Introduction
Acute myeloid leukemia (AML) is characterized by
proliferation of immature myeloid leukocytes in the bone marrow as
a consequence of genetic insults, such as mutations (1,2). To
improve the probability of remission in AML patients, clinical
researchers have employed a dose-intensification strategy using
combined chemotherapy with idarubicin or daunorubicin and
cytarabine (AraC) (3,4). Although such efforts have been shown to
increase overall survival, up to 30% of adults and 70% of elderly
individuals (>65 years old) with AML fail to achieve complete
remission after induction chemotherapy, a condition termed
relapsed/refractory (R/R) AML (5,6).
Efforts have been made to develop novel, targeted
drugs for uncontrolled R/R AML, but such efforts are hampered by
our lack of understanding of the pathogenesis and molecular
mechanism underlying R/R AML. Intensive combination chemotherapy
with high-dose AraC, with or without gemtuzumab ozogamicin, has
been shown to achieve a complete remission rate of only ~30% in R/R
AML patients (7). Recently,
fms-related tyrosine kinase 3 (FLT3) containing an internal tandem
duplication (FLT3-ITD) has been identified as a prognostic
molecular marker in patients with a high recurrence rate and poor
outcome, and is considered a treatment target for R/R AML patients
(8). The pan-kinase inhibitor
midostaurin, a staurosporine derivative, has recently been approved
by the US Food and Drug Administration for patients with
FLT3-mutant AML, but the variability of genetic mutations in AML
makes it difficult to reduce the recurrence rate of R/R AML by
targeting specific genes (9,10).
A leukemia stem cell (LSC)-positive population of
cells that is dependent on oxidative respiration rather than
glycolysis for energy generation has been suggested to be a major
cause of R/R AML (11). Notably, a
recent report demonstrated that tigecycline, which targets
mitochondrial translation, selectively kills leukemic stem and
progenitor cells in vivo in AML and chronic myeloid leukemia
(CML) (12,13). However, the association of genes that
regulate intracellular signaling pathways and mitochondrial
oxidative phosphorylation/glycolysis with AML drug resistance is
poorly understood at present.
Phosphatase and tensin homolog (PTEN) is a
phosphatase of the lipid signaling intermediate,
phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which regulates
phosphoinositide 3-phosphate (PI3K) and protein kinase B (AKT)
signaling. The PI3K/AKT pathway is known to promote the survival
and growth of human solid tumors and hematological malignancies in
response to extracellular signals, such as growth factors, hormones
and cytokines (14–16). Moreover, loss of PTEN activity through
mutations, deletions or promoter methylation is frequently found in
solid tumors and hematological malignancies (17–19).
Although mutations in the PTEN gene are rarely observed in AML
patients (~1 in 59 patients) (20),
PTEN expression is generally downregulated in these patients
(20). Moreover, PTEN is considered
to be a biomarker in elderly patients with R/R AML (21). PTEN is crucial for the proliferation
and differentiation of hematopoietic stem cells (HSCs), and
PTEN-null mice initially develop a myeloproliferative disorder,
followed by leukemia (22,23). In addition, PTEN acts as a tumor
suppressor in LSCs without damaging normal HSCs or compromising
hematopoiesis (24). However, whether
PTEN mutations are associated with AML cell proliferation and
changes in metabolic flow during the refractory period remains
unknown.
PTEN-null mouse embryonic fibroblasts exhibit
metabolic changes in association with downregulation of pyruvate
kinase 2, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
(PFKFB3) and glutaminase, consistent with an anti-Warburg effect
(25). In addition, mitochondrial
mass is increased and glycolysis is induced in PTEN-null
hepatocytes via estrogen-related receptor α (26). The present study, using these reports
as a takeoff point, focused on the involvement of PTEN as a
stemness gene in leukemic cell proliferation and induction of
metabolic flow changes in the context of R/R AML. KG1α refractory
AML cells and HL60 wild-type control cells were used to investigate
the association between PTEN/AKT signaling and metabolic shifts in
R/R AML.
Materials and methods
Chemicals, reagents and
antibodies
Oligomycin (O4876), carbonyl cyanide m-chlorophenyl
hydrazone (CCCP; C2759), rotenone (R8875), and AKT inhibitor
(A6730) were purchased from Sigma-Aldrich; Merck KGaA. TRIzol was
purchased from Invitrogen; Thermo Fisher Scientific, Inc. The Cell
Counting Kit-8 (CCK8) was purchased from Dojindo Molecular
Technologies, Inc. Anti-actin (rabbit polyclonal), anti-PARP
(rabbit polyclonal), anti-GAPDH (rabbit polyclonal) and
anti-hexokinase (HK)2 (rabbit polyclonal) antibodies were purchased
from Santa Cruz Biotechnology, Inc. Antibodies against p-AKT
(Thr308) (rabbit polyclonal), total AKT (rabbit polyclonal) and
caspase-3 (rabbit polyclonal) were from Cell Signaling Technology,
Inc. Antibody against oxidative phosphorylation I subunit, NADH
dehydrogenase (ubiquinone) 1 beta subcomplex subunit 8
(anti-NDUFB8) was obtained from Invitrogen; Thermo Fisher
Scientific, Inc., and antibody against NADH dehydrogenase
(ubiquinone) 1 alpha subcomplex 9 (anti-NDUFA9) was purchased from
Abcam.
Human leukemia cell lines
HL60 (ATCC® CCL-240™) and KG1α
(ATCC® CCL-246.1™) cells were purchased from American
Type Culture Collection and cultured in Iscove's Modified
Dulbecco's Medium (Welgene, Inc.) supplemented with 20% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C in a humidified 5% CO2 atmosphere. The two
cell lines were authenticated by STR genotyping. Plasmocin™
Prophylactic (InvivoGen) was used as a routine addition in liquid
media to prevent mycoplasma contamination in cell cultures. All
cell lines were used in our laboratory for <3 months after
resuscitation.
Cell proliferation and cytotoxicity
measurement
For proliferation rate assays, HL60 and KG1α cells
in complete medium were counted and seeded in 96-well culture
plates at 2×103 cells per well. For measurement of drug
cytotoxicity, cells were seeded at 100% confluence in 96-well cell
culture plates and treated with drugs for 24 h. Thereafter, 10 µl
of CCK8 reagent was added to each well and the plates were
incubated for an additional 2 h. Absorbance was then measured at
450 nm using a Multiskan Ascent microplate spectrophotometer
(Thermo Fisher Scientific, Inc.). To quantify apoptosis, necrosis
and late apoptosis, cells were stained using an FITC Annexin V
Apoptosis Detection Kit (BD Bioscience). After seeding at 100%
confluence in 96-well cell culture plates and treating with drugs
for 24 h, the cells were washed with 1 ml PBS and resuspended in
100 µl staining buffer containing 2 µl of Annexin V-FITC and 2 µl
propidium iodide (PI). The cells were then incubated for 15 min at
room temperature, followed by addition of 400 µl of staining buffer
and quantification of fluorescence by flow cytometry. Fluorescence
in Annexin V-FITC (505–560 nm) and PI (595–642 nm) channels was
measured, and at least 10,000 single-cell events per sample were
collected. Using a gating strategy dependent on the fluorescence
intensity of Annexin V-FITC and PI, cell populations were divided
into double-negative (live) cells, Annexin V-positive (apoptotic)
cells, and double-positive (late apoptotic or necroptotic) cells
(27).
Western blot analysis
Whole-cell lysates from HL60 and KG1α cells were
prepared using RIPA lysis buffer [100 mM Tris-HCl pH 8.5, 200 mM
NaCl, 5 mM EDTA, 0.2% sodium dodecyl sulfate (SDS)] containing a
phosphatase and protease inhibitor cocktail (INtRON BioTechnology).
Equal amounts of protein (20 µg), quantified by the Bradford assay
(Bio-Rad Laboratories, Inc.), were resolved by SDS-PAGE
(polyacrylamide 6 or 12% gel electrophoresis) and transferred to an
Amersham Protran nitrocellulose membrane (pore size, 0.2 µm;
Amersham; GE Healthcare Life Sciences). The membranes were blocked
by incubating for 1 h with TBST (10 mM Tris-HCL pH 7.6, 150 mM
NaCl, 0.1% Tween-20) containing 3% skimmed milk, and were then
sequentially incubated with primary antibody (16 h at 4°C) and the
appropriate horseradish peroxidase-conjugated secondary antibody (2
h at room temperature). Specific antibody-labeled proteins were
detected using an enhanced chemiluminescence (ECL) system (WEST-ZOL
plus; INtRON BioTechnology).
