SNU601 (WT), SNU601-cis2 (C2) and SNU601-cis10 (C10) cells were
lysed, and equal amounts of cell extracts were analyzed by
immunoblotting using antibodies against K6, K16, K17, pan-K,
β-actin, and α-tubulin. (B) SNU601 (WT) (a-d), SNU601-cis2 (C2)
(e-h) and SNU601-cis10 (C10) (i-l) cells were reacted with
antibodies against K6 (a, e and i), K16 (b, f and j), K17 (c, g and
k), pan-K (d, h and l), and stained with FITC-conjugated secondary
antibody. Nuclei of the cells were stained with Hoechst 33342.
Images were captured using a confocal microscope (magnification,
×400; FV1000; Olympus). K, keratin. |
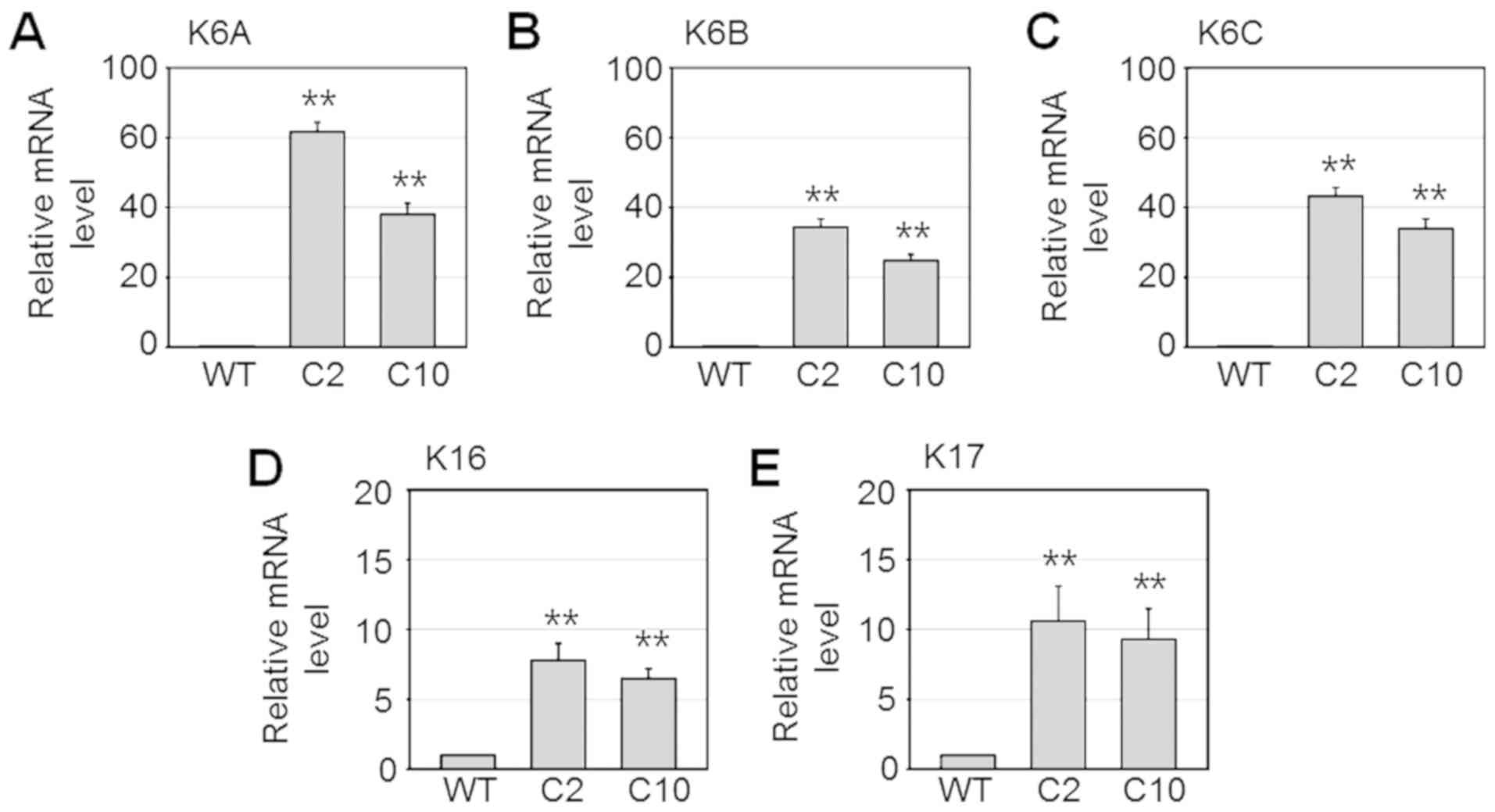 | Figure 2.mRNA levels of K6A, K6B, K6C,
K16, and K17 were upregulated in the cisplatin-resistant
sublines of the SNU601 cells. SNU601 (WT), SNU601-cis2 (C2), and
SNU601-cis10 (C10) cells were harvested and relative mRNA
expression of K6A (A), K6B (B), K6C (C),
K16 (D), and K17 (E) were analyzed by qPCR.
