Introduction
Metastatic melanoma is one of the most challenging
types of cancer to treat due to its aggressiveness and resistance
to treatment. Current progress in the understanding of the
molecular mechanisms responsible for tumor growth and metastasis
has led to the approval of several novel therapies for melanoma;
however, the adverse effects associated with the treatments and the
development of drug resistance in a relative short period of time
have underscored their benefit (1–9). Recent
studies have clearly demonstrated that the inflammatory tumor
microenvironment favors innate and acquired resistance of cancers
cells to cytotoxic drugs altering the treatment outcome (10–13). Thus,
therapeutic approaches based on the anti-neoplastic polarization of
the tumor microenvironment may be promising strategies with which
to overcome drug resistance in melanoma, as well as in general in
solid tumors (14,15).
Among the innate immune cells present in the stroma
of solid tumors, macrophages [tumor-associated macrophages (TAMs)]
are the most abundant, representing up to 50% of the tumor mass
(16). Thus, TAMs are polarized into
the M2 phenotype and are most important in promoting and supporting
tumor inflammation, as well as in all processes dependent on
chronic inflammation, such as angiogenesis, tumor growth and
metastasis, and immunosuppression (17–19).
Several studies have clearly demonstrated the role of TAMs in
promoting tumor angiogenesis via the secretion of several
pro-angiogenic factors, such as vascular endothelial growth factor
(VEGF), platelet-derived growth factor (PDGF) and transforming
growth factor β (TGF-β). Moreover, a high number of TAMs
infiltrated into human tumors is associated with an increased
microvessel density in different types of human cancer (20–24). In
tight association with these findings, we have previously
demonstrated that TAMs are the most important tumor stromal cells
that support melanoma tumor growth in vivo through the
production of pro-angiogenic proteins (25,26).
Consequently, the depletion of TAMs from melanoma tumors markedly
inhibits tumor growth (50% of tumor growth inhibition compared to
the growth of controls) via the suppression of the pro-angiogenic
capacity of these cell types (25,26).
Therefore, anti-inflammatory drugs that disrupt the link between
TAM-induced inflammation/angiogenesis and tumor growth may provide
new therapeutic opportunities against cancer. Thus, the well-known
anti-inflammatory drug, prednisolone disodium phosphate (PLP),
administered in liposomal form, has been shown to exert potent
antitumor effects on B16.F10 melanoma-bearing mice via the
inhibition of the angiogenic/inflammatory capacity of TAMs
(26–28). Additionally, it has been demonstrated
that TAMs can protect tumor cells against the cytotoxicity induced
by chemotherapeutic drugs, such as doxorubicin (DOX) and etoposide
(29). DOX is a potent cytotoxic drug
used in the treatment of various types of cancer (30). Although DOX exerts a potent cytotoxic
effect on several melanoma cell lines, its use as a first-, as well
as second-line therapy for melanoma, is well-tolerated, although it
is limited by a low clinical efficacy, suggesting a modulatory
activity of the tumor microenvironment (31–33).
Based on these findings, the present study aimed to
examine whether prednisolone that targets the antitumor functions
of TAMs, can enhance the cytotoxicity of the DOX on B16.F10
melanoma cells, when both agents are co-administered. To assess the
antitumor efficacy of the combined treatment, we used an in
vitro model for the inflammatory melanoma microenvironment
based on the co-culture of bone marrow-derived macrophages (BMDMs)
and B16.F10 murine melanoma cells at a cell density ratio of 4:1.
This ratio approximates the murine melanoma microenvironment in
vivo that ensures the polarization of BMDMs into TAMs (34). Thus, we examined the effects of the
combined treatment on main processes that can affect melanoma
development and aggressiveness. In this respect, we assessed the
combined therapeutic effects on cancer cell proliferation and
apoptosis, as well as on supportive processes for tumor growth,
such as oxidative stress, as well as on the angiogenic and
inflammatory capacity of the co-culture. To gain insight into the
mechanisms through which this treatment can influence the protumor
functions of TAMs, the effects on key molecules produced by this
cell type and which are involved in the main processes that support
tumor development, such as tumor inflammation and angiogenesis,
were also screened. The results revealed that co-treatment with DOX
and PLP exerted enhanced antitumor effects compared to treatment
with the cytotoxic drug alone, mainly due to the PLP-induced
inhibition of the angiogenic activity of TAMs.
