Introduction
Colorectal cancer (CRC) is a leading cause of
morbidity and mortality worldwide (1). The most common treatment options for CRC
are surgery and chemotherapy, with the third-generation platinum
drug Oxaliplatin (Oxa) being a chief therapeutic strategy (2). However, reports of Oxa-resistance in CRC
therapy are gradually increasing, and this acquired resistance has
become a major obstacle to patient survival in CRC (3).
Epithelial-mesenchymal transition (EMT) is an
essential phenotypic conversion in embryonic development, tissue
remodeling, wound healing, tumor invasion and metastasis. As a
dynamic and reversible process, EMT often occurs at the invasive
front of many metastatic cancers. The loss of E-cadherin is
considered as a vital event in the EMT process (4). Emerging evidence suggests that EMT plays
an important role in acquisition of chemotherapy resistance in
various cancer cells (5). Excision
repair cross-complementing group 1 protein (ERCC1) plays a key role
in nucleotide excision repair and in removing platinum-induced DNA
adducts (6). Previous studies have
revealed that ERCC1 expression is negatively correlated to
E-cadherin in lung cancer (7).
Furthermore, overexpression of snail, ZEB1/2, EMT-related
transcription factors, can contribute to chemoresistance by
promoting ERCC1 expression in head and neck squamous cell carcinoma
cells and lung cancer cells (8,9). In
addition, there are some studies indicating that p53 can promote
collective cellular migration by inducing EMT (10).
Dehydrogenase/reductase SDR family member 2 (DHRS2)
was first identified as a nuclear protein in the sodium
butyrate-treated human hepatoblastoma cell line HepG2 (11). DHRS2 belongs to the short-chain
dehydrogenase/reductase (SDR) family of enzymes that are present in
all life forms and are mainly NAD/NADP-dependent oxido-reductases
that are active on a large and heterogeneous set of substrates
including steroids, retinoids, prostaglandins, metabolites, and
xenobiotics (12,13). DHRS2 maps to chromosome
14q11.2, which is a region characterized by high-frequency loss of
heterozygosis in many different tumors (14–16), and
high levels of DHRS2 indicate a possible function in tumorigenesis
(17). In fact, DHRS2 has been
revealed to be highly concentrated in several types of cancer cells
and could be a prognostic marker of prostate cancer (18), bladder carcinomas (19), and sporadic breast cancer (20). DHRS2 is also aberrantly expressed in
esophageal squamous cell carcinoma (ESCC) where its overexpression
enhanced cancer cell aggressiveness (21). Further characterization of DHRS2
revealed that its aberrant expression was associated with the
mechanisms of drug resistance in acute myelogenous leukemia and
gastric carcinogenesis (22,23). However, it is not known whether there
is an underlying relationship between DHRS2 and chemotherapy
resistance in CRC.
In the present study, proteomics was used to compare
protein expression profiles between the parental colon cancer cell
line HCT116 and the Oxa-resistant subline HCT116/Oxa cells.
Notably, it was revealed that DHRS2 protein levels were
significantly upregulated in HCT116/Oxa cells compared with
parental cells. Furthermore, silencing of DHRS2 sensitized
HCT116/Oxa cells to Oxa by downregulating ERCC1 through a
p53-dependent pathway, and reversed EMT. This finding revealed that
DHRS2 could act as an important regulator of Oxa-resistance
associated with EMT in CRC, which may suggest novel strategies for
defeating Oxa-resistance.
Materials and methods
Antibodies and reagents
Primary antibodies against ABCB1 (cat. no. 13342),
PARP (cat. no. 9532) and LC3-I/II (cat. no. 12741) were obtained
from Cell Signaling Technology, Inc. Primary antibodies against
ABCC1 (cat. no. ab24102), ABCC2 (cat. no. ab203397), and ABCG2
(cat. no. ab24115) were obtained from Abcam. Primary antibodies
against E-cadherin (cat. no. 20874-1-AP), vimentin (cat. no.
60330-1-Ig), Bcl-2 (cat. no. 12789-1-AP), p53 (cat. no.
10442-1-AP), ERCC1 (cat. no. 14586-1-AP), CA9 (cat. no.
11071-1-AP), DHRS2 (cat. no. 15735-1-AP), GAPDH (cat. no.
60004-1-Ig) and β-actin (cat. no. 60008-1-Ig) were obtained from
Proteintech Group. The PrimeScripts RT reagent kit and SYBRs Premix
Ex Taq™ were obtained from Takara (Takara Biotechnology Co.,
Ltd.).
Cell culture and the establishment of
an Oxa-resistant cell line
The HCT116 colorectal carcinoma cell line was
obtained from the Type Culture Collection of the Chinese Academy of
Sciences. HCT116 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM)/F12 culture medium (Gibco-BRL; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (FBS), 100 U/ml
penicillin, and 100 mg/ml streptomycin. The Oxa-resistant CRC cell
line HCT116/Oxa was established in our laboratory. Briefly, HCT116
cells were exposed to a series of stepwise-increased concentrations
of Oxa to develop an HCT116/Oxa-resistant cell line. Both cell
lines were cultured in a 5% CO2 atmosphere at 37°C.
Proteomic sample preparation
Cells were sonicated three times on ice using a high
intensity ultrasonic processor (Ningbo Scientz Biotechnology Co.,
Ltd.) in lysis buffer (8 M urea, 1% protease inhibitor cocktail).
After 12,000 × g at 4°C for 10 min, protein concentration was
determined with a BCA kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. For digestion, the
protein solution was reduced with 5 mM dithiothreitol at 56°C for
30 min and alkylated with 11 mM iodoacetamide for 15 min at room
temperature in darkness. Triethylammonium bicarbonate buffer (TEAB)
was added to dilute samples. Finally, trypsin was added at a 1:50
trypsin: Protein mass ratio for the first digestion overnight and a
1:100 ratio for a second 4 h-digestion. Subsequently, peptides were
then desalted by a Strata X C18 SPE column (Phenomenex),
vacuum-dried, dissolved in 0.5 M TEAB, and labeled based on the
tandem mass tag (TMT) kit. Briefly, 1 U of TMT reagent was
dissolved in acetonitrile and added to peptides for incubation for
2 h at room temperature, followed by pooling, desalting, and drying
by vacuum centrifugation. Agilent 300Extend C18 column (5 µm
particles, 4.6 mm ID, 250 mm length) was used to fractionate
peptides. Briefly, peptides were first separated with a gradient of
8 to 32% acetonitrile (pH 9.0) over 60 min into 60 fractions. Then,
they were combined into 18 fractions and subjected to
vacuum-drying.