**P<0.001 vs. WT cells. K, keratin. |
Knockdown of K6 partially restores
drug sensitivity in the cisplatin-resistant SNU601 cells
In the present study, we explored the significance
of the marked increase of a particular set of keratins in the
drug-resistant SNU601 sublines. It was initially suspected that
keratin overexpression is likely to contribute to the resistance of
cells to cytotoxic stimuli. Here, we performed the gene
interference study to examine whether the most highly induced
keratin, K6, was associated with the drug-resistant phenotype of
SNU601-cis2 (C2) cells. The cells were transfected with two
different K6-specific siRNAs (K6RNAi #1 and K6RNAi #2) and treated
with L-OHP, and then, apoptotic sensitivity was assessed. The
silencing effect was confirmed by the decreased expression of K6 as
determined by immunoblot assay (Fig.
3A). The silenced cells were treated with L-OHP, a platinum
drug having stronger anticancer effect compared to cisplatin, and
stained with Hoechst 33342. Then, fragmented or condensed apoptotic
bodies were detected under a fluorescence microscope (Fig. 3B). As shown in Fig 3B, treatment with L-OHP slightly
increased the apoptotic body formation in both K6 siRNA transfected
cell lines compared to the control RNA-transfected cells
(C2/CTLRNAi). The apoptotic body counts showed that silencing of K6
using both K6RNAi #1 and K6RNAi #2 significantly increased the
percentages of apoptotic bodies after treatment with 50 and 100 µM
L-OHP, although the percentage of apoptotic bodies induced by L-OHP
was much lower than that in the WT cells (Fig. 3C and D). In agreement with this
result, biochemical evidence also indicated that knockdown of K6
increased L-OHP-induced apoptosis in the resistant cells. Knockdown
of K6 decreased the protein level of procaspase-8 and increased
levels of active caspase-3 and cytoplasmic cytochrome c in
the SNU601-cis2 (C2) cells compared to the control RNA-transfected