Oxygen consumption rate (OCR) and
extracellular acidification rate (ECAR)
OCR and ECAR were measured using an xf24
analyzer (Seahorse Bioscience). On day-1, the xf24 biosensor
cartridge was pre-incubated with 1 ml of xf24 calibration
buffer and then incubated at 37°C overnight without CO2.
The cells were acutely treated as described with AKT inhibitor,
idarubicin and/or AraC, delivered via port A, for 24 h. For
measurement of OCR, HL60 and KG1α cells were seeded onto
xf24 cell culture microplates at 1×104 cells per
well in 0.5 ml xf24 medium consisting of Dulbecco's modified
Eagle's medium (pH 7.4) supplemented with 10 mM glucose, 1 mM
sodium pyruvate and 2 mM L-glutamine (without sodium bicarbonate),
and then incubated at 37°C without CO2 for 1 h. The
xf24 analyzer was operated according to the manufacturer's
basic protocol at 37°C with real-time injection of drugs. After
equilibrating for 20 min, each well of the xf24 cartridge
was sequentially injected with the ATPase inhibitor oligomycin (2
µg/ml), the uncoupling agent CCCP (5 µM) and the mitochondrial
electron transport inhibitor rotenone (2 µM), and OCR and ECAR were
measured in real time. For glycolysis stress tests, HL60 and KG1α
cells (1×104 cells per well) were seeded onto
xf24 cell culture microplates containing 0.5 ml Agilent
Seahorse XF Base Medium (Seahorse Bioscience) and 1 mM L-glutamine,
and then incubated at 37°C without CO2 for 45 min. Each
well of the xf24 cartridge was sequentially injected with
glucose (10 mM) for glycolysis, oligomycin (1 µM) for glycolytic
capacity and 2-deoxy glucose for glycolytic reserve
measurement.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. cDNA was synthesized from 20 µg of
total RNA using M-MLV Reverse Transcriptase and oligo-dT primers
(Invitrogen; Thermo Fisher Scientific, Inc.). RT-qPCR was performed
using cDNA, SYBR Green PCR Master Mix (iCycler iQ Real-Time PCR
Detection System; Bio-Rad Laboratories, Inc.) and the following
primer pairs: HK1, 5′-ggccacgatgtagtcacctt-3′ (forward) and
5′-cacgtccaggtcaaattcct-3′ (reverse); HK2,
5′-ccacctttgtgaggtccact-3′ (forward) and 5′-gtcctcagggatggcataga−3′
(reverse); SLC2A1, 5′-cccagatctttggtctggaa-3′ (forward) and
5′-gactttcagggcaaaatgga-3′ (reverse); PFK1,
5′-agagcgtttcgatgatgctt-3′ (forward) and 5′-gttgtaggcagctcggagtc-3′
(reverse); PDK3, 5′-ctaggtggtggtgtcccact-3′ (forward) and
5′-taaccaaatccagccaaagg-3′ (reverse); and 18s ribosomal RNA,
5′-ctggttgatcctgccagtag-3′ (forward) and 5′-cgaccaaaggaaccataact-3′
(reverse). The relative expression of target mRNAs was quantified
and normalized with respect to that of 18S rRNA, which was used as
an endogenous control. Rotor-Gene 6000 real-time rotary analyzer
software (version 1.7; Corbett Life Science-Qiagen) was utilized
along with the 2−ΔΔCq method (28).
Statistical analysis
Results are presented as mean ± standard error of
the mean from at least three independent experiments. A two-tailed
unpaired Student's t-test or one-way analysis of variance with
Friedman test were performed using Graph Pad InStat software
(GraphPad Software, Inc.). A P-value <0.05 was considered to
indicate statistically significant differences.
Results
Downregulation of PTEN and
phosphorylated AKT is associated with refractory AML
LSCs contribute to the central pathogenesis of R/R
AML by sustaining stemness properties and continued propagation of
leukemic cells after induction therapy. Using the KG1α leukemia
cell line as an LSC-like cell model (29) with potential relevance to refractory
AML (30), we first investigated
responsiveness to the AML chemotherapeutic agents, idarubicin and
AraC, compared with HL60 cells. Interestingly, treatment with
idarubicin or AraC alone differentially affected cell viability,
causing ~30% less cytotoxicity against KG1α cells compared with
HL60 cells. Notably, combined treatment with idarubicin and AraC
for 24 h also decreased the viability of HL60 cells without
affecting KG1α cells (Fig. 1A). These
changes in cell viability induced by idarubicin and AraC treatment
were associated with diminished induction of cleaved
poly(ADP-ribose) polymerase (PARP) and cleaved caspase-3 in KG1α
cells, indicating that the remaining KG1α cells are resistant to
and less damaged by idarubicin and AraC (Fig. 1B). To determine the death rate in HL60
cells treated with idarubicin, AraC and idarubicin plus AraC,
Annexin V/PI staining was performed with quantification by flow
cytometry. This analysis revealed that idarubicin, AraC and
idarubicin/AraC increased Annexin V staining in HL60 cells (CTL,
0.5%; idarubicin, 66.3%; AraC, 1.3%; idarubicin/AraC, 63.86%),
which is indicative of early apoptosis. Moreover, idarubicin, AraC
and idarubicin/AraC increased the percentage of Annexin V/PI
double-stained HL60 cells (CTL, 0.6%; idarubicin, 12.5%; AraC,
8.7%; idarubicin/AraC, 31.6%), which is indicative of late
apoptosis and necroptosis (Fig.
1C-E). However, KG1α cells exhibited a slight increase in the
ratio of Annexin V/PI double-stained cells by idarubicin/AraC.
These results suggest that idarubicin and AraC were associated with
a higher apoptotic ratio in HL60 cells compared with KG1α cells.
Next, in order to determine the differences in PTEN expression in
refractory AML, PTEN protein expression was assessed in KG1α and
HL60 cells, and PTEN protein levels were found to be downregulated
in KG1α cells compared with HL60 cells (Fig. 1F). Given previous reports suggesting
that Thr308 phosphorylation of AKT is correlated with poor overall
survival of AML patients (31), the
AKT phosphorylation status was confirmed in the two cell lines. AKT
was found to be constitutively phosphorylated at Thr308 in KG1α
cells (i.e., in the absence of a stimulus) (Fig. 1F). Collectively, these findings
indicate that low expression of PTEN and high
expression/phosphorylation of AKT are correlated with a refractory
AML phenotype against combined treatment with idarubicin and
AraC.
