Introduction
Lung cancer is one of the most common causes of
cancer-related morbidity and mortality, and approximately 1.5
million new cases of lung cancer are diagnosed annually worldwide.
Furthermore, lung cancer accounts for 13% of all newly diagnosed
cancers worldwide (1,2). Approximately 85% of lung tumors are
non-small cell lung cancers (NSCLCs), and adenocarcinoma is the
most prevalent cancer subtype (3).
The high incidences of lung cancer recurrence and metastasis are
the two main causes of the failure to successfully treat NSCLC.
Standard therapies for patients with locally advanced or metastatic
lung cancer consist of radiotherapy and chemotherapy. However, most
patients fail to achieve long-term survival with such treatments as
they become resistant to chemotherapy (4). Chemotherapy resistance plays an
important role in the recurrence and metastasis of NSCLC.
Cisplatin, approved by the FDA in 1978, is one of
most effective chemotherapy drugs commonly used in the treatment of
various types of solid tumors, including testicular, head and neck,
ovarian, esophageal, cervical, and non-small cell lung cancer
tumors (5,6). Cisplatin binds to DNA in cellular nuclei
and mitochondria to form cisplatin-DNA adducts, which exert
cytotoxic effects by blocking the replication and transcription of
DNA in cancer cells (7). As
cisplatin-based chemotherapy is commonly used to treat patients
with advanced NSCLC, cisplatin resistance leads to the failure of
this type of chemotherapy (8).
Therefore, it is critical that we gain a better understanding of
the mechanisms of acquired cisplatin resistance, and develop safe
methods for reversing drug resistance. Apurinic/apyrimidinic
endonuclease 1 (APE1) is a key, multi-functional DNA repair enzyme
that plays important roles in repairing DNA damage, controlling
protein reduction/oxidation, and modulating the activity of
transcription factors (9,10). The mitochondrial intermembrane space
assembly (MIA) pathway and Mia40 protein play pivotal roles in
trafficking APE1 into mitochondria via the formation of disulfide
bonds (11). APE1 provides most of
the total AP endonuclease activity found in human cells, and is
essential for proper functioning of the base excision repair (BER)
pathway. APE1 also helps to regulate gene transcription, and
participates in many important pathways, (e.g. the NF-κB, AP-1,
Myb, HIF-1α and p53 pathways) via redox-dependent mechanisms
(10,12). Importantly, APE1 accumulates in the
cell nuclei and cytoplasm of various tumors, including NSCLC
tumors. Abnormal APE1 expression in cancer patients is associated
with a poor prognosis, tumor cell invasion, metastasis and
angiogenesis, as well as resistance to radiotherapy and
chemotherapy (13–15). Previous research has shown that serum
APE1 levels in patients with NSCLC are inversely associated with
progression-free survival after platinum-containing doublet
chemotherapy, and can serve as a biomarker for predicting disease
prognosis and treatment efficacy (16). However, the role played by APE1 in the
resistance of lung cancer to cisplatin remains unclear.
To explore the role of APE1 in cisplatin resistance,
the levels of APE1 and Mia40 expression were assessed in a
cisplatin-resistant A549 cell line. Moreover, the cellular
behaviors of cisplatin-resistant A549 cells were examined after
altering their levels of APE1 and Mia40 expression, and a
subsequent Parkin-mediated autophagy analysis was also performed.
We believe that our results provide new insights into the cisplatin
resistance of NSCLC cells, and will facilitate the development of
new clinical therapies.
Materials and methods
Cell culture and establishment of a
cisplatin-resistant subline
A549 lung cancer cells were obtained from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China) and
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% heat-inactivated fetal bovine serum
(FBS; HyClone; GE Healthcare), 100 U/ml penicillin, and 100 mg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified atmosphere containing 5% CO2. A
cisplatin-resistant A549 cell line (A549/DDP) was derived from A549
cells by incubating A549 cells with progressively increasing
concentrations of cisplatin. After each treatment, the surviving
cells were re-expanded and conventionally propagated for 4
generations in cisplatin-free medium. The relative levels of
cisplatin resistance were determined using the clonogenic
assay.
CCK-8 assay
Viability of the treated cells was determined by
using a CCK-8 assay kit (Dojindo, Japan) according to the
manufacturer's instructions. Briefly, 5,000 cells in 5 or 100 µl of
medium were seeded into each well of a 96-well culture plate. Next,
10 µl of CCK-8 reagent was added to each well and incubated for 2 h
at 37°C. After incubation, the absorbance of each well at 450 nm
was measured with a microplate reader (Thermo Fisher, Finland).
Colony formation assay
Cells were seeded into the wells of a 12-well plate
(1×103 cells per well) and cultured for 10–14 days.
After culture, the cells were fixed with 20% methanol and stained
with 0.1% crystal violet. Representative microscopic fields of the
stained cells were photographed, and the numbers of colonies were
counted.
Quantitative real-time polymerase
chain reaction (qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract the total RNA from cells
according to the manufacturer's instructions; after which, a
reverse transcription reagent kit (Takara, Tokyo, Japan) was used
to reverse transcribe the total RNA into cDNA. The cDNA was
detected according to the protocol used for SYBR Green Master Mix
(Thermo Fisher Scientific, Inc.). qPCR analysis for relative
expression level of mRNA was determined according to the procedures
as follows: 95°C for 2 min, followed by 40 cycles of 94°C for 20
sec, 58°C for 20 sec, 72°C for 20 sec, and finally extension at
72°C for 4 min. The Agilent Stratagene Mx3000P Sequence Detection
system (Agilent Technologies) was used to perform qPCR analysis.
The primers used for qPCR were synthesized by Invitrogen/Thermo
Fisher Scientific, Inc. and their sequences were as follows: APE1
forward, 5′-CCAGCCCTGTATGAGGACC-3′ and reverse,
5′-GGAGCTGACCAGTATTGATGAGA-3′; Mia40 forward,
5′-ATGACCCCAACGATCCATACG-3′ and reverse,
5′-GGGGATAGAGGTCTGGGTATTT-3′; GAPDH forward,
5′-TGTGGGCATCAATGGATTTGG-3′ and reverse,
5′-ACACCATGTATTCCGGGTCAAT-3′. GAPDH was used as an internal
reference gene. The relative levels of gene expression were
calculated using the 2−∆∆Cq method (17).
Cell apoptosis assay
The A549 cells (5×105) were seeded in
6-well plates (Corning Inc.) and cultured for 24 h, then the A549
cells were transfected as described below. Forty-eight hours later,
the treated cells were collected, washed, fixed, and permeabilized;
after which, they were stained using Annexin V-FTIC/PI (KeyGen)
according to the manufacturer's instructions. After 15 min of
staining, the number of apoptotic cells was determined by flow
cytometry (BD Biosciences).