(CTLRNAi) cells, upon exposure to L-OHP (Fig. 3E). Based on this result,
overexpression of K6 in the resistant cells appears to contribute,
at least partially, to the acquisition of drug resistance.
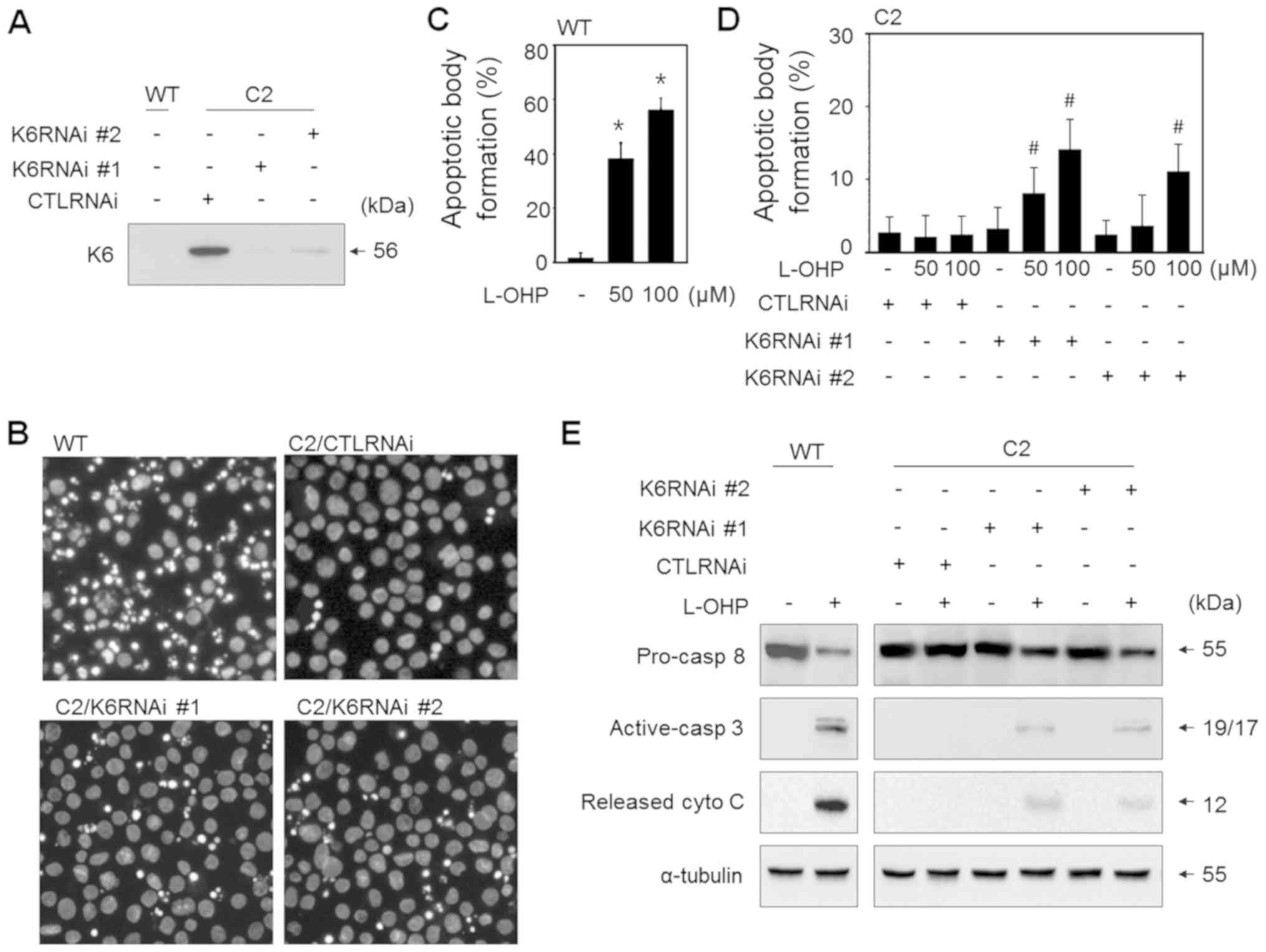 | Figure 3.Knockdown of K6 partially restores the
drug sensitivity of SNU601-cis2 cells. (A) SNU601 cells (WT) and
SNU601-cis2 cells (C2), transfected with scrambled small
interfering RNA (CTLRNAi), K6RNAi #1 and K6RNAi #2, were collected
and the silencing effect of K6 was confirmed by immunoblot assay of
cells not treated with L-OHP. (B-E) SNU601 cells (WT) and
SNU601-cis2 cells (C2), transfected with scrambled small
interfering RNA (CTLRNAi), K6RNAi #1 and K6RNAi #2, were exposed to
100 µM L-OHP for 48 h (B and E), or 50 and 100 µM L-OHP for 48 h (C
and D). The treated cells were then subjected to apoptotic analysis
(B-D) or immunoblotting to detect apoptotic proteins (E). For
apoptosis analysis, the treated cells were stained with Hoechst
33342 and nuclear images were captured under a fluorescence
microscope (B). Apoptotic body formation rates were calculated (C
and D). For immunoblotting analysis, antibodies against
pro-caspase-8, activated caspase-3, and cytochrome c were used.
Total cell lysates were used to detect pro-caspase-8 (pro-casp-8),
activated caspase-3 (active-casp-3) and non-mitochondrial cytosolic
lysates were used to detect released cytochrome c (released
cyto C) (E). *P<0.05 vs. not treated cells;
#P<0.05 vs. CTLRNAi and L-OHP treated. K, keratin;
L-OHP, oxaliplatin. |
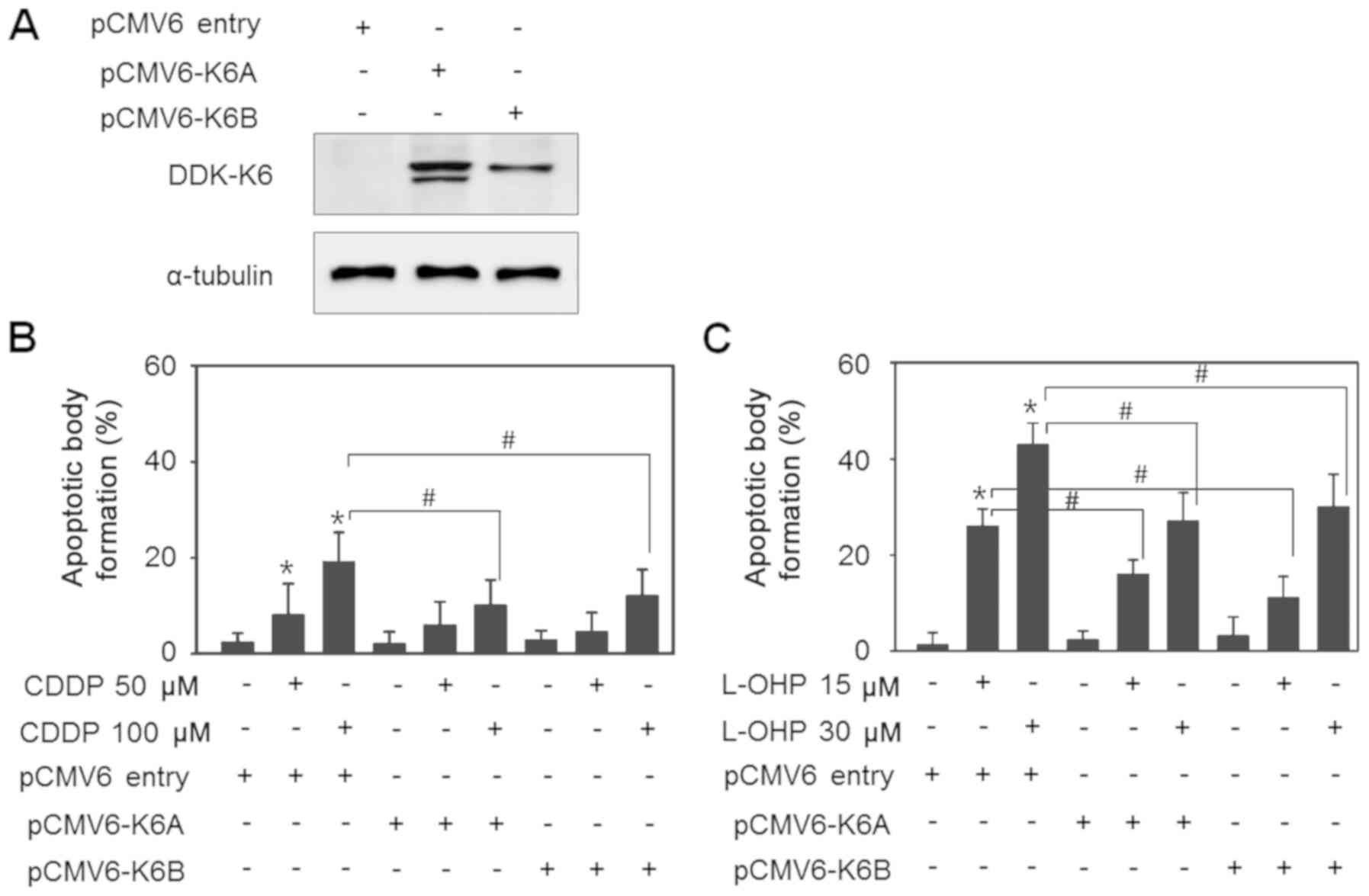
Overexpression of K6A and K6B
decreases drug responsiveness of wild-type cells
In the next experiment, to determine the effect of
K6 on drug resistance, K6 expression was induced by introducing K6
overexpression vectors in the SNU601-WT cells and alteration in the
cellular susceptibility was investigated. The cells were stably
transfected with pCMV6-K6A and pCMV6-K6B, followed by selection