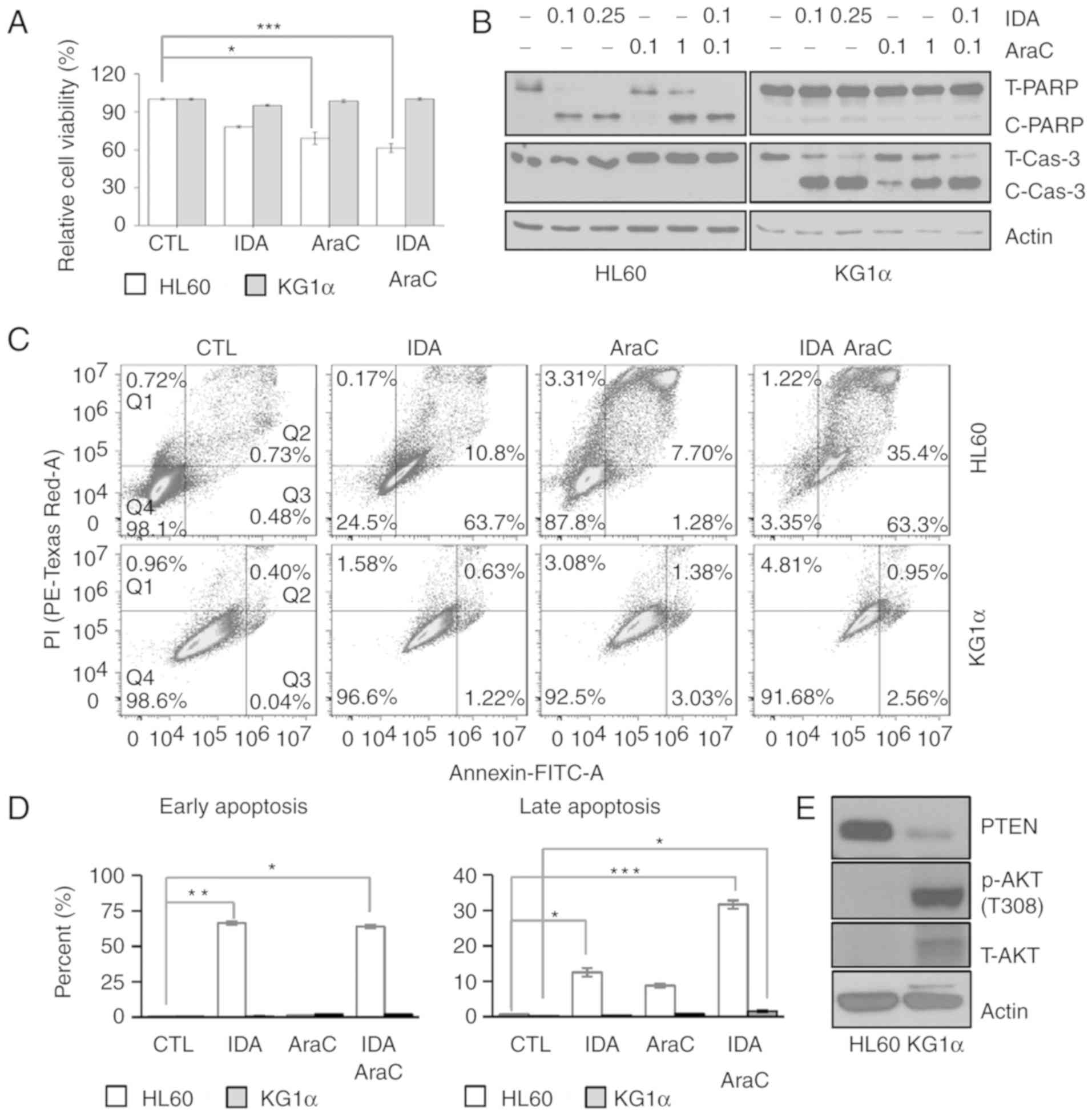 | Figure 1.Differences in drug sensitivity and
PTEN/AKT levels between HL60 and KG1α cells. (A) The viability of
HL60 and KG1α cells, with/without treatment with 0.1 and 0.25 µM
idarubicin and 0.1 and 1 µg/ml AraC, was measured by CCK8 assays
(n=8, *P<0.05, ***P<0.005 vs. CTL). (B) Western blot analysis
of the apoptosis marker proteins PARP and cleaved caspase-3; actin
was used as an internal control. (C and D) Dot plot for flow
cytometric analysis of apoptotic cells after Annexin V-FITC/PI
staining in HL60 and KG1α cells treated with 0.1 µM idarubicin and
0.1 µg/ml AraC. Living cells (Q4) tested negative for both Annexin
V-FITC and PI. Populations testing Annexin V-positive/PI-negative
were classified as early-stage apoptotic cells (Q3), and
double-positive cells were classified as late-stage apoptotic cells
(Q2). (E) Percentage of early and late apoptotic HL60 and KG1α
cells following idarubicin and AraC treatment. (n=3, *P<0.05,
**P<0.01, ***P<0.005 vs. CTL). (F) Western blot analysis of
PTEN, p-AKT (Thr308) and total AKT in HL60 and KG1α cells. PTEN,
phosphatase and tensin homologue; AKT, protein kinase B; PARP,
poly(ADP-ribose) polymerase; PI, propidium iodide; AraC,
cytarabine; IDA, idarubicin; CTL, control. |
The levels of glycolytic
metabolism-related enzymes are higher in KG1α cells compared with
HL60 cells
AKT is involved in the regulation of cell
proliferation and death. Notably, it was recently demonstrated that
overexpression of a constitutively active AKT mutant in cancer
cells upregulates glycolysis by inducing glucose transporter
(GLUT)4 and HK, thereby leading to sustained growth and enhanced
cancer cell survival (32). Moreover,
activation of the AKT cascade effectively induced GLUT1 gene
expression in mouse hepatoma Hepa1c1c7 cells (33). Accordingly, as AKT levels were higher
in refractory AML KG1α cells compared with HL60 cells, we assessed
the expression of genes involved in regulating glucose uptake and
metabolic flow, including mitochondrial enzymes, by RT-qPCR and
western blotting. First, we found that the mRNA expression levels
of GLUT1/SLC2A1 were increased in KG1α cells compared with HL60
cells (Fig. 2A). Moreover, HK1 and
HK2, the initial enzymes of glycolysis, were expressed at higher
levels in KG1α compared with HL60 cells (Fig. 2B and C). In addition, the expression
of phosphofructokinase 1, the most important regulatory enzyme in
this process, was increased more than two-fold in KG1α compared
with HL60 cells. (Fig. 2D).
Consistent with these mRNA expression data, the GLUT1 and HK2
proteins were also more highly expressed in KG1α compared with HL60
cells (Fig. 2F). The final product of
glycolysis is pyruvate, which is translocated to the mitochondria
for the generation of mitochondrial complex I and II substrates via
the tricarboxylic acid cycle. The pyruvate dehydrogenase complex
converts pyruvate to acetyl CoA, which is downregulated by pyruvate
dehydrogenase kinase (PDK) in the mitochondria. As expected, the
PDK3 expression levels were lower in KG1α cells compared with HL60
cells, thereby enhancing mitochondrial respiration by supplying the
substrates, NADH and FADH, despite the absence of a change in the
expression levels of mitochondrial complex subunit proteins
(Fig. 2E).