Mitochondrial membrane potential
The mitochondrial membrane potential was detected by
using the JC-1 staining method. Briefly, the A549 cells
(5×105) were seeded in 6-well plates (Corning Inc.) and
cultured for 24 h. Then the A549 cells were transfected as
described below. Forty-eight hours later, the cells were harvested,
washed with PBS, and then incubated for 30 min with 5 µM JC-1 (Cell
Signaling Technology). After incubation, the cells were analyzed by
flow cytometry (BD Biosciences).
Isolation of subcellular
fractionation
The cytosolic fractions from the cultured A549 cells
were isolated using a biochemical fractionation method using
Mitochondria Isolation Kit for Cultured Cells (Pierce; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, the cells after the indicated treatment were
lysed in lysis buffer A [20 mmol/l Tris (pH 7.4), 150 mmol/l NaCl,
2 mmol/l EDTA, and 1 % Triton X-100 with protease and phosphatase
inhibitors] for 20 min at 4°C, and mitochondria were extracted in a
Dounce homogenizer in mitochondrial buffer. The supernatant was
further centrifuged to pellet the mitochondria, and the resulting
supernatant was stored as the cytosolic fraction. Subcellular
fractionation and western blot analysis were used to detect the
cytochrome c content in the cytosol and mitochondria.
Western blot analysis
Cells (2×106) were collected and lysed in
RIPA buffer. A BCA Protein Assay Kit (Pierce Biotechnology; Thermo
Fisher Scientific, Inc.) was used to measure the protein
concentrations in the lysates. Next, 50 µg samples of total protein
were separated by 12% SDS-PAGE, and the separated protein bands
were transferred onto PVDF membranes (EMD Millipore). The membranes
were first incubated with primary antibodies against APE1 (Abcam,
cat. no. ab137708; dilution 1:1,000), Mia40 (Abcam; cat. no.
ab87033, dilution 1:1,000), GAPDH (Abcam; ab8245, dilution
1:5,000), COX4 (Abcam; cat. no. ab33985, dilution 1:1,000), LC3
(Abcam; cat. no. ab48394, dilution 1:1,000), cytochrome c
(Abcam; cat. no. ab133504, dilution 1:1,000), and Parkin (Abcam;
cat. no. ab77924, dilution 1:1,000), followed by incubation with an
HRP-conjugated goat anti-rabbit antibody (Wuhan Boster Biological
Technology, Ltd.; cat. no. BA1054, dilution 1:20,000) or
HRP-conjugated goat anti-mouse antibody (Wuhan Boster Biological
Technology, Ltd.; cat. no. BA1051, dilution 1:20,000).
Immunostaining of the protein bands was detected by enhanced
chemiluminescence (ECL) reaction (Kibbutz Beit Haemek), and
staining intensity was analyzed with an Alpha Innotech Flour
Chem-FC2 imaging system (Alpha Innotech).
Cell transfection
To force overexpression of APE1 and Mia40 in cells,
pcDNA 3.1 vectors (Genechem) containing APE1 or Mia40 plasmids were
co-transfected into the A549 cells using Lipofectamine 2000
transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
An empty vector served as a negative control. Specific small
interfering RNAs (siRNAs) purchased from RiboBio, Inc. were used to
knock down APE1 and Parkin expression in the cells. The siRNAs used
were si-APE1 (5′-UACUCCAGUCGUACCAGACCUdTdT-3′), si-Parkin
(5′-AUUUCUUGACCUUUUCUCCACdTdT-3′), and a scrambled control siRNA
(5′-CCAUGAGGUCAUGGUCUGdTdT-3′). Lipofectamine 2000 transfection
reagent was used for all siRNA transfections. After 48 h of
transfection with plasmid or siRNA, the transfected A549 cells were
used for subsequent experiments.
Immunofluorescence and confocal
microscopy
Briefly, treated cells were fixed in 4%
paraformaldehyde and then permeabilized with 0.5% Triton X-100 for
15 min. Next, the cells were washed with PBS, blocked with 5% BSA
in PBS, and then incubated with the primary antibody (anti-APE1,
dilution 1:500), overnight at 4°C, followed by incubation with a
secondary antibody that was conjugated with Alexa Fluorescence 568
(Invitrogen, Thermo Fisher Scientific, Inc.; cat. no. A-11011;
dilution 1:1,000.). DAPI was used for nuclear staining
(Sigma-Aldrich, Merck KGaA; cat. no. D9542, dilution 1:5,000). The
stained cells were visualized by confocal fluorescence
microscopy.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5.0 (GraphPad Software, Inc.) and SPSS version 16.0
software (SPSS, Inc.). Results are presented as the mean ± SEM of
values obtained from at least three independent experiments.
Statistical significance was determined by one-way analysis of
variance (ANOVA) followed by Dunnett's test. A P-value <0.05 was
considered statistically significant.
Results
Cisplatin-resistant A549 cells exhibit
high levels of APE1 and autophagy
First, we established a cisplatin-resistant A549
cell line (A549/DDP) by incubating A549 cells with progressively
higher concentrations of cisplatin. The A549 cells were assessed
for viability after they had been treated with 0, 1, 2, 3, 4, 5 and
6 µmol/l cisplatin, respectively (Fig.
1A). A549/DDP cells were assessed for viability after treatment
with 0, 10, 20, 30, 40, 50, and 60 µmol/l cisplatin, respectively.
The A549/DDP cell line was significantly more resistant to
cisplatin, as the IC50 values for the A549 and A549/DDP
cells were 4.10 and 25.91 µmol/l, respectively. Next, we examined
the levels of mitochondrial APE1 (m-APE1) and total APE1 (t-APE1)
in both cell lines. We found that both the m-APE1 and t-APE1
protein levels were increased in the A549/DDP cells when compared
with those in the A549 cells (Fig. 1B and
C). Immunofluorescence assays confirmed the increased t-APE1
protein levels in A549/DDP cells (Fig.
1E), suggesting that APE1 might be transported into the
mitochondria. As the MIA pathway is responsible for APE1
trafficking into the mitochondria, and Mia40 is directly involved
in APE1's mitochondrial translocation, we analyzed the levels of
Mia40 protein. We found that the levels of Mia40 protein were also
increased in the A549/DDP cells when compared with those in the
A549 cells (Fig. 1C). Furthermore,
our findings suggest that the increased m-APE1 protein levels in
A549/DDP cells are mediated by Mia40.