with G418, and the expression of the introduced gene was confirmed
by detection of the DDK-tag by immunoblotting (Fig. 4A). Then, control cells, and pCMV6-K6A-
and pCMV6-K6B-expressing cells were exposed to CDDP or L-OHP, and
the percentages of apoptotic cells were detected. CDDP was less
toxic than L-OHP and it induced apoptosis in 19.7% cells after a 48
h treatment at a concentration of 100 µM in the control cells. On
the other hand, the degree of apoptosis decreased to 11.2 and 13.6%
in the K6A- and K6B-overexpressing cells, respectively (Fig. 4B). When treated with 30 µM L-OHP, the
control group exhibited 46.8% apoptotic cells, which was
significantly higher than the percentage in the K6A- and
K6B-overexpressing groups, which exhibited 26.1 and 29.5% apoptotic
cells, respectively (Fig. 4C).
Therefore, the increased expression of K6 seems to have a reducing
effect on the reactivity to the drugs.
K6 overexpression inhibits lipid raft
formation and localization of death receptor 5 (DR5)
Platinum drugs have been shown to promote the
extrinsic apoptotic pathway by forming lipid rafts and transferring
death receptors into lipid rafts in cancer cells (23,24). In
the present study, we investigated whether this membrane event was
affected by K6 expression in SNU601 cells. Lipid rafts can be
stained using CTxB, which exhibits specific affinity to ganglioside
GM1, a component of lipid rafts. When SNU601-WT cells were exposed
to L-OHP, lipid rafts visualized by CTxB-FITC were clearly observed
at the cell membrane, indicating that lipid rafts were actively
formed (Fig. 5A). However, in
SNU601-cis2 cells, FITC-stained lipid raft structure was barely
detected. When K6 expression was suppressed in the resistant cells
by gene interference using two types of siRNAs (C2/K6RNAi #1 and
C2/K6RNAi #2), the size and frequency of fluorescence spots visible
as lipid rafts increased compared to the control RNA-transfected
resistant (C2/CTLRNAi) cells (Fig.