Liquid chromatography-tandem mass
spectrometry
The peptides were dissolved in 0.1% formic acid
(solvent A), and separated using EASY-nLC 1000 ultra-performance
liquid chromatography UPLC (Thermo Fisher Scientific, Inc.). The
gradient was an increase from 6 to 23% solvent B (0.1% formic acid
in 98% acetonitrile) over 26 min, 23–35% in 8 min, climbing to 80%
in 3 min, and holding at 80% for the last 3 min at a constant flow
rate of 400 nl/min.
The peptides were subjected to a nano electrospray
ionization source followed by tandem mass spectrometry (MS/MS) in Q
Exactive™ Plus (Thermo Fisher Scientific, Inc.) coupled online to
the UPLC. The MS scan was set as 350–1,800 m/z and 70,000
resolution; the MS/MS scan was set as 100 m/z and 17,500
resolution. The automatic gain control was set at 5E4, and the
data-dependent acquisition procedure was applied to data
acquisition.
Data analysis
The resulting MS/MS data were processed using the
Maxquant search engine (v.1.5.2.8). Searches were performed against
SwissProt human database (20,130 sequences) and reverse decoy
database. Trypsin/P was specified as a cleavage enzyme allowing up
to two missing cleavages. For precursor ions, the mass tolerances
of the first search and main search were 20 and 5 ppm,
respectively. The mass tolerance of fragment ions was 0.02 Da. The
false discovery rate was adjusted to <1% and the minimum score
for peptides was set as >40.
MS analysis of TMT-labeled samples was performed on
Q Exactive Protein intensities resulting from average single TMT
reporter ion intensities obtained for each peptide associated with
a specific protein. The average ratio of differential TMT 127/126
expression (1.5-fold increase or decrease) represents the ratio of
two samples. Specifically, differentially expressed proteins were
identified in our TMT experiment using 1.5 and 0.67 as upregulation
and downregulation cutoff points, respectively.
Bioinformatics analysis (gene ontology
and pathway analysis)
Gene Ontology (GO) annotation and enrichment
analyses were performed using DAVID (the Database for Annotation,
Visualization and Integrated Discovery). GO with a corrected
P<0.05 was considered to indicate a statistically significant
difference. Additionally, the Kyoto Encyclopedia of Genes and
Genomes (KEGG) database was used to identify enriched pathways by a
two-tailed Fisher's exact test to assess the enrichment of
differentially expressed proteins against all identified proteins.
Pathways with a corrected P<0.05 were considered significant.
These pathways were classified into hierarchical categories
according to the KEGG website.
Oxaliplatin sensitivity assay
The viability of HCT116/Oxa cells and parental cell
was measured by the CCK-8 assay (Dojindo Molecular Technologies,
Inc.). Briefly, cells were seeded in 96-well plates at a density of
1.0×104 cells and treated with different concentrations
of Oxa for 48 h. Then 10 µl CCK-8 reagent was added to each well
and incubated at 37°C for another 1 h. The absorbance was measured
for each well at a wavelength of 450 nm. All assays were performed
in triplicate.
Cell migration experiment
The cell migration assay was performed using a
Transwell system (8 µm; Corning Inc.) according to the
manufacturer's protocol. Approximately 5×104 cells were
re-suspended in serum-free medium in the top chamber of the insert,
and the bottom chamber was filled with DMEM/F12 containing 10% FBS.
After 24 h of incubation, the cells that had migrated to the lower
side of the membrane were fixed with 4% paraformaldehyde and
stained with 0.1% crystal violet for 20 min at room temperature.
The images were captured using a fluorescence microscope (E1000M
Eclipse; Nikon Corp., Tokyo, japan) and the migrated cells was
counted.
RNA interference
The siRNA against human ERCC1, DHRS2, p53 and
control siRNA were synthesized by Shanghai GenePharma Co., Ltd. The
sequence of the control siRNA was: 5′-UUCUCCGAACGUGUCACGUTT-3′. The
target sequences for ERCC1 siRNA were: siRNA1,
5′-GCCAAGCCCUUAUUCCGAUTT-3′; and siRNA2,
5′-GCGACGUAAUUCCCGACUATT-3′. The target sequences for DHRS2 siRNA
were: siRNA1, 5′-GCGUGGUUCCAGGAAUUAUTT-3′; and siRNA2,
5′-GCUGUCAUCCUGGUCUCUUTT-3′. The target sequences for p53 siRNA
were: siRNA1, 5′-CCACCAUCCACUACAACUATT-3′; and siRNA2,
5′-CCACUGGAUGGAGAAUAUUTT-3′. Cells (1×106/well) were
seeded on a 6-well plate and cultured until the next day. They were
then transfected with 100 pmol siRNA oligomer mixed with
Lipofectamine 2000 reagent (Life Technologies; Thermo Fisher
Scientific, Inc.) in serum-reduced medium according to the
manufacturer's instructions. After 6 h, the medium was changed to
complete culture medium, and the cells were incubated at 37°C in a
CO2 incubator for another 24–48 h before harvest.
Quantitative real-time PCR
Total RNA from cell lines was extracted using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's guidelines. A total of 500 ng RNA was converted
into cDNA using the PrimeScript™ kit (Takara Bio, Inc.) under the
following conditions: 37°C for 15 min, 85°C for 5 sec, and were
held at 4°C. Quantification of target genes and the reference gene
(GAPDH) was studied in triplicate on the ABI-7500 system (Applied
Biosystems, Inc.; Thermo Fisher Scientific, Inc.) using SYBR-Green
fluorescent-based assay (Takara Bio Inc.). The reaction conditions
were as follows: Pre-denaturation at 95°C for 30 sec, followed by
40 cycles at 95°C for 5 sec and 60°C for 34 sec. The primers used
were as follows: DHRS2 forward, 5′-TCACAGAAAGCCTAGCACAG-3′ and
reverse, 5′-TGAGACCATCACCAAGCG-3′; GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′;
ERCC1 forward, 5′-CTCAAGGAGCTGGCTAAGATGT-3′ and reverse,
5′-CATAGGCCTTGTAGGTCTCCAG-3′; vimentin forward,
5′-TGAGTACCGGAGACAGGTGCAG-3′ and reverse,
5′-TAGCAGCTTCAACGGCAAAGTTC-3′; and E-cadherin forward,
5′-TACACTGCCCAGGAGCCAGA-3′ and reverse, 5′-TGGCACCAGTGTCCGGATTA-3′.