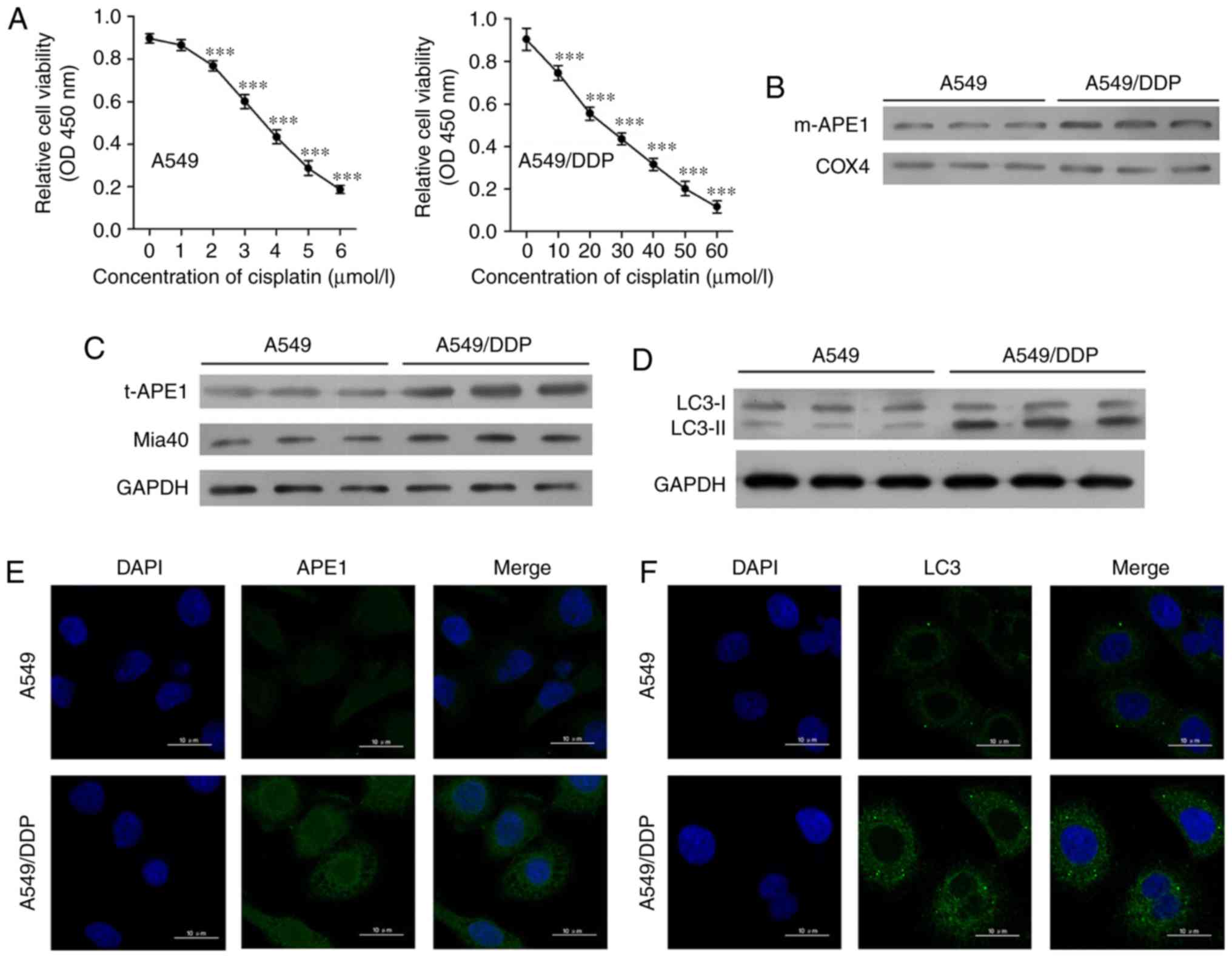 | Figure 1.Cisplatin-resistant A549 cells
exhibit high levels of APE1 and autophagy. (A) A549 cells were
treated with 0, 1, 2, 3, 4, 5 and 6 µmol/l cisplatin for 24 h, and
the A549/DDP cells were treated with 0, 10, 20, 30, 40, 50 and 60
µmol/l cisplatin for 24 h. Cell viability was assessed by the CCK-8
assay. (B) Western blot analysis was performed to analyze the
levels of mitochondrial APE1 (m-APE1) protein in A549/DDP and A549
cells. COX4 was used as loading control for the mitochondrial APE1
protein. (C) Western blot analysis of the total APE1 (t-APE1) and
Mia40 protein levels in A549/DDP and A549 cells. GAPDH was used as
a loading control. (D) Western blot analysis of the LC3 protein
levels in A549/DDP and A549 cells. (E and F) Immunofluorescence
assays were performed to assess the total APE1 and total LC3 levels
in A549 and A549/DDP cells. Data represent results obtained from
three independent experiments (mean ± SEM of triplicate samples).
***P<0.001, vs. 0 µmol/l cisplatin-treated A549 or A549/DDP
cells. APE1, apurinic/apyrimidinic endonuclease 1; LC3,
microtubule-associated protein 1A/1B-light chain 3. |
Accumulating evidence suggests that the autophagy
process in cancer cells is involved in their resistance to
chemotherapy (18–20). As shown in Fig. 1D and F, the levels of LC3II were
increased in the A549/DDP cells. This finding suggests that
cisplatin-resistant A549 cells have higher levels of autophagy.
APE1 and Mia40 overexpression enhances
the cisplatin resistance of A549 cells
To investigate the roles of APE1 and Mia40 in A549
cells, we performed transfection studies that forced the
overexpression of APE1 and Mia40 in A549 cells. The transfection
efficiencies are shown in Fig. 2A and
Fig. S1A. Interestingly, we found
that overexpression of Mia40 increased the levels of mitochondrial
APE1 (m-APE1) protein in the A549 cells (Fig. 2B). Moreover, the levels of t-APE1 and
Mia40 protein in the A549 cells were increased after the cells were
transfected with APE1 and Mia40 plasmids, respectively (Fig. 2C). These results indicate that Mia40
promotes the translocation of APE1 into mitochondria.
Immunofluorescence assays confirmed the increase in APE1 expression
(Fig. 2D). We also assessed the
viability of A549 cells overexpressing APE1 and Mia40, and found
that overexpression of both APE1 and Mia40 enhanced the cisplatin
resistance of the A549 cells (Fig.
2E). Colony-forming assays showed that overexpression of both
APE1 and Mia40 enhanced the colony formation efficiency of the A549
cells (Fig. 2F). In addition, APE1
and Mia40 overexpression decreased the apoptosis rate of the A549
cells after treatment with cisplatin (Fig. 2H and J). As APE1 and Mia40 interact
with each other via disulfide bond formation, and Mia40 levels
directly affect the translocation of APE1 into mitochondria, where
it helps to maintain the integrity of mitochondrial DNA (11), it is possible that the overexpression
of Mia40 enhances cisplatin resistance by promoting the
translocation of APE1 into mitochondria. We next used flow
cytometry to assess the mitochondrial membrane potential of
transfected A549 cells. As shown in Fig.