5A). Then, we separated the lipid raft fraction from whole cell
lysates and detected the protein levels of DR5 and Fas-associated
protein with death domain (FADD) by immunoblotting. Upon exposure
to 100 µM L-OHP, high levels of DR5 and FADD were detected in the
raft fraction, indicating primary localization of DISC factors in
the lipid rafts in the wild-type (WT) cells, but much less
localization in lipid rafts in the resistant cells. However, the
localization of DR5 and FADD in the lipid raft region was higher in
the K6 RNAi transfected SNU601-cis2 (siK6 #1 and siK6 #2) cells
compared to the scrambled RNA transfected SNU601-cis2 (CTLRNAi)
cells (Fig. 5B). Therefore, it was
suggested that the increase in intracellular K6 in the resistant
cells acts as a potential factor that decreases the lipid raft
formation and inhibits translocation of death receptor and adaptor
protein into lipid rafts.
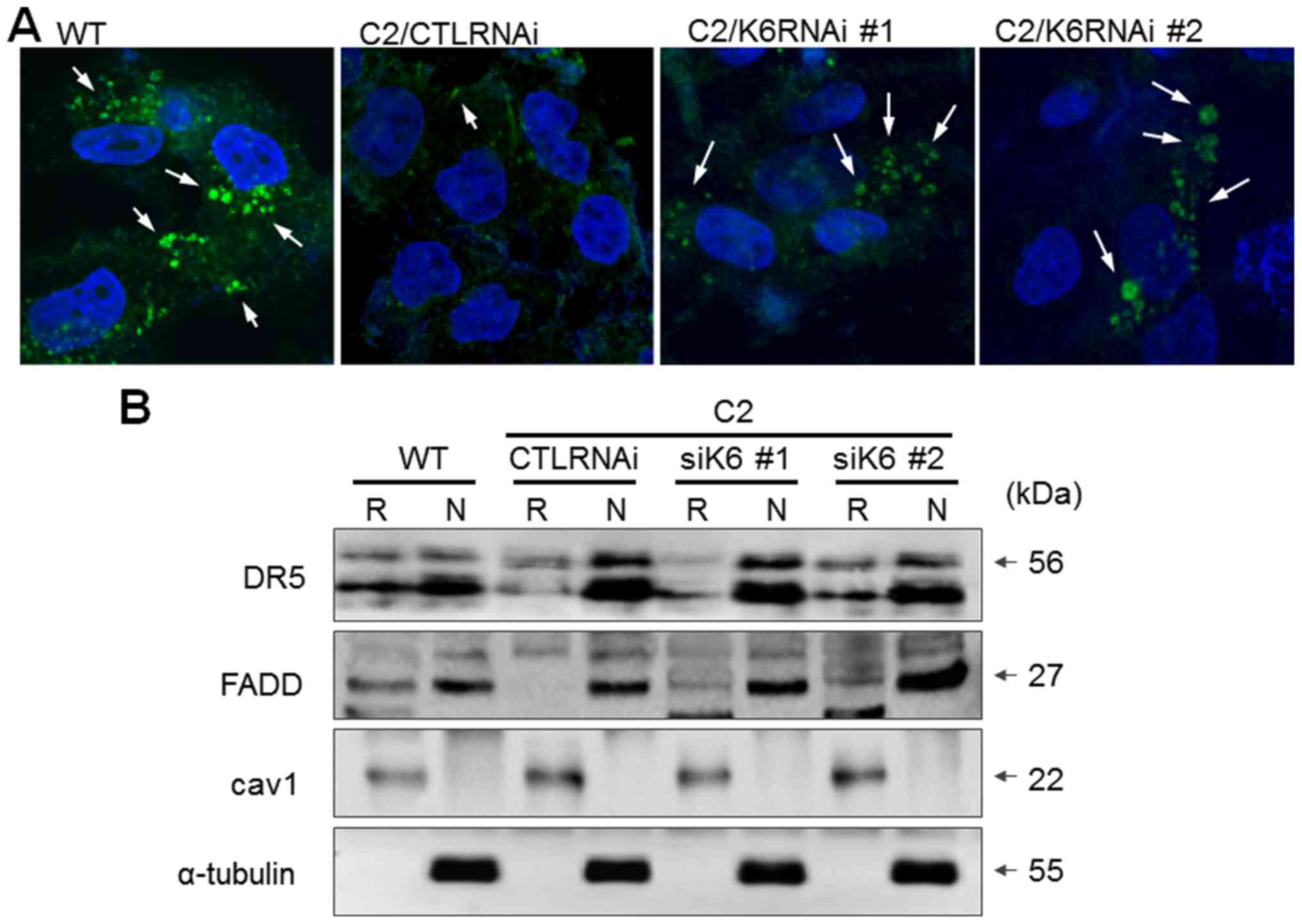 | Figure 5.Knockdown of K6 in SNU601-cis2 cells
restores lipid-raft structures and DR5 translocation into lipid
raft fraction. (A) SNU601 (WT) and SNU601-cis2 cells (C2),
transfected with scrambled small interfering RNA (C2/CTLRNAi),
either K6RNAi #1 or K6RNAi #2, were exposed to 100 µM L-OHP for 6
h. After fixation, cells were stained with CTxB-FITC and observed
under a confocal microscope (magnification, ×400; FV1000; Olympus).