Materials and methods
Cell types and culture conditions
B16.F10 murine melanoma cells (CRL-6475; American
Type Culture Collection) were cultured in Dulbecco's modified
Eagle's medium (DMEM, Lonza Group Ltd.), supplemented with 10%
heat-inactivated fetal bovine serum (HyClone; GE Healthcare Life
Sciences), 100 IU/ml penicillin, 100 µg/ml streptomycin and 4 mM
L-glutamine (Lonza Group Ltd.) as monolayer at 37°C in a 5%
CO2 humidified atmosphere.
Bone marrow cells were isolated from the femurs of
male C57BL/6 mice (Cantacuzino Institute, Bucharest, Romania)
following a previously published protocol (35). The adherent BMDMs were harvested after
7 days of cultivation in medium supplemented with 10 ng/ml
macrophage colony-stimulating factor (M-CSF; Cell Signaling
Technology). For the co-culture model, the BMDMs were cultured with
B16.F10 cells at a cell density ratio of 4:1 for an optimal
cytokine exchange and interaction specific for in vivo
melanoma microenvironment (34,36). When
used in monoculture, the differentiated macrophages were polarized
toward the M2 phenotype by supplementation of the growth medium
with 20 ng/ml interleukin (IL)-4 (Cell Signaling Technology) for 24
h. Experiments were performed according to the European and
national regulations and were approved by the Committee on the
Ethics of Animal Experiments of the Babes-Bolyai University
(registration no. 31444/27.03.2017).
Stock solutions of DOX and PLP
DOX (Sigma-Aldrich, cat. no. D2975000) and PLP)
(Sigma-Aldrich, cat. no. 1557000) were dissolved in sterile water
to prepare stock solutions of 10 and 100 mM, respectively. Working
solutions were prepared directly into the culture media.
Cell proliferation assay
To determine the effects of DOX, administered alone
or in combination with PLP, on tumor cell proliferation, the
B16.F10 melanoma cells (1,000 cells/well) co-cultured with BMDMs
(4,000 cells/well) were seeded in a 96-well plate for 24 h. Various
concentrations of DOX (ranging from 0.007–0.5 µM) and PLP (ranging
from 2.5–20,000 µM) were administered alone and tested in
triplicate to assess the IC50 values. Based on our
previous studies (28,37), the concentration of 410 µM of PLP was
selected to be administered in combination with various
concentrations of DOX on the cell co-culture. Although this
concentration of PLP does not exert any significant
anti-proliferative effects on melanoma cells, it has been proven to
exhibit antitumor activity mediated by its inhibitory effects on
the angiogenic and inflammatory proteins produced by TAMs (28,37). Cell
co-culture incubated only with medium was used as a control. The
proliferative activity of the cells following treatment with the
test agents was examined by ELISA, BrdU-colorimetric immunoassay
[Cell Proliferation ELISA, BrdU (colorimetric); Roche Applied
Science] according to the manufacturer's instructions. This method
is based on the incorporation of the pyrimidine
analogue-bromodeoxyuridine (BrdU)-instead of thymidine into the DNA
of proliferating cells. The B16.F10 melanoma cells in the
co-culture model were incubated with BrdU solution for 24 h and the
culture medium was completely removed from each well. Following
this step, the cells were fixed and the DNA was denatured. A
monoclonal antibody conjugated with peroxidase (anti-BrdU-POD, cat.
no. 11647229001, Roche Applied Science, dilution, 1:100; part of
Cell Proliferation ELISA, BrdU kit) was added in each well, in
order to detect the incorporated BrdU in the newly synthesized
cellular DNA. The antibody was removed after 1 h of incubation at
room temperature, and the cells were washed 3 times with
phosphate-buffered saline. A peroxidase substrate
(tetramethyl-benzidine) was added to each well, and the immune
complexes were detected by measuring the absorbance of the reaction
product at 450 nm with a reference wavelength of 655 nm using a
microplate reader (BMG Labtech, serial no. 415-1324), as previously
described (38).