2G, the overexpression of both APE1 and Mia40 enhanced the
mitochondrial membrane potential. Furthermore, overexpression of
APE1 and Mia40 also promoted autophagy in A549 cells by
upregulating LC3-II expression, and decreasing the levels of
cytochrome c protein in the cytosolic fraction (Fig. 2I). It is possible that APE1 induced
Parkin-mediated mitophagy, because as shown in Fig. 2I, overexpression of APE1 increased
Parkin expression, and overexpression of Mia40 further increased
the levels of Parkin protein in the A549 cells.
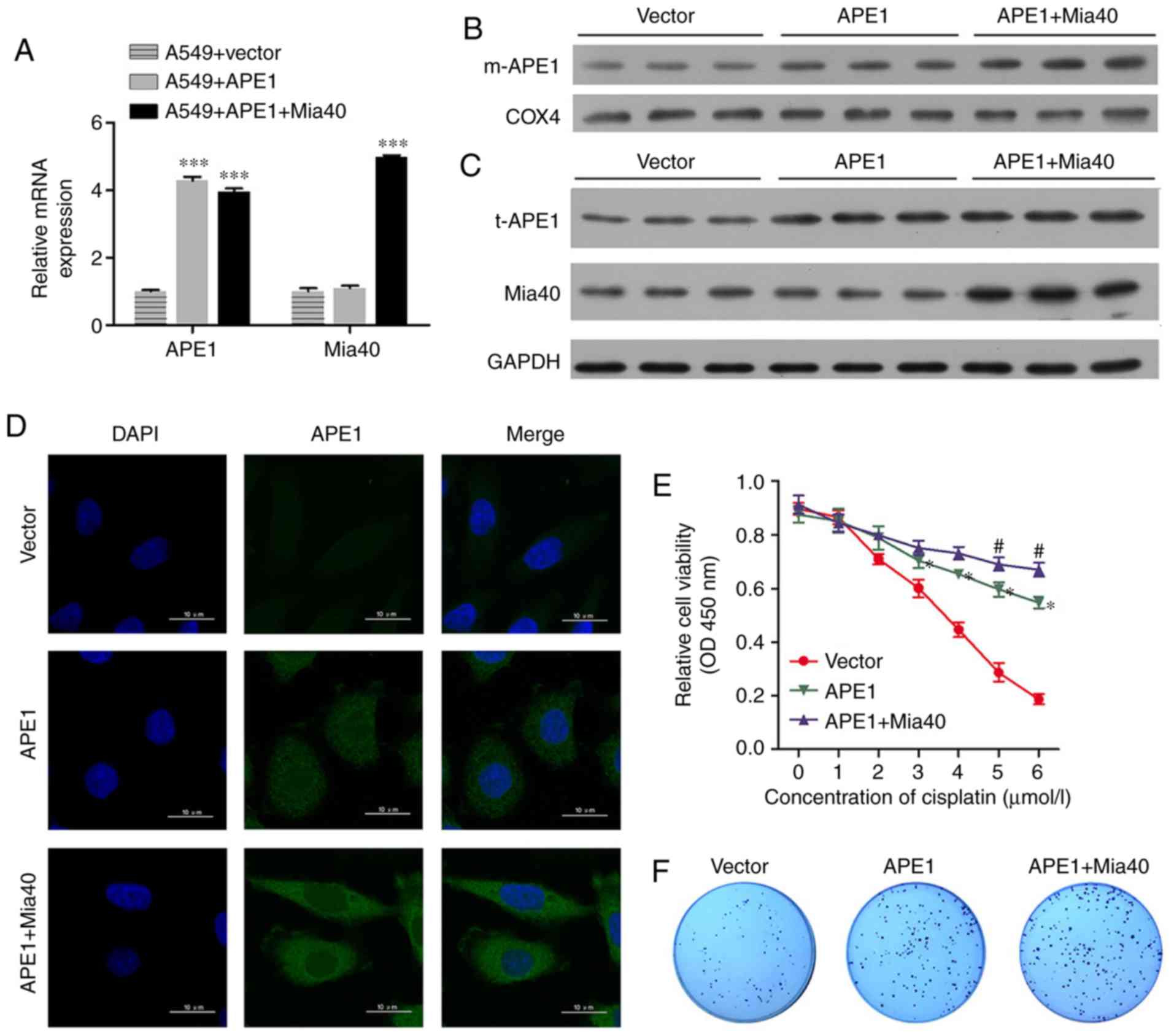 | Figure 2.APE1 and Mia40 overexpression
increases the cisplatin resistance of A549 cells. A549 cells were
transfected with vectors containing an APE1 overexpression plasmid
and Mia40 overexpression plasmid as indicated. (A) Transfection
efficiency was assessed by qPCR. (B) Western blot analysis of the
mitochondrial APE1 (m-APE1) protein levels in A549 cells after
transfection. COX4 was used as loading control for mitochondrial
APE1. (C) Western blot analysis was used to analyze the total APE1
(t-APE1) and Mia40 protein levels in A549 cells after transfection.
GAPDH was used as a loading control. (D) Immunofluorescence assays
were performed to assess the total APE1 levels in A549 cells after
transfection. (E) The transfected A549 cells were treated with 0,
1, 2, 3, 4, 5, and 6 µmol/l cisplatin for 24 h, and their viability
was assessed by the CCK-8 assay. (F) Colony-formation assays were
performed to analyze the colony formation efficiency of the
transfected A549 cells. *P<0.05, ***P<0.001, vs. the vector
group. #P<0.05, vs. the APE1 group. APE1,
apurinic/apyrimidinic endonuclease 1; LC3, microtubule-associated
protein 1A/1B-light chain 3. (G) Flow cytometry was used to analyze
the cellular distribution of JC-1 after transfection. (H)
Transfected A549 cells were treated with cisplatin, and their rate
of apoptosis was analyzed by flow cytometry. (I) The transfected
A549 cells were subjected to fractionation to obtain the cytosolic
fraction. Western blot analysis was performed to analyze the levels
of LC3, total cytochrome c, and Parkin in the transfected
A549 cells. (J) The cell apoptosis rate is shown in a histogram.