The quantification was based on ΔΔCq calculations (24) and was normalized to GAPDH as a loading
control.
Immunofluorescence
Treated cells were washed with ice-cold
phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for
20 min, and permeabilized with 0.3% Triton X-100 for 10 min. After
blocking with 2% bovine serum albumin for 30 min at room
temperature, cells were incubated with primary antibodies against
E-cadherin or vimentin (1:100 dilution) at 4°C overnight. Slides
were then washed three times with PBS and incubated with Alexa
Fluor 488 (1:1,000 dilution; cat. no Ab150077) or Alexa Fluor
594-conjugated secondary antibodies (1:1,000 dilution; cat. no.
ab150120; both Abcam) for 1 h at room temperature. Nuclei were
stained with 4′,6-diamidino-2-phenylindole (10 µg/ml) for 10 min.
Samples were examined to analyze the expression of E-cadherin and
vimentin.
Western blotting
After washing three times with ice-cold PBS, cells
were lysed by western blot lysis buffer containing 50 mM Tris-HCl
(pH 7.6), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% Na-deoxycholate, 5
µg/ml aprotinin, 5 µg/ml leupeptin, and 1mM phenylmethylsulfonyl
fluoride on ice. Lysates were centrifuged at 12,000 × g for 20 min
at 4°C and denatured at 100°C for 10 min. Equal amounts of protein
samples (30 µg/sample) were loaded in each well of a 10% sodium
dodecyl sulfate polyacrylamide gel, then electrophoretically
transferred onto 0.45 µm polyvinylidene difluoride membranes.
Following blocking with 5% non-fat milk at room temperature for 2
h, the membranes were incubated with primary antibodies (1:1,000
dilution) at 4°C overnight, then with horseradish
peroxidase-conjugated goat anti-mouse IgG (1:5,000 dilution; cat.
no. SA00001-1) or horseradish peroxidase-conjugated goat
anti-rabbit IgG (1:5,000 dilution; cat. no. SA00001-2; both Wuhan
Sanying Biotechnology) for 2 h at room temperature.
Chemiluminescence reagent (Pierce; Thermo Fisher Scientific, Inc.)
was used to detect specific immune complexes. Protein gray value
detection was performed using ImageJ software (version 1.4.3.67;
National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Results were expressed as the means ± SD of three
independent experiments unless otherwise specified. Data from two
groups were compared using two-tailed unpaired Student's t-test.
One-way analysis of variance (ANOVA) with Tukey's post hoc test was
used to assess differences among groups. These analyses were
performed using GraphPad Prism Software version 5.0 (GraphPad
Software Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
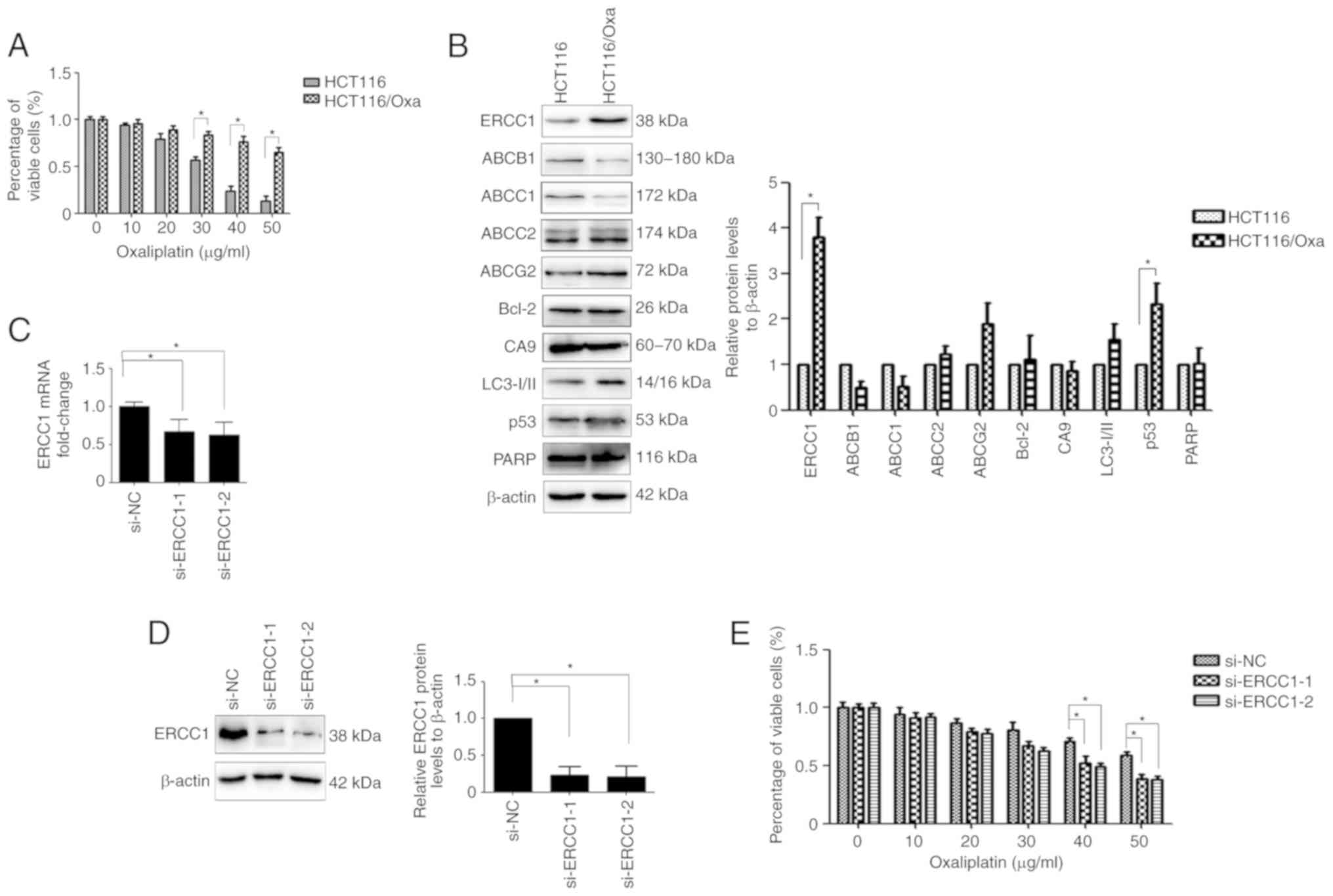
ERCC1 is overexpressed in HCT116/Oxa
cells and contributes to their resistance to Oxa
As revealed in Fig.