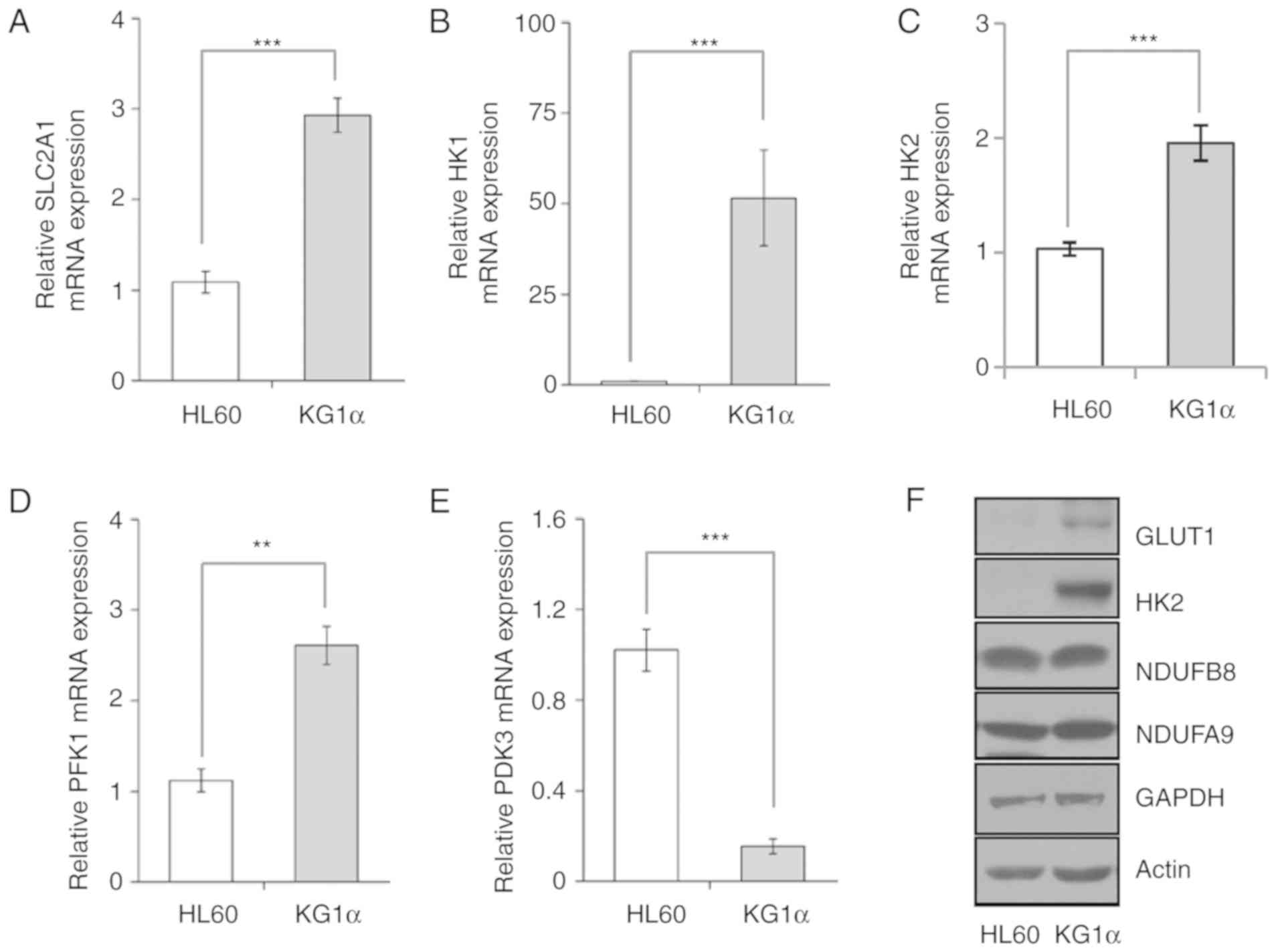 | Figure 2.Expression of glycolysis-related
genes in HL60 and KG1α cells. (A-E) mRNA levels of (A)
SLC2A1/GLUT1, (B) HK1, (C) HK2, (D) PFK1 and (E) PDK3, as
determined by quantitative polymerase chain reaction analysis (n=5
or 6, **P<0.01, ***P<0.005 vs. HL60). (F) Western blot
analysis of GLUT1, HK2, NDUFB8, NDUFA9 and GAPDH; actin was used as
a loading control. GLUT, glucose transporter; HK, hexokinase; PFK,
phosphofructokinase; PDK, pyruvate dehydrogenase kinase. |
Mitochondrial respiration and
glycolysis capacity are higher in KG1α compared with HL60
cells
To verify the correlation between upregulated
expression of glycolytic genes and physiological changes in
glycolysis rate in KG1α cells, glycolytic activity was assessed by
measuring the ECAR and OCR using an xf24 analyzer.
Consistent with the observed gene expression profile, a glucose
stress test using glucose, oligomycin and 2-deoxyglucose showed
higher glycolytic capacity in KG1α cells compared with that in HL60
cells (Fig. 3A). Next, cellular acid
production was quantified by measuring the hydrogen concentration
in the medium. Compared with HL60 cells, KG1α cells maintained a
low-pH environment from day 2 onwards (Fig. 3B). Interestingly, basal and maximal
OCRs were also higher in KG1α cells compared with those in HL 60
cells (Fig. 3C). Taken together,
these data indicate that PTEN-mutant KG1α cells are characterized
by more prominent mitochondrial respiration and glycolytic pathway
flux compared with HL60 cells.
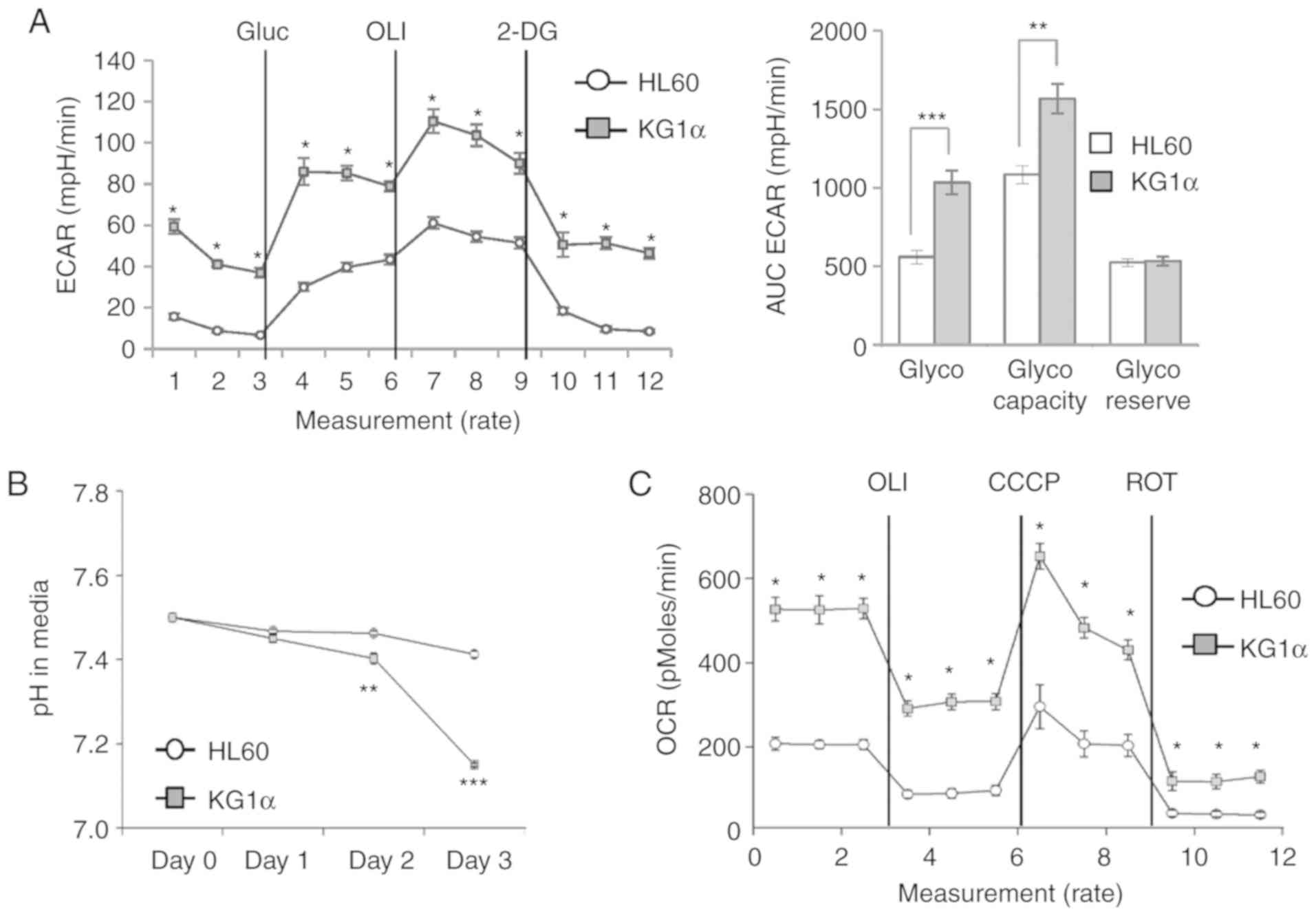 | Figure 3.Metabolic phenotype in HL60 and KG1α
cells. (A) ECAR in HL60 and KG1α cells, determined using an
xf24 analyzer. Glucose (Gluc; 10 mM) was used for
glycolysis; oligomycin (OLI; 1 µM) was used for glycolytic
capacity; and 2-deoxyglucose (2-DG; 50 mM) was used for calculation
of the glycolytic reserve. Left panel, real-time ECAR; right panel,
quantification of each step of ECAR (n=4, *P<0.05, **P<0.01,
***P<0.005 vs. HL60). (B) pH levels in media after culturing
HL60 and KG1α cells for 48 h (n=4, **P<0.01, ***P<0.005 vs.
HL60). (C) OCR in HL60 and KG1α cells, determined using an
xf24 analyzer (n=7 or 8, *P<0.05 vs. HL60). OLI 2 µg/ml
was used for uncoupling; CCCP (5 µM) was used to induce maximal
OCR; and rotenone (ROT, 2 µM) was used to assess non-mitochondrial
OCR. ECAR, extracellular acidification rate; OCR, oxygen
consumption rate. |
Inhibition of AKT inhibits refractory
AML cell growth through downregulation of glycolysis
To confirm the role of AKT in refractory AML cell
proliferation, mitochondrial OCR and ECAR were measured in HL60 and
KG1α cells in real time using an xf24 analyzer following
introduction of an AKT inhibitor into an xf24 cartridge. As
expected, acute injection of the AKT inhibitor reduced ECAR and
enhanced maximal and basal OCR (Fig.