GAPDH was used as loading control. Data represent results obtained
from three independent experiments (mean ± SEM of triplicate
samples). ****P<0.0001 vs. the vector group. APE1,
apurinic/apyrimidinic endonuclease 1; LC3, microtubule-associated
protein 1A/1B-light chain 3. |
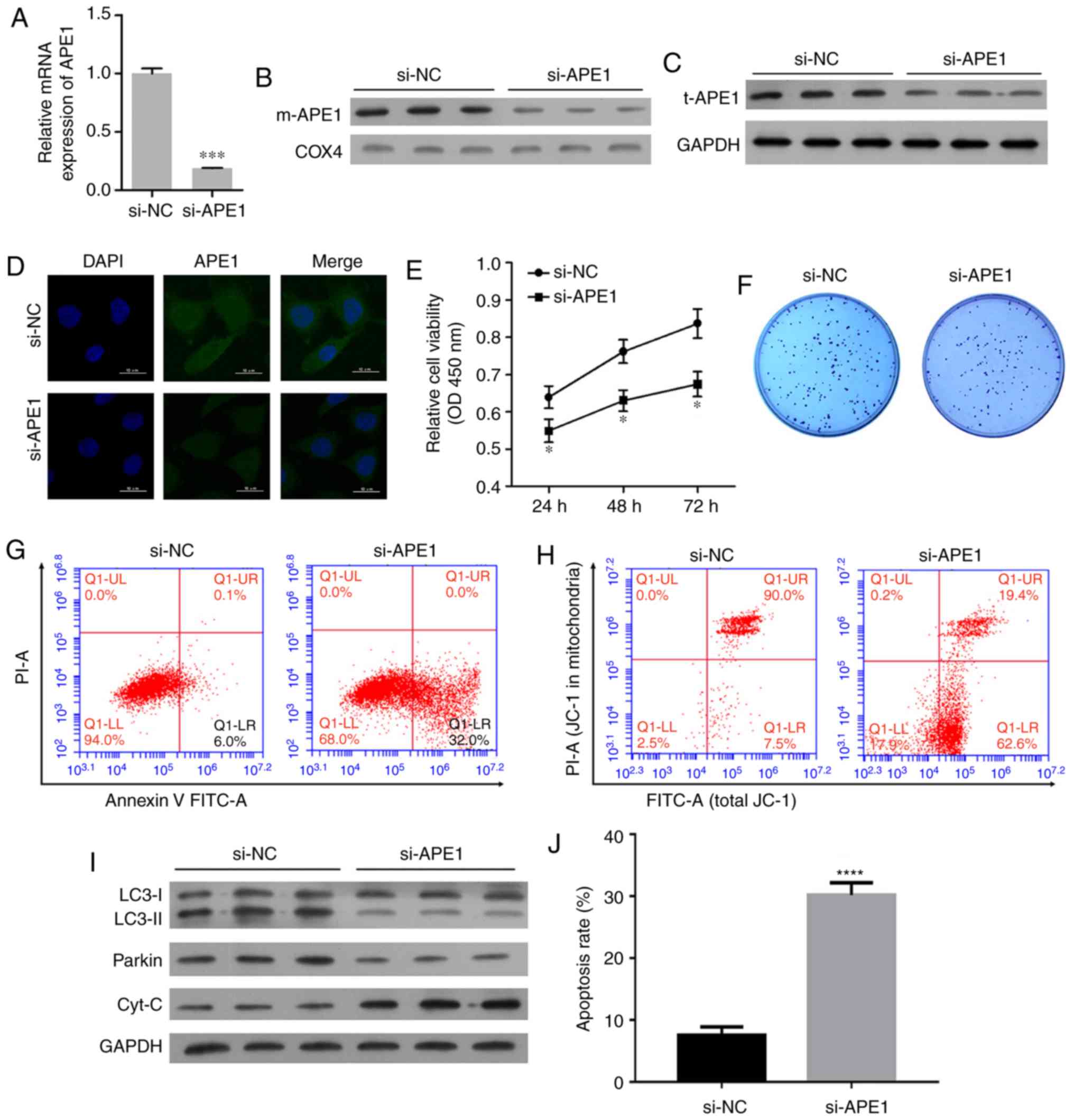
APE1 knockdown promotes the cisplatin
sensitivity of A549/DDP cells
To further investigate the role of APE1 in A549/DDP
cells, we performed transfections to knock down APE1 expression
those cells. The transfection efficiency is shown in Fig. 3A. Both the m-APE1 and total APE1
(t-APE1) protein levels in A549/CDD cells were significantly
decreased by APE1 knockdown (Fig. 3B and
C). The decrease in t-APE1 protein was confirmed by an
immunofluorescence assay (Fig. 3D).
APE1 knockdown increased the cisplatin sensitivity of A549/DDP
cells (Fig. 3E). In addition, APE1
knockdown reduced the colony formation efficiency and promoted the
apoptosis of A549/DDP cells (Fig. 3F, G
and J). The mitochondrial membrane potential of A549/DDP cells
was significantly downregulated after APE1 knockdown (Fig. 3H). With regards to cell mitophagy,
APE1 knockdown reduced the levels of LC3-II and Parkin in the
A549/DDP cells; however, it significantly upregulated the levels of
cytochrome c protein in the cytosolic fraction (Fig. 3I). Collectively, APE1 was shown to
play an important role in the cisplatin resistance of A549 cells,
and regulate Parkin-mediated mitophagy.