Assessment of apoptosis/necrosis in
the co-culture model
To determine the capacity of the combination therapy
to induce the apoptosis of melanoma cells co-cultured with murine
macrophages, we used the Annexin V-fluorescein isothiocyanate
(FITC) assay (Cayman Chemical Co.). The principle of this protocol
is based on the externalization of phosphatidylserine and
phosphatidylethanolamine on the outer leaflet of the plasma
membrane of the apoptotic cells. The redistribution of the
phospholipids is measured after high-affinity binding to Annexin V
conjugated with FITC. Thus, 5×104 cells/well
(1×104 melanoma cells and 4×104 macrophages)
were seeded in a 96-well black culture plate for 24 h. Several
concentrations of DOX were added in triplicate at concentrations
ranging from 0.03 to 0.5 µM. PLP was used at the concentration of
410 µM based on our previous reported data showing that this is the
maximum concentration that could be achieved in tumors (37). At the end of the incubation period, a
double staining with Annexin V-FITC and propidium iodide (PI) was
performed at room temperature for 10 min as described previously
(39). Fluorescence was determined
using a fluorescence plate reader (BMG Labtech, serial no.
415-1324). The fluorescence emitted by early apoptotic cells
(Annexin V FITC+/PI−) was measured at 535 nm
with an excitation wavelength of 485 nm, whereas fluorescence
determined by late apoptotic and necrotic cells was measured at 640
nm with excitation at 540 nm. 5-Fluorouracil (Sigma-Aldrich) (25
mg/ml)-treated cells were used as a positive control for late
apoptosis. Cells cultured only in cell culture medium were used as
a negative control, as previously described (39).
Preparation of cell lysates
To obtain cell lysates, melanoma cells co-cultured
with macrophages, as well as IL-4-polarized macrophages following
24 h of incubation with the different treatments, were washed with
PBS and viable (adherent) cells were mechanically detached and
lysed with cell lysis buffer (10 mM Hepes, 200 mM NaCl, 1% Triton
X, 10 mM MgCl2, 1 mM DTT), following 30 min of
incubation on ice. Complete Protease Inhibitor Cocktail tablets
(Roche Applied Science) were added to the lysis buffer. Cell
lysates were cleared by centrifugation at 18,000 × g for 10 min at
4°C and the supernatant was collected. The protein content of the
cell lysates was determined by Bradford assay (Sigma-Aldrich).
HPLC determination of malondialdehyde
levels in cell co-culture
Malondialdehyde (MDA) is the main product of lipid
peroxidation mediated by reactive oxygen species (ROS) and
therefore, it is a good indicator of the overall levels of
oxidative stress. MDA levels in the cell lysates were determined
according to the method employed by Karatas et al (2002)
(40) through high-performance liquid
chromatography (HPLC) as previously described (38). The column type was RP18 (5 µm)
(Supelco) and the mobile phase consisted of 30 mM
KH2PO4/methanol in a volume ratio of 65:35.
The flow rate was set at 0.5 ml/min and MDA was measured using a UV
detector (Jasco Corp.) set at 254 nm. Each sample was tested in
duplicate. The retention time of MDA was approximately 5.4 min.
Data were expressed as µmoles of MDA/mg of protein.
Angiogenic and inflammatory protein
array
To assess whether the combination therapy alters the
cell production of proteins involved in angiogenesis and
inflammation, a screening for 24 proteins involved in angiogenesis
and inflammation was performed as previously described by using a
protein array of RayBio® Mouse Angiogenic protein
Antibody Array membranes 1.1 (RayBiotech Inc.) (39). One array membrane containing 24 types
of primary antibodies against specific mouse proteins was used per
cell lysate. The array membranes were incubated with 200 µg of
proteins of cell lysates, overnight at 4°C. A mixture of secondary
biotin-conjugated antibodies against the same angiogenic proteins
as those for primary antibodies (included in the array), was added
on the membranes and incubated for 2 h at room temperature,
followed by incubation with HRP-conjugated streptavidin for an
additional 2 h at room temperature. Each incubation step was
followed by 5 washing steps. Thereafter, the membranes were
incubated with a mixture of two detection buffers for 1 min,
exposed to an X-ray film (Kodak) for 4 min and then the films were
developed. The protein expression level was quantified by measuring
the intensity of the color of each spot on the membranes, in
comparison to the positive control spots already bound to the
membranes, using TotalLab Quant Software version 12 for Windows.