4A-C). Due to the low levels of AKT phosphorylation in HL60
cells, the effect of AKT inhibitors on AKT phosphorylation was
confirmed by basal or pharmacological stimulation. Since it is
known that insulin and insulin growth factor (IGF)2 promote AKT
phosphorylation and affect cell survival in AML (34,35), we
investigated AKT phosphorylation following treatment with insulin
and IGF2 in HL60 and KG1α cells. Pretreatment with AKT inhibitor
was applied 4 h prior to the addition of 100 nM insulin, IGF2 (100
ng/ml) or vehicle for 2 h. As a result, AKT inhibitors reduced AKT
phosphorylation under basal and pharmacological-stimulated
conditions in HL60 and KG1α cells (Fig.
4D). Under these conditions, ECAR was prolonged and decreased
in KG1α cells, but OCR remained unchanged (Fig. 4E and F). Inhibition of AKT ultimately
reduced cell proliferation only in AKT-hypersensitive KG1α cells
(Fig. 4G). These results suggest that
AKT activation affects refractory AML cell proliferation in a
glycolysis-dependent manner.
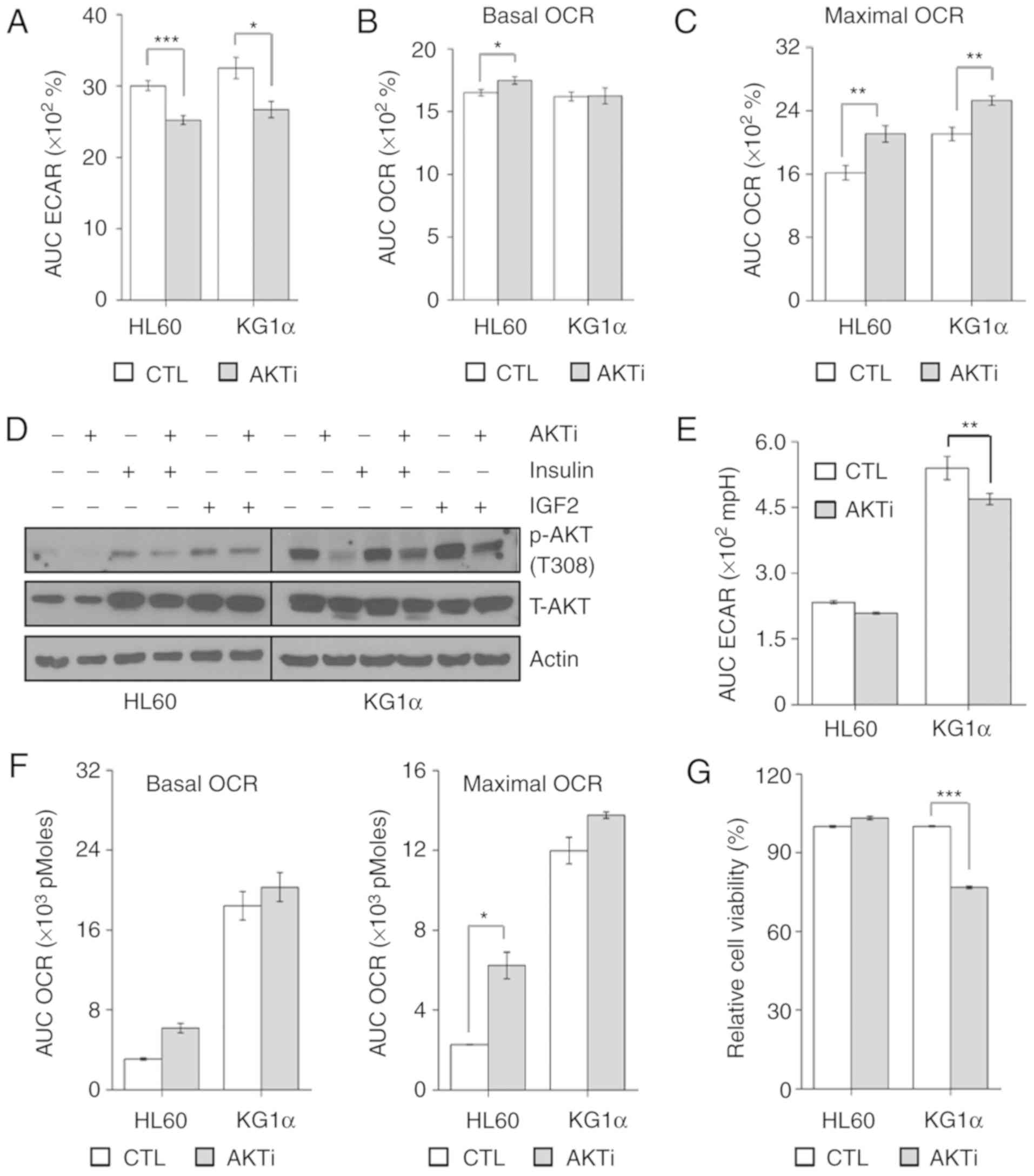 | Figure 4.AKT inhibition reduces the
proliferation of KG1α cells. (A and B) Measurement of (A) ECAR and
(B) OCR in KG1α cells treated with AKT inhibitor (AKTi) for 20 min
(n=4, *P<0.05, ***P<0.005 vs. CTL). (C) CCCP (5 µM) induces
maximal OCR in KG1α cells treated with AKTi for 40 min (n=4,
**P<0.001 vs. CTL). (D) Immunoblotting of total cell lysates for
p-AKT (Thr308) and total AKT after treatment with AKTi (1 µM) for 6
h with/without insulin (100 nM) or IGF2 (100 ng/ml) for 2 h. Actin
was used as a loading control. (E) Measurement of ECAR in HL60 and
KG1α cells treated with AKTi (n=4, **P<0.001 vs. CTL). (F)
Measurement of OCR in HL60 and KG1α cells treated with AKTi (n=4,
*P<0.05 vs. CTL). CCCP (5 µM) was used to induce maximal OCR.
(G) Viability of HL60 and KG1α cells after treatment with 1 µM AKTi
for 24 h, determined by CCK8 assay (n=8, ***P<0.005 vs. CTL).
ECAR, extracellular acidification rate; OCR, oxygen consumption
rate; CTL, control; IGF, insulin-like growth factor. |
Targeting the PTEN/AKT pathway by
inhibiting AKT enhances the drug sensitivity of refractory AML
cells
As noted above (see Fig.
1A), refractory AML KG1α cells exhibited diminished
responsiveness to chemotherapy (idarubicin and AraC). To test the
role of AKT in drug sensitivity, KG1α cells were treated with
idarubicin plus Ara-C, idarubicin plus Ara-C and an AKT inhibitor,
or AKT inhibitor alone for 24 h, and ECAR and OCR were assessed
using an xf24 analyzer; cell viability was also measured
using Annexin V/PI staining and CCK8 assays. Consistent with the
results presented in Fig. 4, we found
that treatment with AKT inhibitor decreased glycolysis, whereas
treatment with idarubicin and AraC did not (Fig. 5A). In addition, co-treatment with AKT
inhibitor and idarubicin and AraC further reduced glycolysis,
glycolytic capacity, and glycolytic reserve. Interestingly, the
three-drug combination (idarubicin, AraC and AKT inhibitor) reduced
basal OCR, whereas treatment with individual drugs produced little
change in OCR (Fig. 5B). To determine
the cell apoptotic ratio, Annexin V/PI staining was used in
conjunction with flow cytometry to quantify the apoptotic rate of
KG1α cells following treatment with idarubicin, AraC, and AKT
inhibitor. Although CCK8 assays revealed that cell viability was
reduced by treatment with idarubicin and AraC (4.2%) or AKT
inhibitor (14.4%), co-treatment with AKT inhibitor and idarubicin
and AraC further reduced viability (26.6%) (Fig. 5C), without increasing Annexin
V-stained and Annexin V/PI double-stained apoptotic cells (Fig. 5D). In conclusion, AKT inhibition
enhances chemotherapeutic susceptibility in KG1α cells by
suppressing the AKT signaling pathway and decreasing proliferative
potential without inducing apoptosis.