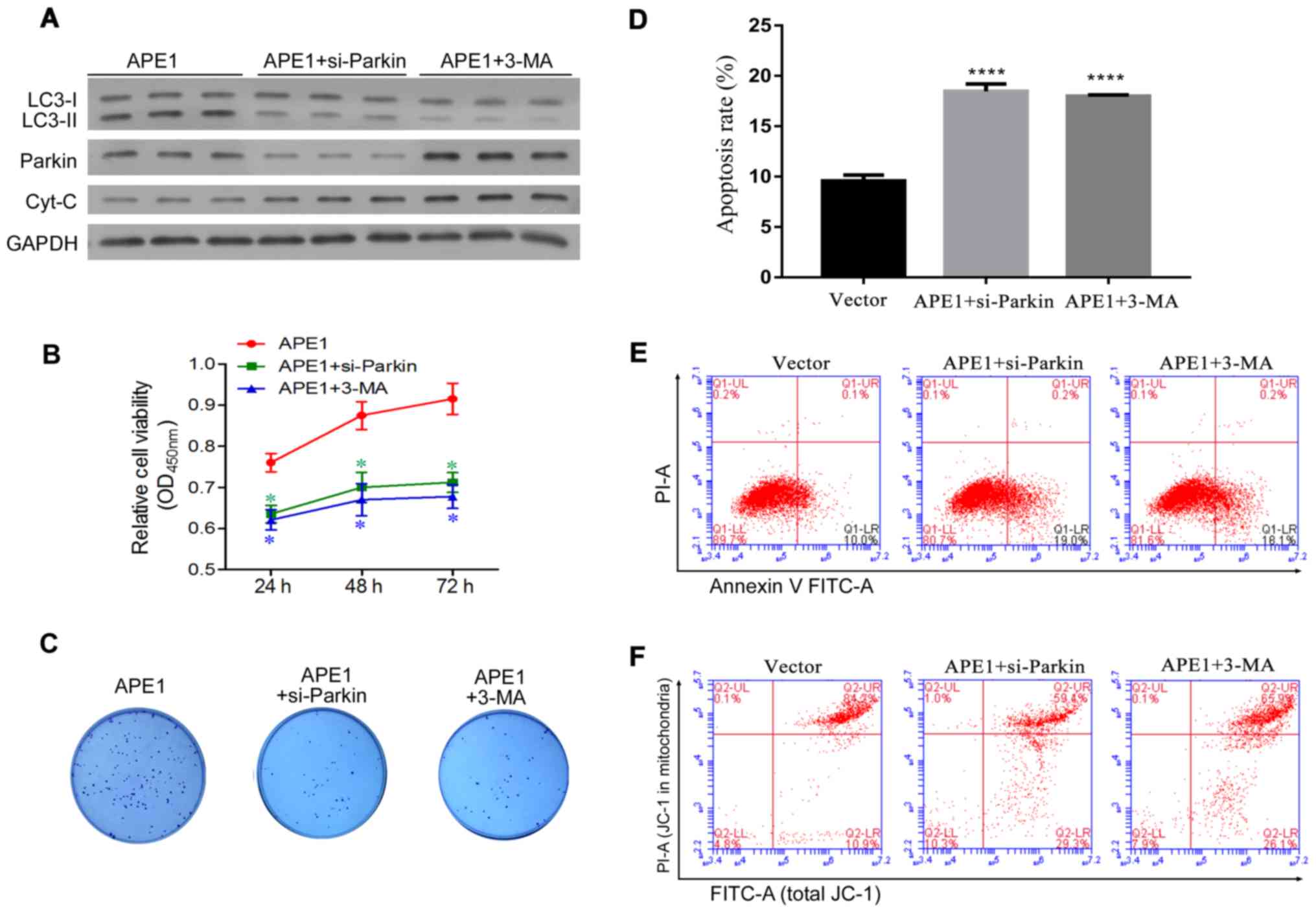
Parkin-mediated mitophagy plays an
important role in the APE1-induced cisplatin resistance of A549
cells
Given that APE1 overexpression and knockdown were
shown to regulate Parkin-mediated mitophagy, we next investigated
whether Parkin-mediated mitophagy is involved in cisplatin
resistance. Here, the study was divided into three groups as
follows: a) APE1 overexpression group; b) APE1
overexpression+Parkin siRNA group; c) APE1 overexpression+3-MA
group. 3-MA (5 mM) (Sigma-Aldrich; Merck KGaA, M9298) was used as
an inhibitor of autophagy according to the product introduction. In
A549 cells, Parkin was significantly knocked down by si-Parkin
transfection (Fig. S1B). As shown in
Fig. 4A, Parkin knockdown and 3-MA
treatment significantly decreased the levels of autophagy in the
APE1-overexpressing A549 cells, and also increased the levels of
cytochrome c protein in these cells (Fig. 4A). With regards to cisplatin
resistance, both Parkin knockdown and 3-MA treatment significantly
increased the cisplatin sensitivity of the APE1-overexpressing A549
cells (Fig. 4B). Moreover, Parkin
knockdown and 3-MA treatment reduced the colony formation
efficiency and significantly promoted the apoptosis of the
APE1-overexpressing A549 cells (Fig.
4C-E). We also assessed the mitochondrial membrane potential of
these cells, and found that their mitochondrial membrane potential
was reduced by Parkin knockdown and 3-MA treatment (Fig. 4F). In brief, Parkin-mediated mitophagy
was shown to play an important role in the APE1-induced cisplatin
resistance of A549 cells.
Discussion
Autophagy is a selective autophagic process that
provides metabolites needed for biosynthesis and energy production
in response to stressful conditions (21). Autophagy induced by metabolic and
therapeutic stress can play a dual role (22). For its pro-death role, excessive
autophagy in cancer cells induces ‘autophagic cell death’ or ‘type
II programmed cell death.’ For its pro-survival role, autophagy
eliminates damaged organelles and recycles cellular degradation
products (23,24).
Mitochondrial-specific autophagy (mitophagy) is an
important mitochondrial quality control system that selectively
degrades excess or damaged mitochondria via an autophagic process.
Similarly, mitophagy plays a dual role in cancer cell drug
resistance that depends on the specific micro-environmental
condition and cell type (25).
Mitophagy can promote the survival of various types of cancer cells
and tumors exposed to cytotoxic stress by degrading damaged
mitochondria and reducing the levels of reactive oxygen species in
mitochondria (26,27). Paradoxically, excessively damaged
mitochondria may induce cell metabolic disorders and force cancer
cells to undergo ‘autophagic cell death.’ While chemotherapeutic
agents such as cisplatin induce oxidative stress and mitochondrial
dysfunction in cancer cells, mitophagy selectively degrades
excessive or damaged mitochondria via autophagy (28). Therefore, the progress of mitophagy
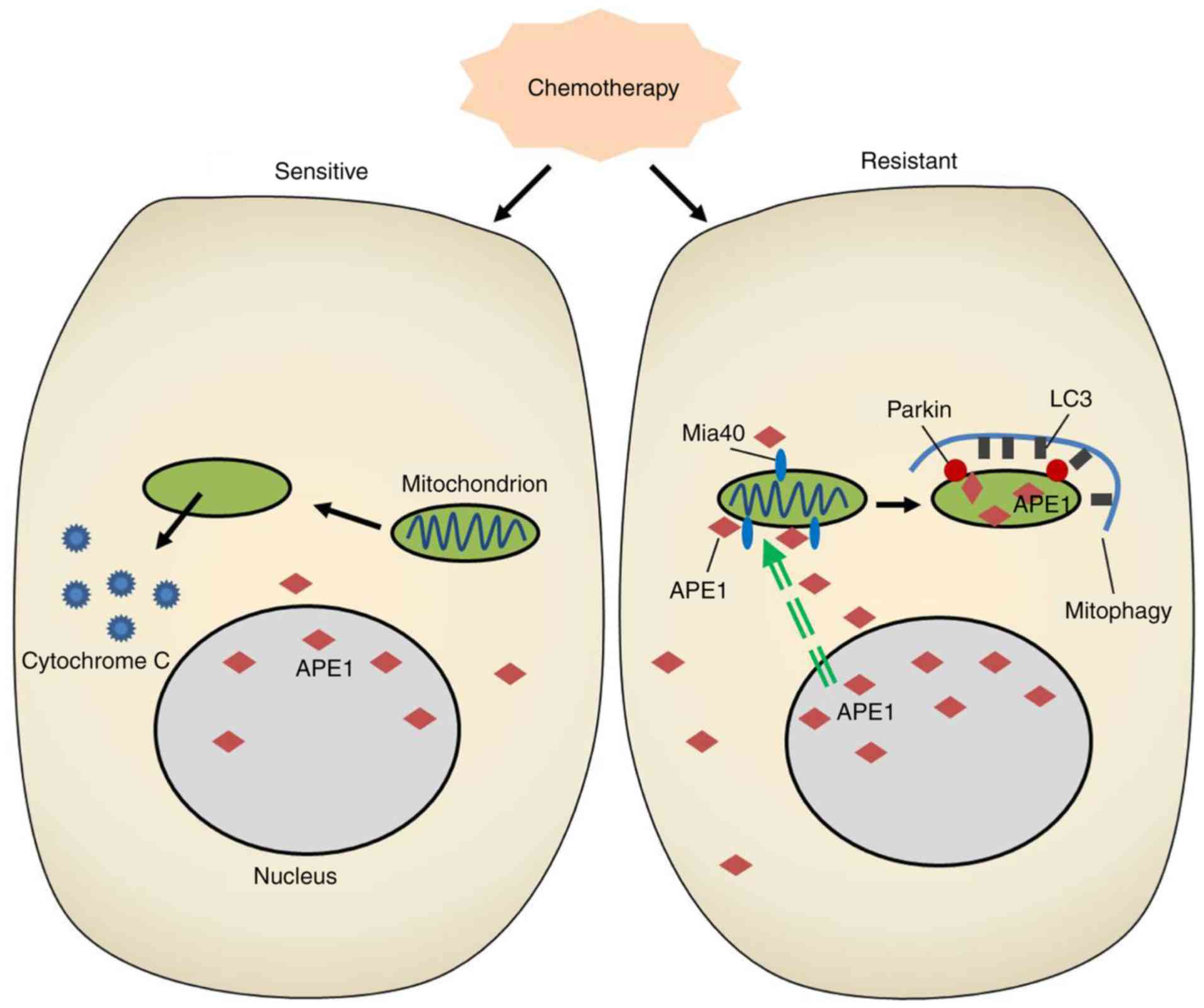
helps to mediate drug resistance in cancer cells. In our study, the
cisplatin-resistant A549 cell line displayed an increased level of
mitophagy when compared with the classical A549 cell line, and
inhibition of autophagy by 3-MA treatment or Parkin knockdown
reduced the cisplatin resistance of A549 cells. Interestingly, we
found accumulations of APE1 throughout cisplatin-resistant A549
cells, and in the mitochondria of those cells. In addition, our
findings demonstrated for the first time that APE1 can induce
Parkin-mediated mitophagy in A549 cells (Fig. 5).
Parkin, an E3 ubiquitin ligase, selectively degrades
damaged mitochondria via mitophagy in mammalian cells (29,30). For
Parkin-mediated mitophagy, Parkin requires PINK1, a Ser/Thr kinase,
to assist in sensing a mitochondria's functional status (31–33). The
efficiency of mitophagy depends on the levels of ubiquitin and
deubiquitinases present in mammalian cells (33–35).