1A, HCT116/Oxa cells revealed increased resistance to Oxa
compared with their parental cells. To explore which protein was
responsible for Oxa resistance in HCT116/Oxa cells, western blot
analysis was used. Elevated levels of ERCC1, a major protein
involved in the nucleotide excision repair pathway, were observed
in HCT116/Oxa cells compared with HCT116 cells (Fig. 1B). To confirm whether ERCC1 expression
is involved in Oxa-resistance, the HCT116/Oxa cell sensitivity to
Oxa was analyzed after ERCC1 knockdown. ERCC1 mRNA and protein
expression levels were significantly suppressed by si-ERCC1
transfection in HCT116/Oxa cells (Fig. 1C
and D), and ERCC1 knockdown effectively attenuated
Oxa-resistance in HCT116/Oxa cells (Fig.
1E).
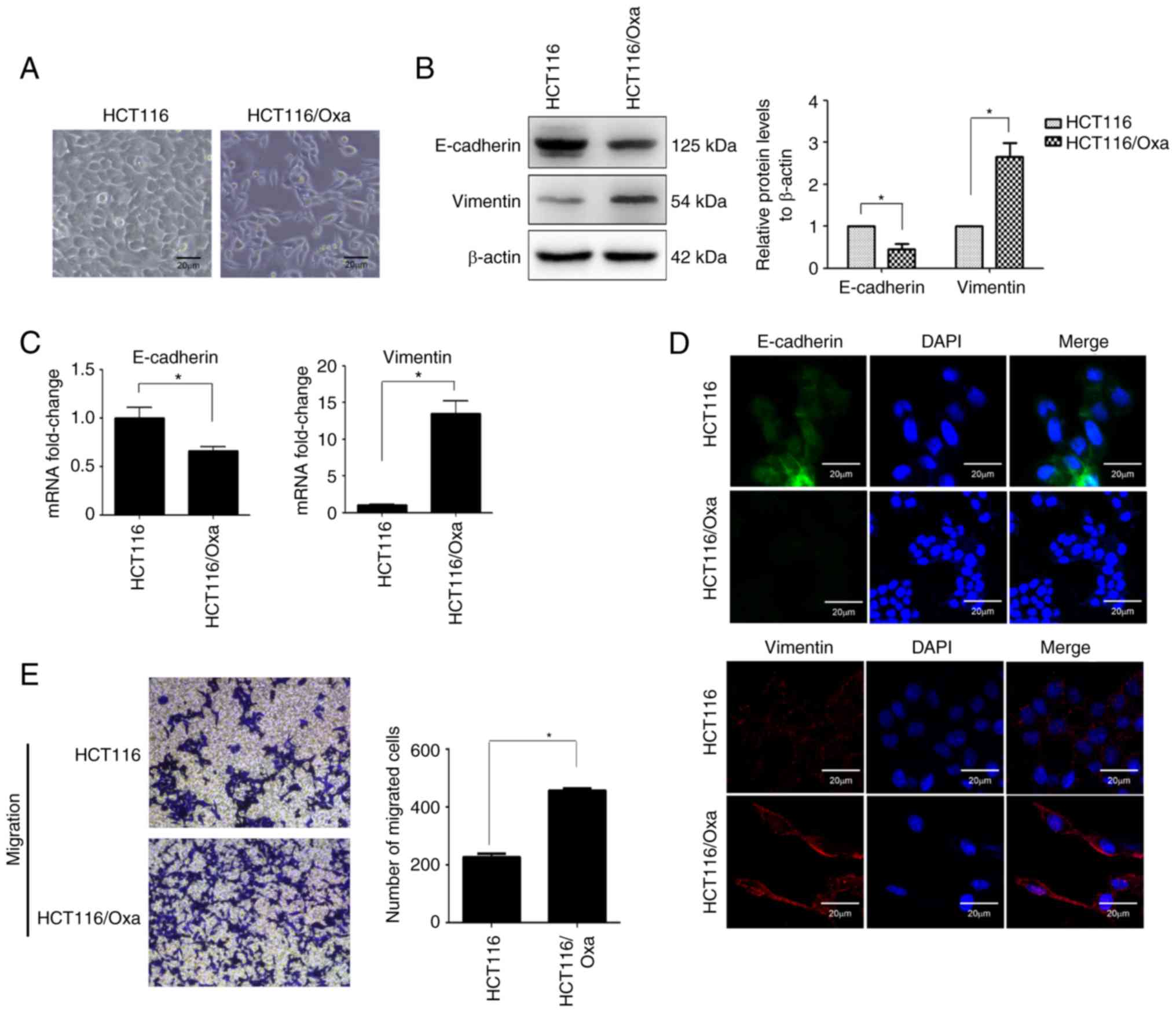
HCT116/Oxa cells exhibit an EMT
phenotype
EMT plays a critical role in drug resistance.
Morphological analysis under an inverted phase contrast microscope
revealed that HCT116 cells had a round morphology, while HCT116/Oxa
cells were of irregular shapes including long strips, fusiform, and
polygonal (Fig. 2A). These
morphological differences indicated that HCT116/Oxa cells have a
mesenchymal-like phenotype as evidenced by the relative
downregulation of the epithelial biomarker E-cadherin and
upregulation of the mesenchymal biomarker vimentin (Fig. 2B and C). The EMT phenotype of
HCT116/Oxa cells was confirmed by immunofluorescence staining
(Fig. 2D). The Transwell assay
revealed that HCT116/Oxa cells had a higher capacity for migration
relative to HCT116 cells (Fig.
2E).
Identification of differentially
expressed proteins and enrichment analysis
To gain further understanding of the molecular
mechanisms involved in Oxa-resistance in HCT116/Oxa cells,
TMT-based quantitative proteomic analysis was performed. A total of
5,345 proteins were identified in this study, of which 4,599 had
available quantitative information. A full list of proteins and
relevant MS data are presented in Supplementary Data I. Of the
4,599 commonly expressed proteins, 64 were upregulated (fold ratio
>1.5) and 50 were downregulated (fold ratio <0.67) in
HCT116/Oxa cells compared with HCT116 cells (a full list of
proteins is presented in Supplementary Data II). The differential
expression of proteins commonly expressed in parental and
Oxa-resistant cell lines was further visualized using an
expression-based heatmap (Fig.