Arrows indicate lipid-raft structures. (B) SNU601 (WT) and
SNU601-cis2 cells (C2), transfected with scrambled small
interfering RNA (CTLRNAi), either K6RNAi #1 (siK6 #1) or K6RNAi #2
(siK6 #2), were exposed to 100 µM L-OHP for 24 h and lipid raft
fractions or non-lipid raft fractions were obtained and analyzed by
immunoblotting to detect DR5 and FADD. caveolin-1 (cav1), lipid
raft loading control; α-tubulin, non-raft protein loading control.
R, raft; N, non-raft; L-OHP, oxaliplatin; DR5, death receptor 5;
FADD, Fas-associated protein with death domain. |
Discussion
In the present study, it was demonstrated that the
expression of specific keratin members, K6, K16 and K17, were
significantly upregulated in the cisplatin-resistant variants of
SNU601 gastric cancer cells compared to those in the parental
cells. However, the overall intracellular keratin level was not
significantly different in the resistant cells when pan-K was
detected using western blot and immunofluorescence analyses,
indicating that K6, K16, and K17 are specifically induced with
little effect on other keratins. The expression of keratins is
generally coordinated with specific partners of the opposite type.
Thus, the increase in a keratin stimulates induction of the partner
keratin, as well as the downregulation of a keratin leads to the
degradation of its partner through ubiquitination. Among these
three keratins, K6 is a type II keratin and the isoforms of K6 were
shown as pairing partners with type I keratins, K16 and K17,
enabling complete keratin filament formation. Here, we focused on
the role of the most highly expressed keratin, K6, since it may
represent the effects of K6/K16 and K6/K17.
In general, K6 filaments are characteristically
expressed in hair follicles, oral epithelia, and palmoplantar
epidermis (25). In addition to its
constitutive expression, K6 is also elevated in the proliferative
basal cells in wound edges of the skin where it appears to regulate
cell growth in injured epithelial tissue during wound repair
(26). K6/K16 and K6/K17 pairs are
also elevated in certain skin diseases that induce keratinocyte
hyperproliferation, such as psoriasis or skin tumors, and thus, it
is frequently referred to as hyperproliferation- or
activation-associated keratin (27,28).
Therefore, overexpression of K6 seems to be associated with the
regulation of cell functions, such as proliferation or wound
healing apart from simple structural and mechanical support.
However, the significance of its induction in cancer has not yet
been evaluated. Here, we modulated the expression of K6 to
understand the role of the overexpression of K6 intermediate
filaments. Gene silencing of K6 in the resistant cells restored
anticancer drug-mediated apoptosis, as detected by enhancement of
apoptotic body formation, caspase-8 and −3 activation, and
cytochrome c release. With a similar tendency,
overexpression of K6A and K6B vectors in the parental cells reduced
apoptosis upon exposure to cisplatin and oxaliplatin. Hence, an
elevated level of K6 appears to be associated with cellular
resistance to apoptosis. To date, the association of other subtypes
of keratin, especially K8, with resistance to chemotherapeutic
drugs has been suggested in several studies. Hepatocytes from
K8-null mice were found to be more sensitive to Fas-mediated
apoptosis than their wild-type counterparts (29), and the role of K8 in the protection
from apoptosis mediated by toxic agents was found to be related to
c-FLIP content and ERK1/2 signaling (14). Transfection of K8 and K18 in mouse L
cells and ectopic expression of K8/K18 in NIH3T3 fibroblasts led to
resistance against multiple anticancer drugs (30,31).
Suppression of K8 was found to be associated with the sensitivity
of nasopharyngeal carcinoma cells to cisplatin (32). In addition, association of K5
overexpression with chemoresistance of ovarian cancer has been
suggested (33). However, to the best
of our knowledge, the present study is the first report on the role
of K6 in drug tolerance.