Defects of Parkin are often associated with various pathologies,
including cancer. A previous study showed that Parkin deficiency
promoted tumorigenesis in a mouse model. However, Parkin is often
inactivated in many tumors, and its role remains unclear. Parkin
was reported to promote resistance to apoptosis independent of
mitophagy (36). However, Villa et
al (25) reported that E3
ubiquitin ligase (ARIH1/HHARI) protects against
chemotherapy-induced cell death by Parkin-mediated mitophagy
(25). Our study showed that the
levels of Parkin in cisplatin-resistant A549 cells were increased,
and that Parkin knockdown increased the cisplatin sensitivity of
APE1-overexpressing A549 cells. Thus, Parkin-mediated mitophagy in
A549 cells promoted cisplatin resistance of these cells. Our
results are in accordance with the conclusion previously reached by
Villa et al (25).
As we found that Parkin-mediated mitophagy in A549
cells promotes the cisplatin resistance of these cells, strategies
for inhibiting mitophagy should receive increased attention as
methods for overcoming cisplatin resistance and improving the
efficacy of cancer therapy. However, the functional outcomes
produced by mitophagy-induced cell death or survival can depend on
the type of cancer treatment administered, and the results achieved
can be confusing. In breast cancer, inhibition of mitophagy by
liensinine markedly promoted the sensitivity of the cancer cells to
classical chemotherapeutic agents, including doxorubicin,
paclitaxel, vincristine, and cisplatin (37). Moreover, treatment with liensinine
significantly inhibited late-stage mitophagy and reduced the
viability of breast cancer cells, but increased their rate of
apoptosis. An in vivo assay showed that inhibition of
mitophagy inhibited tumor growth in the MDA-MB-231 ×enograft model
(37). Our study demonstrated that
inhibition of mitophagy by Parkin knockdown and 3-MA treatment
significantly enhanced the cisplatin sensitivity of tumor cells.
Our findings provide a potential therapeutic strategy for treating
lung cancer.
Our study has some limitations that should be
mentioned. First, due to limited research funds, only one cell line
was used in the study, and this may weaken the reliability of our
findings. Thus, it is important to verify our findings in other
NSCLC cell lines before reaching a final conclusion. Second,
although we had intended to clarify the molecular mechanism of APE1
in a cisplatin-resistant cell model, some differences exist between
the results shown by in vitro and in vivo assays.
Hence, some further in vivo assays should be conducted to
confirm our findings. Despite these weaknesses, our study provides
novel information concerning cisplatin resistance in NSCLC.
In conclusion, our study revealed a novel mechanism
by which APE1 promotes cisplatin resistance in lung cancer cells.
In the cisplatin-resistant cell line, APE1 was highly expressed and
was translocated into the mitochondria via its interaction with
Mia40. This mitochondrial translocation of APE1 increased the
mitochondrial membrane potential, decreased the levels of
cytochrome c protein, and induced Parkin-mediated mitophagy,
resulting in the cisplatin resistance of lung cancer cells.
Supplementary Material
Supporting Data
Acknowledgements
L.A.S. edited and revised the manuscript.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81501993).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
ZL and NL conceived and designed the research
methods, drafted and revised the manuscript. ZL, YW, LW and YD
performed experiments. ZL, JZ and FC analyzed the data and prepared
the figures. WX and JH assisted with the analysis of the data and
interpreted the results of the experiments. All authors approved
the final version of the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren J, Nie Y, Lv M, Shen S, Tang R, Xu Y,
Hou Y, Zhao S and Wang T: Estrogen upregulates MICA/B expression in
human non-small cell lung cancer through the regulation of ADAM17.