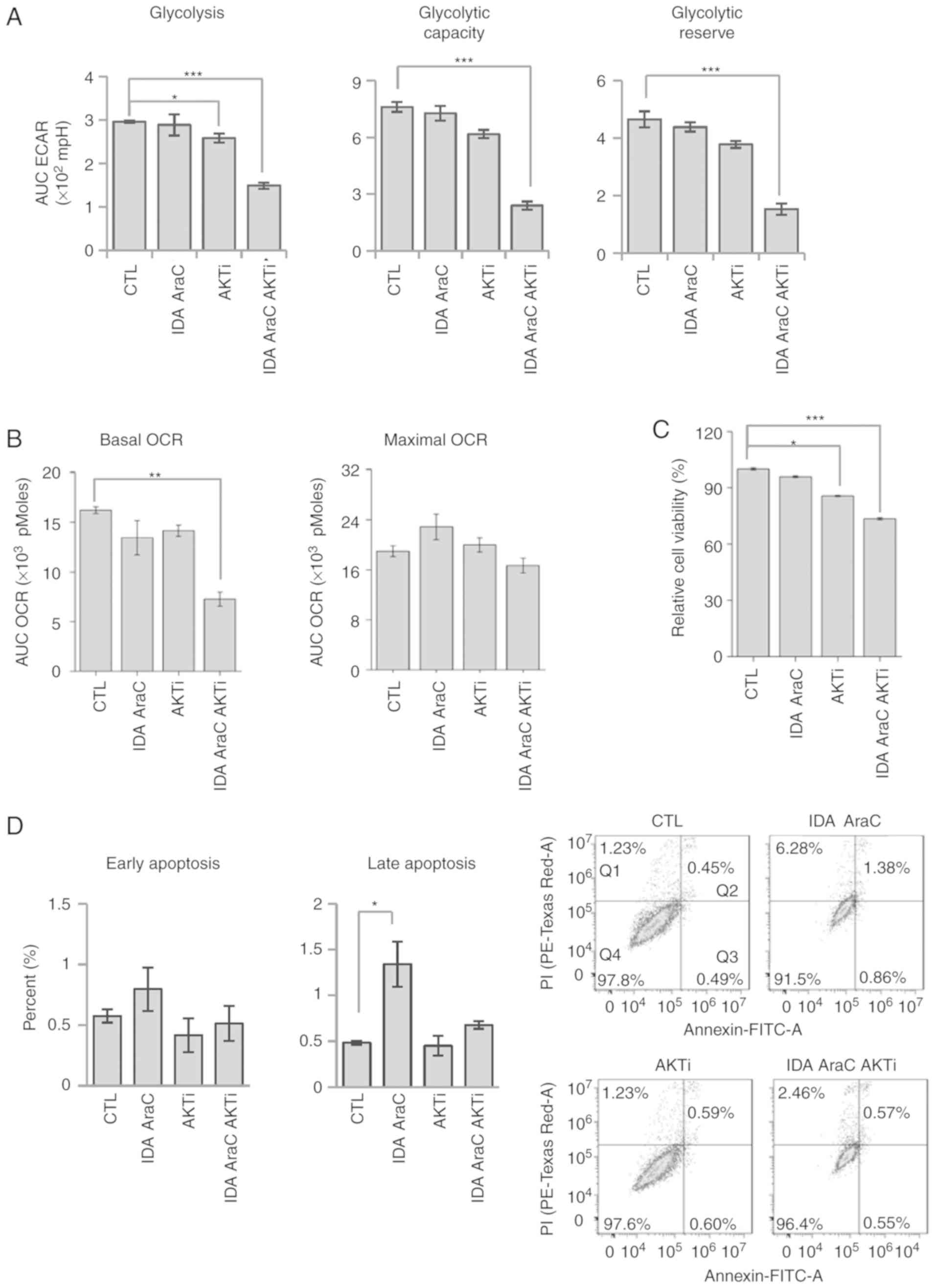 | Figure 5.AKT inhibition reduces the viability
of chemoresistant KG1α cells. (A) Measurement of ECAR in KG1α cells
treated with IDA, AraC, and AKT inhibitor (AKTi) for 24 h.
Quantification of glycolysis, glycolytic capacity, and glycolytic
reserve. (n=4, *P<0.05, ***P<0.005 vs. CTL). (B) Measurement
of OCR in KG1α cells treated with IDA, AraC, and AKTi for 24 h
(n=4, **P<0.01 vs. CTL). CCCP (5 µM) was used to induce maximal
OCR. (C) Viability of KG1α cells treated with IDA, AraC and AKTi
for 24 h, as determined by CCK8 assays. (n=8, *P<0.05,
***P<0.005 vs. CTL). (D) Percentage of early and late apoptotic
KG1α cells following treatment with IDA, AraC and AKTi for 24 h
(n=3, *P<0.05 vs. CTL). Living cells (Q4) tested negative for
both Annexin V-FITC and PI. Populations testing Annexin
V-positive/PI-negative were classed as early-stage apoptotic cells
(Q3), whereas double-positive cells were classed as late-stage
apoptotic cells (Q2). ECAR, extracellular acidification rate; OCR,
oxygen consumption rate; AraC, cytarabine; IDA, idarubicin; PI,
propidium iodide; CTL, control. |
Discussion
Resistance to intensive therapy is a major hurdle in
the treatment of AML (36). Several
new therapeutic approaches targeting mutant genes have been tested
in an effort to overcome drug resistance (36,37).
Despite such efforts, however, the molecular mechanisms responsible
for refractory drug responses remain unidentified. Importantly,
treatments based on the different properties of R/R AML cells must
be developed as a common chemotherapy for AML. The results of the
present study suggest that inhibiting the activity of AKT, which is
a prominent metabolic modulator in R/R AML, is a promising new
therapeutic strategy.
Inhibition of anaerobic glycolysis is important in
relapse and chemoresistance. For example, chemoresistant colon
cancer cells exhibit defective mitochondrial ATP production and
enhanced aerobic glycolysis (38). It
has also been demonstrated that glycolysis is increased and the
efficiency of mitochondrial oxidative phosphorylation is reduced in
drug-resistant HL60 ADR cells compared with the drug-sensitive HL60
cells (39). Consistent with those
findings, the results of the present study demonstrated that
glycolysis and mitochondrial oxidative phosphorylation activity
were both increased in KG1α cells, which exhibited resistance to
idarubicin and AraC, compared with control HL60 cells. Our data
further suggested that this phenotype of KG1α cells may result from
downregulation of PTEN, which is mutated in these cells.
Although PTEN mutations are rare in AML patients,
they have been found to be correlated with refractory AML (20). Clinically, AML patients with a low
level of PTEN and a high level of cyclin D1 expression have a high
rate of relapse within 1 year (40).
In addition, elderly PTEN-positive, CD44-negative AML patients
survive significantly longer compared with PTEN-negative,
CD44-positive patients (21).
Although the association between PTEN mutation/expression and R/R
AML has been investigated, the metabolic status of PTEN-mutated,
refractory AML cells has not been fully elucidated. The present
study demonstrated that PTEN affects metabolic flow and cell
viability in refractory AML cell lines through inhibition of its
downstream target, AKT. These results indicate that PTEN is a
regulator of refractory AML cells through enhancement of
glycolysis.
LSCs, which are capable of producing identical
daughter cells as well as differentiated cells, are considered a
major cause of relapse and a serious obstacle to the successful
treatment of AML (41). As cells of
the KG1α leukemic cell line are considered to be LSC-like, they may
also be used to evaluate whether the differential metabolic flow in
LSCs underlies R/R AML. The metabolism of cancer cells is remodeled
from oxidative phosphorylation to aerobic glycolysis even under
aerobic conditions, a process termed the ‘Warburg effect’, which
serves to support rapid growth and avoid hypoxia (42). The Warburg effect was initially
hypothesized to result from diminished mitochondrial function
(43); however, despite utilizing
aerobic glycolysis, most cancer cells also consume oxygen (44,45).