3A).
To classify categories of commonly expressed
proteins and canonical pathways affected by the up- or
downregulation of these proteins, GO enrichment analysis was
performed using DAVID and KEGG. The enrichment analysis of
dysregulated proteins in cellular component, biological process,
and molecular function is presented in Fig. 3B. In cellular component analysis, the
majority of identified proteins were classified into the
extracellular region, extracellular region part, extracellular
space, and extracellular exosome. Molecular functional
classification revealed that most of these proteins were involved
in oxidoreductase activity, aldehyde dehydrogenase
[NAD(P)+] activity, oxidoreductase activity, acting on
the CH-OH group of donors, and virus receptor activity. Regarding
biological processes, most of the identified proteins were enriched
in small molecule metabolic processes, single organism metabolic
processes, oxidation-reduction processes, and organic acid
metabolic processes. KEGG analysis identified nine significant
pathways (P<0.05; Fig. 3C), with
lysosome and central carbon metabolism in cancer being the most
significantly enriched. Notably, there was a good agreement of
selected validation immunoblots with MS data (Fig. 3D).
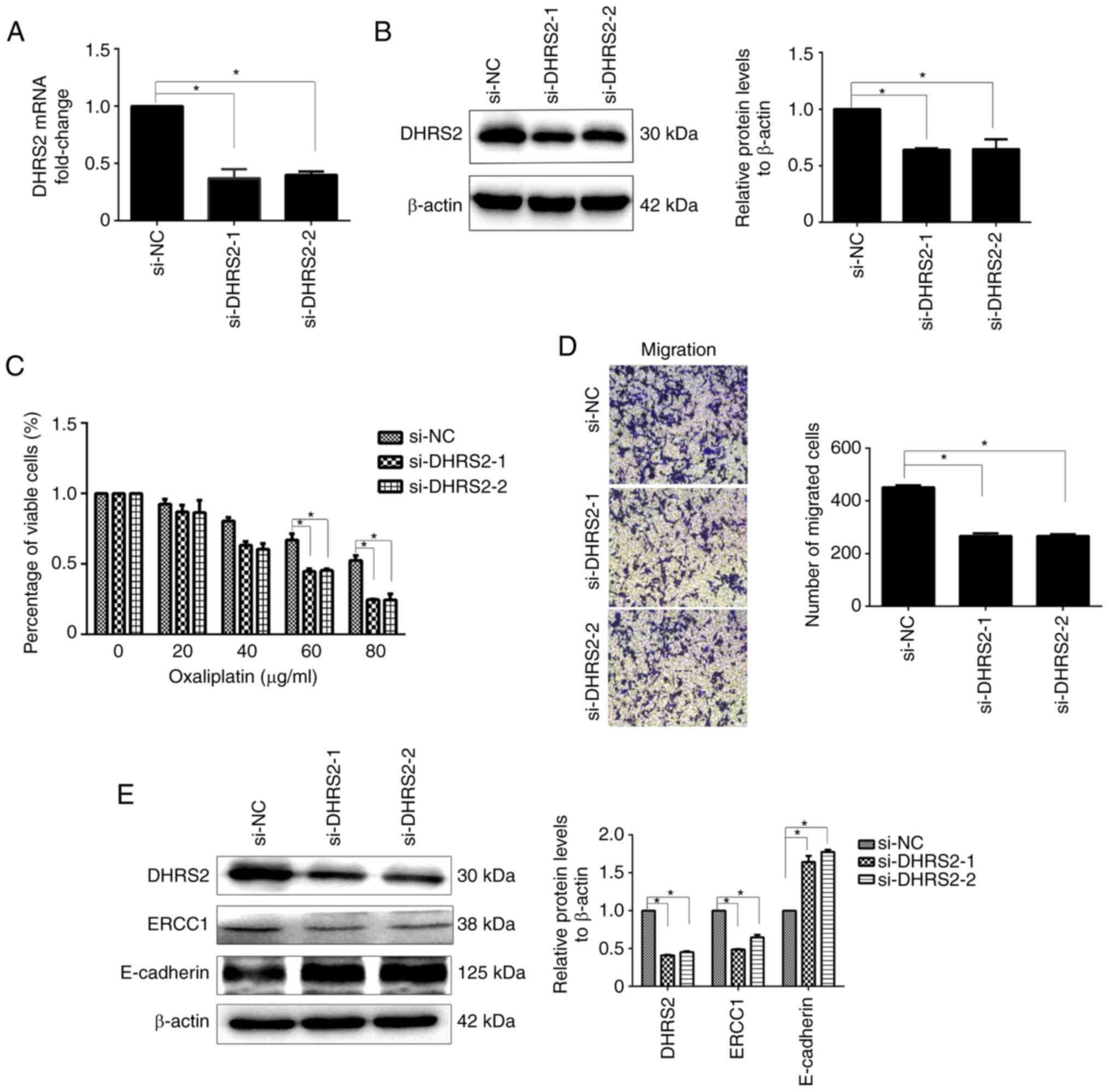
DHRS2 knockdown reverses Oxa
resistance through ERCC1 in HCT116/Oxa cells and inhibits
OXA-induced EMT
To explore the role of DHRS2 in CRC cell
Oxa-resistance, transient knockdown experiments were next
performed. HCT116/Oxa cells transfected with control or DHRS2
siRNAs were treated with different concentrations of Oxa. As
revealed in Fig. 4A and B,
DHRS2-specific siRNA significantly reduced DHRS2 mRNA and protein
expression. The CCK-8 assay revealed that the suppression of DHRS2
decreased the viability of HCT116/Oxa cells compared with cells
transfected with control siRNA (Fig.
4C). Transwell assays revealed that the migration capacities of
HCT116/Oxa cells were reduced in the si-DHRS2 group relative to the
si-NC group (Fig. 4D).
To investigate the function(s) of DHRS2 in
Oxa-resistance in HCT116/Oxa cells with an EMT phenotype, the
protein expression of ERCC1 and E-cadherin was assessed. Western
blotting demonstrated reduced expression of ERCC1 and increased
expression of E-cadherin in the si-DHRS2 group compared with the
si-control group (Fig. 4E).
Collectively, these data revealed that DHRS2 is an important factor
in Oxa resistance and EMT in CRC cells.