Cell Mol Immunol. 12:768–776. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gao L, Zhang H, Zhang B, Zhang L and Wang
C: Prognostic value of combination of preoperative platelet count
and mean platelet volume in patients with resectable non-small cell
lung cancer. Oncotarget. 8:15632–15641. 2017.PubMed/NCBI
|
|
4
|
Proto C, Imbimbo M, Gallucci R, Brissa A,
Signorelli D, Vitali M, Macerelli M, Corrao G, Ganzinelli M, Greco
FG, et al: Epidermal growth factor receptor tyrosine kinase
inhibitors for the treatment of central nervous system metastases
from non-small cell lung cancer: The present and the future. Transl
Lung Cancer Res. 5:563–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jamieson ER and Lippard SJ: Structure,
recognition, and processing of cisplatin-DNA adducts. Chem Rev.
99:2467–2498. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nature Rev Drug Discov. 4:307–320.
2005. View
Article : Google Scholar
|
|
8
|
Gridelli C, Aapro M, Ardizzoni A, Balducci
L, De Marinis F, Kelly K, Le Chevalier T, Manegold C, Perrone F,
Rosell R, et al: Treatment of advanced non-small-cell lung cancer
in the elderly: Results of an international expert panel. J Clin
Oncol. 23:3125–3137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lindahl T: Instability and decay of the
primary structure of DNA. Nature. 362:709–715. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li M and Wilson DM III: Human
apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal.
20:678–707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barchiesi A, Wasilewski M, Chacinska A,
Tell G and Vascotto C: Mitochondrial translocation of APE1 relies
on the MIA pathway. Nucleic Acids Res. 43:5451–5464. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lo YL, Jou YS, Hsiao CF, Chang GC, Tsai
YH, Su WC, Chen KY, Chen YM, Huang MS, Hu CY, et al: A polymorphism
in the APE1 gene promoter is associated with lung cancer risk.
Cancer Epidemiol Biomarkers Prev. 18:223–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Woo J, Park H, Sung SH, Moon BI, Suh H and
Lim W: Prognostic value of human apurinic/apyrimidinic endonuclease
1 (APE1) expression in breast cancer. PLoS One. 9:e995282014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ren T, Qing Y, Dai N, Li M, Qian C, Yang
Y, Cheng Y, Li Z, Zhang S, Zhong Z and Wang D:
Apurinic/apyrimidinic endonuclease 1 induced upregulation of
fibroblast growth factor 2 and its receptor 3 induces angiogenesis
in human osteosarcoma cells. Cancer Sci. 105:186–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang D, Xiang DB, Yang XQ, Chen LS, Li MX,
Zhong ZY and Zhang YS: APE1 overexpression is associated with
cisplatin resistance in non-small cell lung cancer and targeted
inhibition of APE1 enhances the activity of cisplatin in A549
cells. Lung Cancer. 66:298–304. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang S, He L, Dai N, Guan W, Shan J, Yang
X, Zhong Z, Qing Y, Jin F and Chen C: Serum APE1 as a predictive
marker for platinum-based chemotherapy of non-small cell lung
cancer patients. Oncotarget. 7:77482–77494. 2016.PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mowers EE, Sharifi MN and Macleod KF:
Functions of autophagy in the tumor microenvironment and cancer
metastasis. FEBS J. 285:1751–1766. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun T, Liu H and Ming L: Multiple roles of
autophagy in the sorafenib resistance of hepatocellular carcinoma.
Cell Physiol Biochem. 44:716–727. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY,
Hung SC, Hsiao M, Yao CJ and Shieh MJ: Autophagy promotes
resistance to photodynamic therapy-induced apoptosis selectively in
colorectal cancer stem-like cells. Autophagy. 10:1179–1192. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sridhar S, Botbol Y, Macian F and Cuervo
AM: Autophagy and disease: Always two sides to a problem. J Pathol.
226:255–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jarauta V, Jaime P, Gonzalo O, de Miguel
D, Ramírez-Labrada A, Martínez-Lostao L, Anel A, Pardo J, Marzo I
and Naval J: Inhibition of autophagy with chloroquine potentiates
carfilzomib-induced apoptosis in myeloma cells in vitro and in
vivo. Cancer Lett. 382:1–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Villa E, Proics E, Rubio-Patino C, Obba S,
Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragón L, Riley JS,
et al: Parkin-Independent mitophagy controls chemotherapeutic
response in cancer cells. Cell Rep. 20:2846–2859. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chourasia AH, Tracy K, Frankenberger C,
Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale
JW, Karczmar GS and Macleod KF: Mitophagy defects arising from
BNip3 loss promote mammary tumor progression to metastasis. EMBO
Rep. 16:1145–1163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan C, Luo L, Guo CY, Goto S, Urata Y,
Shao JH and Li TS: Doxorubicin-induced mitophagy contributes to
drug resistance in cancer stem cells from HCT8 human colorectal
cancer cells. Cancer Lett. 388:34–42. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan C and Li TS: Dual role of mitophagy in
cancer drug resistance. Anticancer Res. 38:617–621. 2018.PubMed/NCBI
|
|
29
|
Kitada T, Asakawa S, Hattori N, Matsumine
H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y and Shimizu N:
Mutations in the parkin gene cause autosomal recessive juvenile
parkinsonism. Nature. 392:605–608. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clark IE, Dodson MW, Jiang C, Cao JH, Huh
JR, Seol JH, Yoo SJ, Hay BA and Guo M: Drosophila pink1 is required
for mitochondrial function and interacts genetically with parkin.
Nature. 441:1162–1166. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Geisler S, Holmstrom KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biol.
12:119–131. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawajiri S, Saiki S, Sato S, Sato F,
Hatano T, Eguchi H and Hattori N: PINK1 is recruited to
mitochondria with parkin and associates with LC3 in mitophagy. FEBS
Lett. 584:1073–1079. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuda N, Sato S, Shiba K, Okatsu K,
Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al:
PINK1 stabilized by mitochondrial depolarization recruits parkin to
damaged mitochondria and activates latent parkin for mitophagy. J
Cell Biol. 189:211–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vives-Bauza C, Zhou C, Huang Y, Cui M, de
Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:378–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carroll RG, Hollville E and Martin SJ:
Parkin sensitizes toward apoptosis induced by mitochondrial
depolarization through promoting degradation of Mcl-1. Cell Rep.
9:1538–1553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming
QL, Huang C, Li P and Gao N: A novel autophagy/mitophagy inhibitor
liensinine sensitizes breast cancer cells to chemotherapy through
DNM1L-mediated mitochondrial fission. Autophagy. 11:1259–1279.
2015. View Article : Google Scholar : PubMed/NCBI
|