Activation of mitochondrial metabolism through mitochondrial
biogenesis and quality control has been reported to maintain the
supply of macromolecules to cancer cells (46). Cancer cells use glutamate as an
important alternative carbon source to glucose. Previous
experimental studies have demonstrated that cancer cells use
glutamate for protein anabolism, and that Krebs cycle
intermediates, through glutaminolysis, may act as building blocks
for macromolecules necessary for growth and proliferation (47,48).
Moreover, glutamine-driven oxidative phosphorylation supports ATP
production in murine renal epithelial cells (49). In leukemia, LSC-positive cells are
dependent on oxidative respiration rather than glycolysis for
energy generation (11).
AraC-resistant pre-existing and persisting cells display increased
mitochondrial mass and retain active polarized mitochondria; this
is consistent with a high oxidative phosphorylation status and not
an LSC phenotype (13). In addition,
upregulation of oxidative metabolism supports the survival of
primitive CML cells, and combination treatment with imatinib and
tigecycline, an antibiotic that inhibits mitochondrial protein
translation, selectively eradicates CML LSCs in vitro as
well as in vivo (50). KG1α
cells with a highly oxidative respiratory capacity display
characteristics of LSC-like cells and expression of the refractory
AML phenotype. This observation highlights the need for further
studies on how the regulation of mitochondrial function contributes
to the survival of PTEN mutant, refractory leukemia cells.
In clinical trials, AKT inhibitors have proven to be
highly toxic or cause only incomplete response in patients with
advanced acute leukemia (51). The
present study demonstrated that conventional chemotherapy together
with AKT inhibitors, but not AKT inhibitors alone, reduce the
viability of refractory AML cells by reducing mitochondrial OCR and
glycolysis. Taken together, these findings suggest that metabolic
deterioration mediated by AKT inhibition is involved in promoting
susceptibility to the drug-refractory AML phenotype, providing a
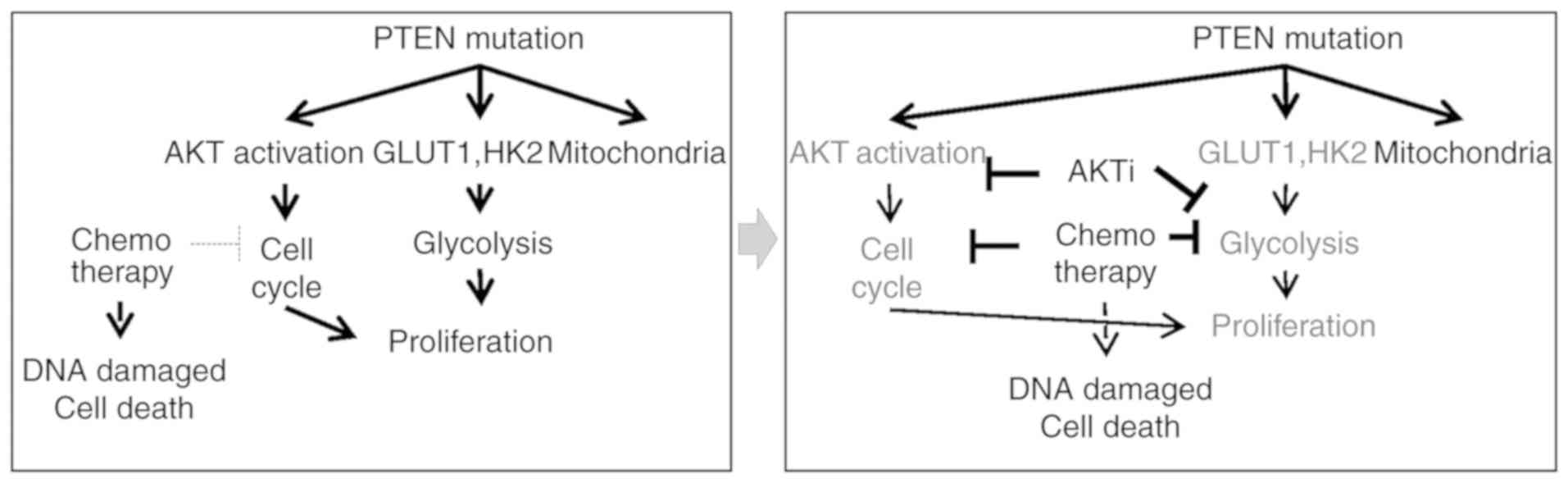
rationale for further study of targeted therapy (Fig. 6).
Acknowledgements
Not applicable.
Funding
The present study was supported by National Research
Foundation of Korea (NRF) grants funded by the Ministry of Science
and ICT (MSIT) (nos. 2016R1A6A3A11935284, 2016R1A2B4010398 and
2017R1A5A2015385 and 2019M3E5D1A02068575) and by the Ministry of
Education (nos. 2014R1A6A1029617 and 2016R1D1A1B03932766).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MJR, JYH and GRK made substantial contributions to
the conception and design of the study. MJR was responsible for the
acquisition of data and JH provided materials for the experiments.
MJR and JYH helped with the analysis and interpretation of the
data. MJR, XJ, WC, JYH and GRK wrote the manuscript. SJK, MJL, YLL,
JHS, JC, YSJ and ICS contributed to the discussion and revised the
article, and approved the final version of the manuscript. GRK and
JYH were responsible for the integrity of the work as a whole. All
the authors have read and approved the final version of the
manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
R/R AML
|
relapsed/refractory acute myeloid
leukemia
|
|
LSC
|
leukemia stem cell
|
|
ECAR
|
extracellular acidification rate
|
|
OCR
|
oxygen consumption rate
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute Myeloid Leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gale RP: Advances in the treatment of
acute myelogenous leukemia. N Engl J Med. 300:1189–1199. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sakashita A, Hattori T, Miller CW,
Suzushima H, Asou N, Takatsuki K and Koeffler HP: Mutations of the
p53 gene in adult T-cell leukemia. Blood. 79:477–480.
1992.PubMed/NCBI
|
|
4
|
Kimby E, Nygren P and Glimelius B;
SBU-group: Swedish Council of Technology Assessment in Health Care:
A systematic overview of chemotherapy effects in acute myeloid
leukaemia. Acta Oncol. 40:231–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mohammadi M, Cao Y, Glimelius I, Bottai M,
Eloranta S and Smedby KE: The impact of comorbid disease history on
all-cause and cancer-specific mortality in myeloid leukemia and
myeloma-a Swedish population-based study. BMC Cancer. 15:8502015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Kouchkovsky I and Abdul-Hay M: Acute
myeloid leukemia: a comprehensive review and 2016 update'. Blood
Cancer J. 6:e4412016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stone RM, Moser B, Sanford B, Schulman P,
Kolitz JE, Allen S, Stock W, Galinsky I, Vij R, Marcucci G, Hurd D,
et al: High dose cytarabine plus gemtuzumab ozogamicin for patients
with relapsed or refractory acute myeloid leukemia: Cancer and
leukemia Group B study 19902. Leuk Res. 35:329–333. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aziz H, Ping CY, Alias H, Ab Mutalib NS
and Jamal R: Gene mutations as emerging biomarkers and therapeutic
targets for relapsed acute myeloid leukemia. Frontiers in
Pharmacology. 8:8972017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levis M: Midostaurin approved for
FLT3-mutated AML. Blood. 129:3403–3406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stein EM, DiNardo CD, Pollyea DA, Fathi
AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn
IW, et al: Enasidenib in mutant IDH2 relapsed or refractory
acute myeloid leukemia. Blood. 130:722–731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lagadinou ED, Sach A, Callahan K, Rossi
RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer
KM, et al: BCL-2 inhibition targets oxidative phosphorylation and
selectively eradicates quiescent human leukemia stem cells. Cell
Stem Cell. 12:329–341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Škrtić M, Sriskanthadevan S, Jhas B,
Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N,
et al: Inhibition of mitochondrial translation as a therapeutic
strategy for human acute myeloid leukemia. Cancer Cell. 20:674–688.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuntz EM, Baquero P, Michie AM, Dunn K,
Tardito S, Holyoake TL, Helgason GV and Gottlieb E: Targeting
mitochondrial oxidative phosphorylation eradicates
therapy-resistant chronic myeloid leukemia stem cells. Nat Med.