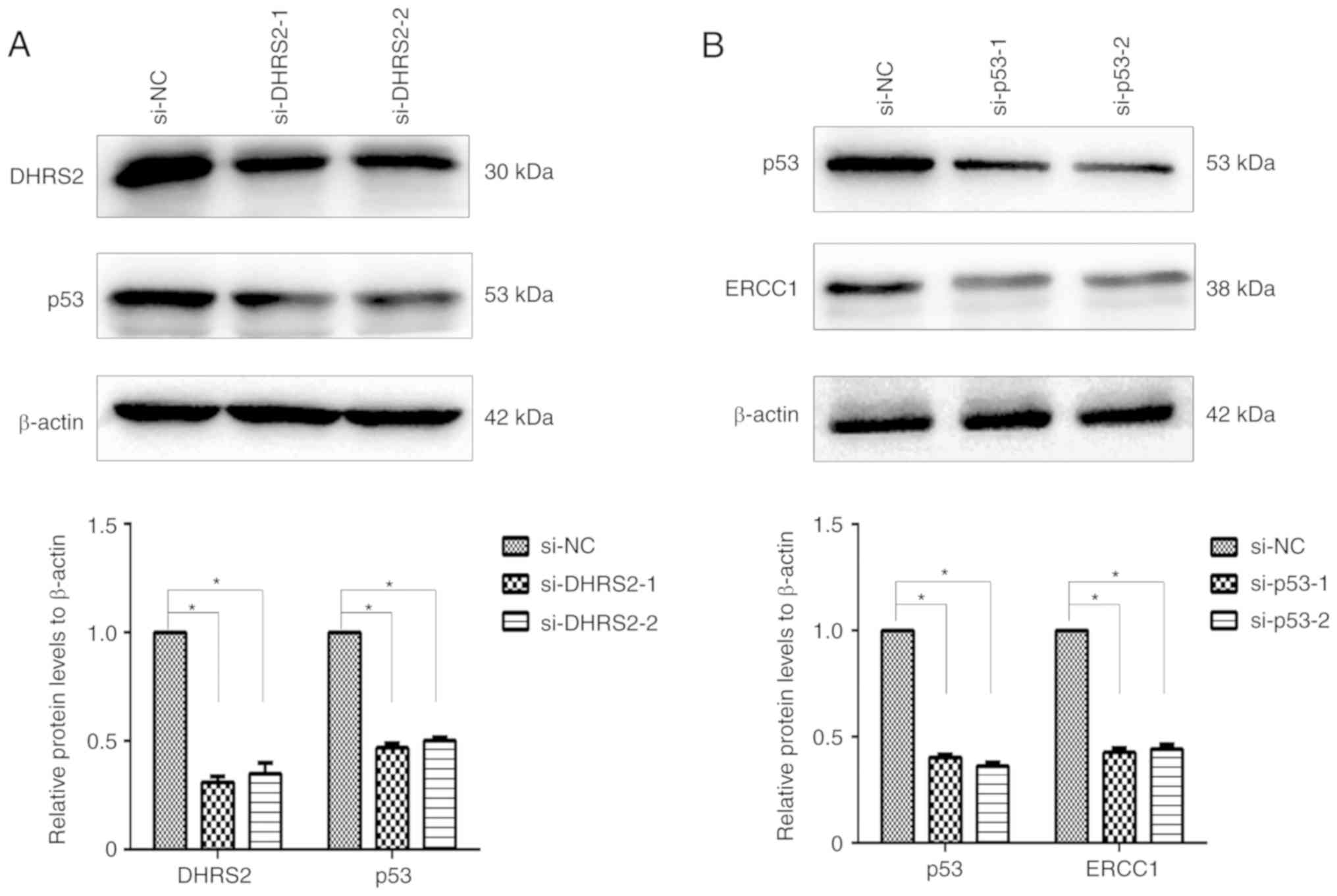
DHRS2 confers Oxa resistance through
the p53/ERCC1 pathway
Next, the mechanism underlying the involvement of
DHRS2 in Oxa-resistance in CRC cells was explored. Because DHRS2
has been reported to stabilize p53 in an osteosarcoma cell line
(25), it was examined whether it was
also stabilized in HCT116/Oxa cells. Our previous results revealed
that DHRS2 and p53 were markedly increased in HCT116/Oxa cells
compared with the parental cell line. Silencing of DHRS2 in
HCT116/Oxa cells by siRNA decreased p53 compared with control cells
(Fig. 5A). To assess whether p53
regulates ERCC1 expression, p53 siRNA was used and the results
revealed a significant decrease of ERCC1 protein expression
(Fig. 5B).
Discussion
Although recent chemotherapy regimens have
significantly improved the survival of patients with metastatic
disease, almost all patients with CRC eventually become
chemo-resistant with distant metastases (26,27).
Therefore, it is critical to delineate drug resistance mechanisms
to commonly used therapeutic agents such as Oxa to improve CRC
patient survival.
CRC cells develop resistance to Oxa through a
reduction of cellular accumulation, intracellular inactivation,
blocking the induction of apoptosis, and the enhancement of DNA
repair. ERCC1 is an important mediator of nucleotide excision
repair (NER), and is widely recognized as a powerful component of
the DNA repair mechanism (28).
Previous studies have revealed that ERCC1 expression levels are
positively correlated with the DNA repair capacity, and are
associated with cellular and clinical resistance to platinum-based
therapy of lung, ovarian, and gastric cancer (6,29,30). Furthermore, several prospective
validation studies revealed that ERCC1 expression may serve as a
biomarker for platinum-based therapy responses (31–33). In
the present study, the Oxa-resistant human CRC cell line HCT116/Oxa
was successfully established. It was revealed that of several
selected drug resistance-associated proteins, the expression of
ERCC1 was the most elevated in these cells, and that reducing ERCC1
expression resulted in loss of the chemo-resistant phenotype. These
results suggested that the aberrant expression of ERCC1 may be a
major cause of chemotherapy resistance in HCT116/Oxa cells.
To further explore the mechanisms of Oxa-resistance
in HCT116/Oxa cells, we searched for proteins associated with drug
resistance in parental HCT116 cells and HCT116/Oxa cells using
TMT-based quantitative proteomics. DHRS2 expression was the most
significantly upregulated in HCT116/Oxa cells, and this was
confirmed by western blotting. Moreover, knockdown of DHRS2 in
HCT116/Oxa cells resulted in an enhancement of Oxa-sensitivity,
while DHRS2 silencing suppressed ERCC1 expression. These results
indicated that DHRS2 facilitates HCT116/Oxa cell chemotherapy
sensitivity to Oxa by promoting the expression of ERCC1.