The mechanisms by which K6 induces resistance in
this system should be studied in more detail, but it seems to
contribute to suppression of the extrinsic apoptotic pathway. We
observed that K6 overexpression reduced lipid raft formation and
the translocation of DR5 into the lipid raft fraction. In line with
our results, there have been reports that the role of keratin in
the protection from apoptosis is linked to the suppression of
membrane events for death receptor activation. K8/K18 was shown to
provide resistance to Fas-induced apoptosis and this protection
occurred through modulation of Fas targeting to the cell surface in
hepatocytes (29). Furthermore, K8
expression has been implicated in the regulation of the size of
lipid rafts and the migration of Fas into lipid rafts through
acid-sphingomyelinase activation (34,35). Thus,
suppression of the lipid raft-mediated apoptotic pathway seems to
be one of the mechanisms involved in keratin-mediated cell
resistance to cytotoxic agents. The results of our study suggest
that K6 contributes to the cell resistance against cytotoxic
stimuli by affecting death receptor redistribution into lipid
rafts. The findings from this study suggest that keratins should be
considered not only as cancer markers, but also as regulators of
cancer cell signaling and drug responsiveness, and the fine-tuning
of the regulatory mechanism of keratin expression may provide an
effective strategy against chemotherapy-resistant cancer.
Acknowledgements
We thank Professor Cheol-Hee Choi for the
cisplatin-resistant SNU601 cells and Ms. Jeong-Eun Choi and Ms.
Yoo-Ri Choi for their excellent technical assistance.
Funding
This study was supported by a research fund from
Chosun University, 2016.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SIH designed and wrote the manuscript. SCL and KRP
performed the experiments and analyzed the data. SIH and SCL
reviewed and edited the manuscript. All authors read and approved
the final manuscript and agree to be accountable for all aspects of
the research in ensuring that the accuracy or integrity of any part
of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Komatsu M, Sumizawa T, Mutoh M, Chen ZS,
Terada K, Furukawa T, Yang XL, Gao H, Miura N, Sugiyama T and
Akiyama S: Copper-transporting P-type adenosine triphosphatase
(ATP7B) is associated with cisplatin resistance. Cancer Res.
60:1312–1316. 2000.PubMed/NCBI
|
|
2
|
Cort A, Ozben T, Saso L, De Luca C and
Korkina L: Redox control of multidrug resistance and its possible
modulation by antioxidants. Oxid Med Cell Longev. 2016:42519122016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–11941. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Islam SU, Shehzad A, Sonn JK and Lee YS:
PRPF overexpression induces drug resistance through actin
cytoskeleton rearrangement and epithelial-mesenchymal transition.
Oncotarget. 8:56659–56671. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Foerster F, Braig S, Moser C, Kubisch R,
Busse J, Wagner E, Schmoeckel E, Mayr D, Schmitt S, Huettel S, et
al: Targeting the actin cytoskeleton: Selective antitumor action
via trapping PKCvarepsilon. Cell Death Dis. 5:e13982014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mukhtar E, Adhami VM and Mukhtar H:
Targeting microtubules by natural agents for cancer therapy. Mol
Cancer Ther. 13:275–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xi J, Zhu X, Feng Y, Huang N, Luo G, Mao
Y, Han X, Tian W, Wang G, Han X, et al: Development of a novel
class of tubulin inhibitors with promising anticancer activities.
Mol Cancer Res. 11:856–864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moll R, Divo M and Langbein L: The human
keratins: Biology and pathology. Histochem Cell Biol. 129:705–733.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moll R, Franke WW, Schiller DL, Geiger B
and Krepler R: The catalog of human cytokeratins: Patterns of
expression in normal epithelia, tumors and cultured cells. Cell.
31:11–24. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fuchs E: Keratins and the skin. Annu Rev
Cell Dev Biol. 11:123–153. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Coulombe PA and Omary MB: ‘Hard’ and
‘soft’ principles defining the structure, function and regulation
of keratin intermediate filaments. Curr Opin Cell Biol. 14:110–122.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galvin S, Loomis C, Manabe M, Dhouailly D
and Sun TT: The major pathways of keratinocyte differentiation as
defined by keratin expression: An overview. Adv Dermatol.
4:277–300. 1989.PubMed/NCBI
|
|
13
|
Van Muijen GN, Warnaar SO and Ponec M:
Differentiation-related changes of cytokeratin expression in
cultured keratinocytes and in fetal, newborn, and adult epidermis.
Exp Cell Res. 171:331–345. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gilbert S, Loranger A and Marceau N:
Keratins modulate c-Flip/extracellular signal-regulated kinase 1
and 2 antiapoptotic signaling in simple epithelial cells. Mol Cell
Biol. 24:7072–7081. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roux A, Loranger A, Lavoie JN and Marceau
N: Keratin 8/18 regulation of insulin receptor signaling and
trafficking in hepatocytes through a concerted
phosphoinositide-dependent Akt and Rab5 modulation. FASEB J.
31:3555–3573. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lahdeniemi IAK, Misiorek JO, Antila CJM,
Landor SK, Stenvall CA, Fortelius LE, Bergström LK, Sahlgren C and
Toivola DM: Keratins regulate colonic epithelial cell
differentiation through the Notch1 signalling pathway. Cell Death
Differ. 24:984–996. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Srivastava SS, Alam H, Patil SJ,
Shrinivasan R, Raikundalia S, Chaudhari PR and Vaidya MM: Keratin
5/14mediated cell differentiation and transformation are regulated
by TAp63 and Notch1 in oral squamous cell carcinomaderived cells.