23:1234–1240. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manning BD and Toker A: AKT/PKB Signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hawkins PT and Stephens LR: PI3K
signalling in inflammation. Biochim Biophys Acta. 1851:882–897.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao HF, Wang J and Tony To SS: The
phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase
signaling in cancer: Alliance or contradiction? (Review). Int J
Oncol. 47:429–436. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang YH, Lee HS and Kim WH: Promoter
methylation and silencing of PTEN in gastric carcinoma. Lab Invest.
82:285–291. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
García JM, Silva J, Peña C, Garcia V,
Rodríguez R, Cruz MA, Cantos B, Provencio M, España P and Bonilla
F: Promoter methylation of the PTEN gene is a common molecular
change in breast cancer. Genes Chromosomes Cancer. 41:117–124.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alvarez-Nunez F, Bussaglia E, Mauricio D,
Ybarra J, Vilar M, Lerma E, de Leiva A and Matias-Guiu X; Thyroid
Neoplasia Study Group, : PTEN promoter methylation in sporadic
thyroid carcinomas. Thyroid. 16:17–23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu TC, Lin PM, Chang JG, Lee JP, Chen TP
and Lin SF: Mutation analysis of PTEN/MMAC1 in acute myeloid
leukemia. Am J Hematol. 63:170–175. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang X, Li D, Li T, Zhao BO and Chen X:
Prognostic value of the expression of phosphatase and tensin
homolog and CD44 in elderly patients with refractory acute myeloid
leukemia. Oncol Lett. 10:103–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yilmaz OH, Valdez R, Theisen BK, Guo W,
Ferguson DO, Wu H and Morrison SJ: Pten dependence distinguishes
haematopoietic stem cells from leukaemia-initiating cells. Nature.
441:475–482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang J, Grindley JC, Yin T, Jayasinghe S,
He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM,
et al: PTEN maintains haematopoietic stem cells and acts in lineage
choice and leukaemia prevention. Nature. 441:518–522. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fragoso R and Barata JT: PTEN and leukemia
stem cells. Adv Biol Regul. 56:22–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L, Xiong H, Wu F, Zhang Y, Wang J,
Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, et al: Hexokinase
2-mediated Warburg effect is required for PTEN- and
p53-deficiency-driven prostate cancer growth. Cell Rep.
8:1461–1474. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, He L, Zeng N, Sahu D, Cadenas E,
Shearn C, Li W and Stiles BL: Phosphatase and tensin homolog
deleted on chromosome 10 (PTEN) signaling regulates mitochondrial
biogenesis and respiration via estrogen-related receptor α (ERRα).
J Biol Chem. 288:25007–25024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pietkiewicz S, Schmidt JH and Lavrik IN:
Quantification of apoptosis and necroptosis at the single cell
level by a combination of Imaging Flow Cytometry with classical
Annexin V/propidium iodide staining. J Immunol Methods. 423:99–103.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
She M, Niu X, Chen X, Li J, Zhou M, He Y,
Le Y and Guo K: Resistance of leukemic stem-like cells in AML cell
line KG1a to natural killer cell-mediated cytotoxicity. Cancer
Lett. 318:173–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wiernik PH, Banks PL, Case DC Jr, Arlin
ZA, Periman PO, Todd MB, Ritch PS, Enck RE and Weitberg AB:
Cytarabine plus idarubicin or daunorubicin as induction and
consolidation therapy for previously untreated adult patients with
acute myeloid-leukemia. Blood. 79:313–319. 1992.PubMed/NCBI
|
|
30
|
Gallay N, Dos Santos C, Cuzin L, Bousquet
M, Simmonet Gouy V, Chaussade C, Attal M, Payrastre B, Demur C and
Récher C: The level of AKT phosphorylation on threonine 308 but not
on serine 473 is associated with high-risk cytogenetics and
predicts poor overall survival in acute myeloid leukaemia.
Leukemia. 23:1029–1038. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and
Thompson CB: Akt stimulates aerobic glycolysis in cancer cells.
Cancer Res. 64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barthel A, Okino ST, Liao J, Nakatani K,
Li J, Whitlock JP Jr and Roth RA: Regulation of GLUT1 gene
transcription by the serine/threonine kinase Akt1. J Biol Chem.
274:20281–20286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu DD, Wang Y, Zhou PJ, Qin SR, Zhang R,
Zhang Y, Xue X, Wang J, Wang X, Chen HC, et al: The
IGF2/IGF1R/nanog signaling pathway regulates the proliferation of
acute myeloid leukemia stem cells. Front Pharmacol. 9:6872018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matkovic K, Brugnoli F, Bertagnolo V,
Banfic H and Visnjic D: The role of the nuclear Akt activation and
Akt inhibitors in all-trans-retinoic acid-differentiated HL-60
cells. Leukemia. 20:941–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thol F, Schlenk RF, Heuser M and Ganser A:
How I treat refractory and early relapsed acute myeloid leukemia.
Blood. 126:319–327. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bose P, Vachhani P and Cortes JE:
Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat
Options Oncol. 18:172017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou Y, Tozzi F, Chen J, Fan F, Xia L,
Wang J, Gao G, Zhang A, Xia X, Brasher H, et al: Intracellular ATP
levels are a pivotal determinant of chemoresistance in colon cancer
cells. Cancer Res. 72:304–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song K, Li M, Xu X, Xuan LI, Huang G and
Liu Q: Resistance to chemotherapy is associated with altered
glucose metabolism in acute myeloid leukemia. Oncol Lett.
12:334–342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tanioka M, Sakai K, Sudo T, Sakuma T,
Kajimoto K, Hirokaga K, Takao S, Negoro S, Minami H, Nakagawa K and
Nishio K: Transcriptional CCND1 expression as a predictor of poor
response to neoadjuvant chemotherapy with trastuzumab in
HER2-positive/ER-positive breast cancer. Breast Cancer Res Treat.
147:513–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jordan CT: The leukemic stem cell. Best
Pract Res Clin Haematol. 20:13–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Weinhouse S: On respiratory impairment in
cancer cells. Science. 124:267–269. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zu XL and Guppy M: Cancer metabolism:
Facts, fantasy, and fiction. Biochem Biophys Res Commun.
313:459–465. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Jiang Y, Meisenhelder J, Yang W,
Hawke DH, Zheng Y, Xia Y, Aldape K, He J, Hunter T, et al:
Mitochondria-translocated PGK1 functions as a protein kinase to
coordinate Glycolysis and the TCA Cycle in tumorigenesis. Mol Cell.
61:705–719. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mates JM, Segura JA, Campos-Sandoval JA,
Lobo C, Alonso L, Alonso FJ and Márquez J: Glutamine homeostasis
and mitochondrial dynamics. Int J Biochem Cell Biol. 41:2051–2061.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Meng M, Chen S, Lao T, Liang D and Sang N:
Nitrogen anabolism underlies the importance of glutaminolysis in
proliferating cells. Cell Cycle. 9:3921–3932. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fan J, Kamphorst JJ, Mathew R, Chung MK,
White E, Shlomi T and Rabinowitz JD: Glutamine-driven oxidative
phosphorylation is a major ATP source in transformed mammalian
cells in both normoxia and hypoxia. Mol Syst Biol. 9:7122013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Farge T, Saland E, de Toni F, Aroua N,
Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M,
Scotland S, et al: Chemotherapy-resistant human acute myeloid
leukemia cells are not enriched for leukemic stem cells but require
oxidative metabolism. Cancer Discov. 7:716–735. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gojo I, Perl A, Luger S, Baer MR,
Norsworthy KJ, Bauer KS, Tidwell M, Fleckinger S, Carroll M and
Sausville EA: Phase I study of UCN-01 and perifosine in patients
with relapsed and refractory acute leukemias and high-risk
myelodysplastic syndrome. Invest New Drugs. 31:1217–1227. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sampath D, Malik A, Plunkett W, Nowak B,
Williams B, Burton M, Verstovsek S, Faderl S, Garcia-Manero G, List
AF, et al: Phase I clinical, pharmacokinetic, and pharmacodynamic
study of the Akt-inhibitor triciribine phosphate monohydrate in
patients with advanced hematologic malignancies. Leuk Res.
37:1461–1467. 2013. View Article : Google Scholar : PubMed/NCBI
|