DHRS2 was previously reported to bind to mouse
double minute 2 homolog (MDM2), resulting in the attenuation of
MDM2-mediated p53 degradation (25).
Consistent with this, in the present study it was revealed that p53
was decreased in si-DHRS2 HCT116/Oxa cells and was increased in
DHRS2-overexpressing HCT116/Oxa cells compared with HCT116 cells.
p53 is one of the most well-studied tumor suppressor genes. It
could be activated through a myriad of cellular stresses ranging
from DNA damage to hypoxia, stress, and a plethora of other causes.
Upon activation, p53 acts as a zinc-containing transcription factor
that regulates downstream genes, including p21 which leads to cell
cycle arrest, as well as PUMA/FAS/BAX which induce apoptosis
(34). Previous studies have
demonstrated that p53 also modulates cisplatin sensitivity and the
NER pathway in response to cisplatin-induced DNA damage. Liu et
al (35) reported that p53
directly bound the ERCC1 promoter and induced gene expression,
which protected cells from cisplatin-induced DNA damage. In the
present study, p53 silencing in HCT116/Oxa cells significantly
reduced ERCC1 expression, indicating that the Oxa-induced DNA
damage function of DHRS2 occurs via stabilization of p53 and
upregulation of ERCC1.
Recent studies have revealed that EMT is a vital
process that modulates cancer progression and metastasis. Moreover,
accumulating evidence implicates EMT in drug resistance (36), while some cultured resistant cells
expressing aggressive phenotypes have the EMT phenotype (37). Similarly, in the present study, it was
demonstrated that HCT116/Oxa cells displayed an EMT phenotype, with
decreased E-cadherin and increased vimentin expression, and
enhanced migratory capacity. Further analyses observed the opposite
effects following the silencing of DHRS2. Recently, it was reported
that DHRS2 inhibits cell growth and motility in ESCC (38). Additionally, the overexpression of
miR-145-3p significantly reduced ESCC cancer cell proliferation,
migration, and invasive abilities by silencing DHRS2 (21). A set of transcriptional factors have
been implicated in the regulation of EMT, including Snail, Slug,
ZEB1, and Twist (39). Among them,
Snail and Slug are degraded by ubiquitination via the proteasome
pathway resulting in E-cadherin expression (40,41). It
has been revealed that MDM2, one of the E3 ligases of p53, reduces
lung cancer mobility and metastasis through mediation of Slug
degradation via the ubiquitination proteasome pathway (41,42).
Accordingly, it was speculated that DHRS2 expression may prompt the
migration of CRC cells by interacting with E3 ubiquitin ligase MDM2
and interfere with its function which suppresses degradation of
EMT-inducing transcriptional factor Slug/Snail. Further experiments
are required to elucidate the exact mechanism of DHRS2 in the
regulation of the EMT process in cancer cells.
In conclusion, it was revealed that HCT116/Oxa cells
acquire EMT features and demonstrate increased migration compared
with parental HCT116 cells. It was also revealed that DHRS2
regulated ERCC1 to promote chemotherapy resistance through a
p53-dependent pathway, and mediated EMT by suppressing E-cadherin
expression. The present study identifies DHRS2 as a potential CRC
therapeutic target for simultaneous addressing of cancer metastasis
and chemoresistance.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81502599), the Key Research
and Development Projects in Anhui province (grant no.
201904a07020082) and the Natural Science Foundation of Anhui
Province (grant nos. 1608085MH229 and 1608085QH217).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YQL and HW conceived and designed the experiments.
JML, LZ, FY and WQY performed the experiments. JML and GMJ analyzed
the data and wrote the paper. YQ and HW revised the paper. All
authors read and approved the final manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saika K and Sobue T: Cancer statistics in
the world. Gan To Kagaku Ryoho. 40:2475–2480. 2013.(In Japanese).
PubMed/NCBI
|
|
2
|
Arredondo J, Baixauli J, Pastor C,
Chopitea A, Sola JJ, González I, A-Cienfuegos J, Martínez P,
Rodriguez J and Hernández-Lizoain JL: Mid-term oncologic outcome of
a novel approach for locally advanced colon cancer with neoadjuvant
chemotherapy and surgery. Clin Transl Oncol. 19:379–385. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olaussen KA, Dunant A, Fouret P, Brambilla
E, André F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH,
et al: DNA repair by ERCC1 in non-small-cell lung cancer and
cisplatin-based adjuvant chemotherapy. N Engl J Med. 355:983–991.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Chen Y, Xiang F, Li M, Li H, Chi J
and Ren K: Suppression of TGF-β1 enhances chemosensitivity of
cisplatin-resistant lung cancer cells through the inhibition of
drug-resistant proteins. Artif Cells Nanomed Biotechnol.
46:1505–1512. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu Y, Jin D, Wang X, Du J, Di W, An J,
Shao C and Guo J: UBE2C induces cisplatin resistance via
ZEB1/2-dependent upregulation of ABCG2 and ERCC1 in NSCLC cells. J
Oncol. 2019:86078592019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu DS, Lan HY, Huang CH, Tai SK, Chang
SY, Tsai TL, Chang CC, Tzeng CH, Wu KJ, Kao JY and Yang MH:
Regulation of excision repair cross-complementation group 1 by
Snail contributes to cisplatin resistance in head and neck cancer.
Clin Cancer Res. 16:4561–4571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
He S, Carman CV, Lee JH, Lan B, Koehler S,
Atia L, Park CY, Kim JH, Mitchel JA, Park JA, et al: The tumor
suppressor p53 can promote collective cellular migration. PLoS One.
14:e02020652019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Donadel G, Garzelli C, Frank R and
Gabrielli F: Identification of a novel nuclear protein synthesized
in growth-arrested human hepatoblastoma HepG2 cells. Eur J Biochem.
195:723–729. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gabrielli F and Tofanelli S: Molecular and
functional evolution of human DHRS2 and DHRS4 duplicated genes.