Oncol Rep. 39:2393–2401. 2018.PubMed/NCBI
|
|
18
|
Chivu-Economescu M, Dragu DL, Necula LG,
Matei L, Enciu AM, Bleotu C and Diaconu CC: Knockdown of KRT17 by
siRNA induces antitumoral effects on gastric cancer cells. Gastric
Cancer. 20:948–959. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu H, Choi SM, An CS, Min YD, Kim KC, Kim
KJ and Choi CH: Concentration-dependent collateral sensitivity of
cisplatin-resistant gastric cancer cell sublines. Biochem Biophys
Res Commun. 328:618–622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwasaki I, Sugiyama H, Kanazawa S and
Hemmi H: Establishment of cisplatin-resistant variants of human
neuroblastoma cell lines, TGW and GOTO, and their drug
cross-resistance profiles. Cancer Chemother Pharmacol. 49:438–444.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun CL and Chao CC: Cross-resistance to
death ligand-induced apoptosis in cisplatin-selected HeLa cells
associated with overexpression of DDB2 and subsequent induction of
cFLIP. Mol Pharmacol. 67:1307–1314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu L, Qu X, Zhang Y, Hu X, Yang X, Hou K,
Teng Y, Zhang J, Sada K and Liu Y: Oxaliplatin enhances
TRAIL-induced apoptosis in gastric cancer cells by CBL-regulated
death receptor redistribution in lipid rafts. FEBS Lett.
583:943–948. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang CR, Jin ZX, Dong L, Tong XP, Yue S,
Kawanami T, Sawaki T, Sakai T, Miki M, Iwao H, et al: Cisplatin
augments FAS-mediated apoptosis through lipid rafts. Anticancer
Res. 30:2065–2071. 2010.PubMed/NCBI
|
|
25
|
Rothnagel JA, Seki T, Ogo M, Longley MA,
Wojcik SM, Bundman DS, Bickenbach JR and Roop DR: The mouse keratin
6 isoforms are differentially expressed in the hair follicle,
footpad, tongue and activated epidermis. Differentiation.
65:119–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wong P and Coulombe PA: Loss of keratin 6
(K6) proteins reveals a function for intermediate filaments during
wound repair. J Cell Biol. 163:327–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang CK, Magnaldo T, Ohtsuki M, Freedberg
IM, Bernerd F and Blumenberg M: Epidermal growth factor and
transforming growth factor alpha specifically induce the
activation- and hyperproliferation-associated keratins 6 and 16.
Proc Natl Acad Sci USA. 90:6786–6790. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leigh IM, Navsaria H, Purkis PE, McKay IA,
Bowden PE and Riddle PN: Keratins (K16 and K17) as markers of
keratinocyte hyperproliferation in psoriasis in vivo and in vitro.
Br J Dermatol. 133:501–511. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gilbert S, Loranger A, Daigle N and
Marceau N: Simple epithelium keratins 8 and 18 provide resistance
to Fas-mediated apoptosis. The protection occurs through a
receptor-targeting modulation. J Cell Biol. 154:763–773. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bauman PA, Dalton WS, Anderson JM and
Cress AE: Expression of cytokeratin confers multiple drug
resistance. Proc Natl Acad Sci USA. 91:5311–5314. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anderson JM, Heindl LM, Bauman PA, Ludi
CW, Dalton WS and Cress AE: Cytokeratin expression results in a
drug-resistant phenotype to six different chemotherapeutic agents.
Clin Cancer Res. 2:97–105. 1996.PubMed/NCBI
|
|
32
|
Wang Y, He QY, Tsao SW, Cheung YH, Wong A
and Chiu JF: Cytokeratin 8 silencing in human nasopharyngeal
carcinoma cells leads to cisplatin sensitization. Cancer Lett.
265:188–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ricciardelli C, Lokman NA, Pyragius CE,
Ween MP, Macpherson AM, Ruszkiewicz A, Hoffmann P and Oehler MK:
Keratin 5 overexpression is associated with serous ovarian cancer
recurrence and chemotherapy resistance. Oncotarget. 8:17819–17832.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gilbert S, Loranger A, Lavoie JN and
Marceau N: Cytoskeleton keratin regulation of FasR signaling
through modulation of actin/ezrin interplay at lipid rafts in
hepatocytes. Apoptosis. 17:880–894. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gilbert S, Loranger A, Omary MB and
Marceau N: Keratin impact on PKCδ- and ASMase-mediated regulation
of hepatocyte lipid raft size-implication for FasR-associated
apoptosis. J Cell Sci. 129:3262–3273. 2016. View Article : Google Scholar : PubMed/NCBI
|