Gene. 511:461–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bray JE, Marsden BD and Oppermann U: The
human short-chain dehydrogenase/reductase (SDR) superfamily: A
bioinformatics summary. Chem Biol Interact. 178:99–109. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bjorkqvist AM, Wolf M, Nordling S,
Tammilehto L, Knuuttila A, Kere J, Mattson K and Knuutila S:
Deletions at 14q in malignant mesothelioma detected by
microsatellite marker analysis. Br J Cancer. 81:1111–1115. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Debiec-Rychter M, Sciot R, Pauwels P,
Schoenmakers E, Dal Cin P and Hagemeijer A: Molecular cytogenetic
definition of three distinct chromosome arm 14q deletion intervals
in gastrointestinal stromal tumors. Genes Chromosomes Cancer.
32:26–32. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao JY, Huang XM, Yu XJ, Huang LX, Wu QL,
Xia JC, Wang HY, Feng QS, Ren ZF, Ernberg I, et al: Loss of
heterozygosity and its correlation with clinical outcome and
Epstein-Barr virus infection in nasopharyngeal carcinoma.
Anticancer Res. 21:3021–3029. 2001.PubMed/NCBI
|
|
17
|
Pellegrini S, Censini S, Guidotti S,
Iacopetti P, Rocchi M, Bianchi M, Covacci A and Gabrielli F: A
human short-chain dehydrogenase/reductase gene: Structure,
chromosomal localization, tissue expression and subcellular
localization of its product. Biochim Biophys Acta. 1574:215–222.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peterson LE, Ozen M, Erdem H, Amini A,
Gomez L, Nelson CC and Ittmann M: Artificial neural network
analysis of DNA microarray-based prostate cancer recurrence.
Proceedings of the IEEE Symposium on Computational Intelligence in
Bioinformatics and Computational Biology. 1–8. 2005.
|
|
19
|
Shedden KA, Taylor JM, Giordano TJ, Kuick
R, Misek DE, Rennert G, Schwartz DR, Gruber SB, Logsdon C, Simeone
D, et al: Accurate molecular classification of human cancers based
on gene expression using a simple classifier with a pathological
tree-based framework. Am J Pathol. 163:1985–1995. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bhattacharyya C, Grate LR, Rizki A,
Radisky D, Molina FJ, Jordan MI, Bissell MJ and Mian IS:
Simultaneous classification and relevant feature identification in
high-dimensional spaces: Application to molecular profiling data.
Signal Processing. 83:729–743. 2003. View Article : Google Scholar
|
|
21
|
Shimonosono M, Idichi T, Seki N, Yamada Y,
Arai T, Arigami T, Sasaki K, Omoto I, Uchikado Y, Kita Y, et al:
Molecular pathogenesis of esophageal squamous cell carcinoma:
Identification of the antitumor effects of miR145-3p on gene
regulation. Int J Oncol. 54:673–688. 2019.PubMed/NCBI
|
|
22
|
Yamada H, Arakawa Y, Saito S, Agawa M,
Kano Y and Horiguchi-Yamada J: Depsipeptide-resistant KU812 cells
show reversible P-glycoprotein expression, hyper-acetylated
histones, and modulated gene expression profile. Leukemia research.
30:723–734. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han Y, Song C, Wang J, Tang H, Peng Z and
Lu S: HOXA13 contributes to gastric carcinogenesis through DHRS2
interacting with MDM2 and confers 5-FU resistance by a
p53-dependent pathway. Mol Carcinog. 57:722–734. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deisenroth C, Thorner AR, Enomoto T, Perou
CM and Zhang Y: Mitochondrial Hep27 is a c-Myb target gene that
inhibits Mdm2 and stabilizes p53. Mol Cell Biol. 30:3981–3993.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cunningham D, Atkin W, Lenz HJ, Lynch HT,
Minsky B, Nordlinger B and Starling N: Colorectal cancer. Lancet.
375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bowden NA: Nucleotide excision repair: Why
is it not used to predict response to platinum-based chemotherapy?
Cancer Lett. 346:163–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park DJ and Lenz HJ: Determinants of
chemosensitivity in gastric cancer. Curr Opin Pharmacol. 6:337–344.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Selvakumaran M, Pisarcik DA, Bao R, Yeung
AT and Hamilton TC: Enhanced cisplatin cytotoxicity by disturbing
the nucleotide excision repair pathway in ovarian cancer cell
lines. Cancer Res. 63:1311–1316. 2003.PubMed/NCBI
|
|
31
|
Reynolds C, Obasaju C, Schell MJ, Li X,
Zheng Z, Boulware D, Caton JR, Demarco LC, O'Rourke MA, Shaw Wright
G, et al: Randomized phase III trial of gemcitabine-based
chemotherapy with in situ RRM1 and ERCC1 protein levels for
response prediction in non-small-cell lung cancer. J Clin Oncol.
27:5808–5815. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cobo M, Isla D, Massuti B, Montes A,
Sanchez JM, Provencio M, Viñolas N, Paz-Ares L, Lopez-Vivanco G,
Muñoz MA, et al: Customizing cisplatin based on quantitative
excision repair cross-complementing 1 mRNA expression: A phase III
trial in non-small-cell lung cancer. J Clin Oncol. 25:2747–2754.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simon G, Sharma A, Li X, Hazelton T, Walsh
F, Chiappori CA, Haur E, Tanvetyanon T, Antonia S, Cantor A and
Bepler G: Feasibility and efficacy of molecular analysis-directed
individualized therapy in advanced non-small-cell lung cancer. J
Clin Oncol. 25:2741–2746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu YC, Chang PY and Chao CC: CITED2
silencing sensitizes cancer cells to cisplatin by inhibiting p53
trans-activation and chromatin relaxation on the ERCC1 DNA repair
gene. Nucleic Acids Res. 43:10760–10781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Zhang G, Zhang H, Zhang F, Zhou B,
Ning F, Wang HS, Cai SH and Du J: Acquisition of
epithelial-mesenchymal transition phenotype and cancer stem
cell-like properties in cisplatin-resistant lung cancer cells
through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol.
723:156–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou Y, Wang L, Ban X, Zeng T, Zhu Y, Li
M, Guan XY and Li Y: DHRS2 inhibits cell growth and motility in
esophageal squamous cell carcinoma. Oncogene. 37:1086–1094. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang SP, Wang WL, Chang YL, Wu CT, Chao
YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, et al: p53 controls
cancer cell invasion by inducing the MDM2-mediated degradation of
Slug. Nat Cell Biol. 11:694–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin TY and Hsu HY: Ling Zhi-8 reduces lung
cancer mobility and metastasis through disruption of focal adhesion
and induction of MDM2-mediated Slug degradation. Cancer Lett.
375:340–348. 2016. View Article : Google Scholar : PubMed/NCBI
|