Each protein level from the treated groups was expressed as
percentage of the same protein level from the untreated cells
(controls). Each protein for each experimental group was determined
in duplicate.
RT-qPCR quantification of markers for
the IL-4 polarization of macrophages
Total RNA was isolated from the M2 polarized murine
macrophages following treatment with either 410 µM PLP + 0.06 µM
DOX or 410 µM PLP + 0.5 µM DOX using a RNA kit (peqGOLD Total RNA
kit, PeqLab). Untreated cells were used as a control. To avoid
contamination with genomic DNA, 1 µg of total RNA was digested with
1U of RNase free DNase (Thermo Fisher Scientific, Inc.) for 30 min
at 37°C followed by the addition of EDTA and incubation at 65°C for
10 min. Following DNase digestion, 750 ng of total RNA were
reverse-transcribed into cDNA using the Verso cDNA Synthesis kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Identical samples from each experimental group were
processed in the absence of reverse transcriptase and served as
controls for genomic DNA contamination. Reverse transcription
products (1 µl) were amplified in a 25-µl reaction mix containing
1X Maxima SYBR-Green qPCR Master Mix (Thermo Fisher Scientific,
Inc.), and 0.3 µM of each primer using a Corbett RotorGene
instrument using the following cycling parameters: Pre-incubation
at 95°C for 10 min, then cycling: 95°C for 15 sec, 60°C for 30 sec,
and then 72°C for 30 sec. To examine for primer specificity,
melting curves were generated.
The sequences of the primers were as follows: Mouse
IL-10 forward, 5′-GGTTGCCAAGCCTTATCGGA-3′ and reverse,
5′-ACCTGCTCCACTGCCTTGCT-3′; mouse arginase-1 (Arg-1) forward,
5′-CTCCAAGCCAAAGTCCTTAGAG-3′ and reverse,
5′-AGGAGCTGTCATTAGGGACATC-3; and mouse β-actin forward,
5′-TCTTTGCAGCTCCTTCGTTGCCGGTCC-3′ and reverse,
5′-GTCCTTCTGACCCATTCCCACCATCACAC-3′. Gene expression was calculated
by relative quantitation using the comparative Cq method (ΔΔCq), as
previously described (41). Mouse
β-actin was used as a reference gene. Gene expression was reported
as fold change (2−ΔΔCt), relative to the untreated
control cells, used as a calibrator.
Statistical analysis
Data from different experiments are reported as the
means ± standard deviation (SD). The IC50 values of
different treatments were calculated using non-linear regression of
sigmoidal dose response curves offered by the GraphPad Prism
version 6 for Windows (GraphPad Software, Inc.). The differences
between the effects of treatments on the production of markers for
specific pro-tumor processes were analyzed by one-way ANOVA. To
analyze the treatment effects on cell proliferation, as well as on
the levels of angiogenic/inflammatory proteins in cells, two-way
ANOVA with Bonferroni correction for multiple comparisons was used.
All statistical analyses were performed using the same statistical
software mentioned above. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
Cytotoxic effects of the combination
therapy
To investigate whether PLP can enhance the antitumor
effects of DOX on B16.F10 melanoma cells, various concentrations of
DOX were administered alone, as well as in combination with PLP in
the co-culture model. The cytotoxic effects of the various
treatments on the B16.F0 melanoma cells co-cultured with
macrophages were assessed with regard to cancer cell proliferation
(Fig. 1 and Table I) and the induction of apoptosis in
the cell co-culture (Fig. 2).
 | Table I.Synergistic effect of co-treatment
with PLP and DOX on B16.F10 cell proliferation. |
Table I.
Synergistic effect of co-treatment
with PLP and DOX on B16.F10 cell proliferation.
|
|
|
| Combination index
(CI) |
|---|
|
|
|
|
|
|---|
| Treatment | IC50
(µM) | 95% confidence
interval | CI value | Interpretation |
|---|
| DOX | 0.029 | 0.023 to 0.040 | – | – |
| PLP | 2706.0 | 911.4 to 8035 | – | – |
| 410 µM PLP +
DOX | 0.015 | 0.013 to 0.018 | 0.668 | Synergism |
Synergistic effects of combined
treatment with PLP and DOX on B16.F10 cell proliferation
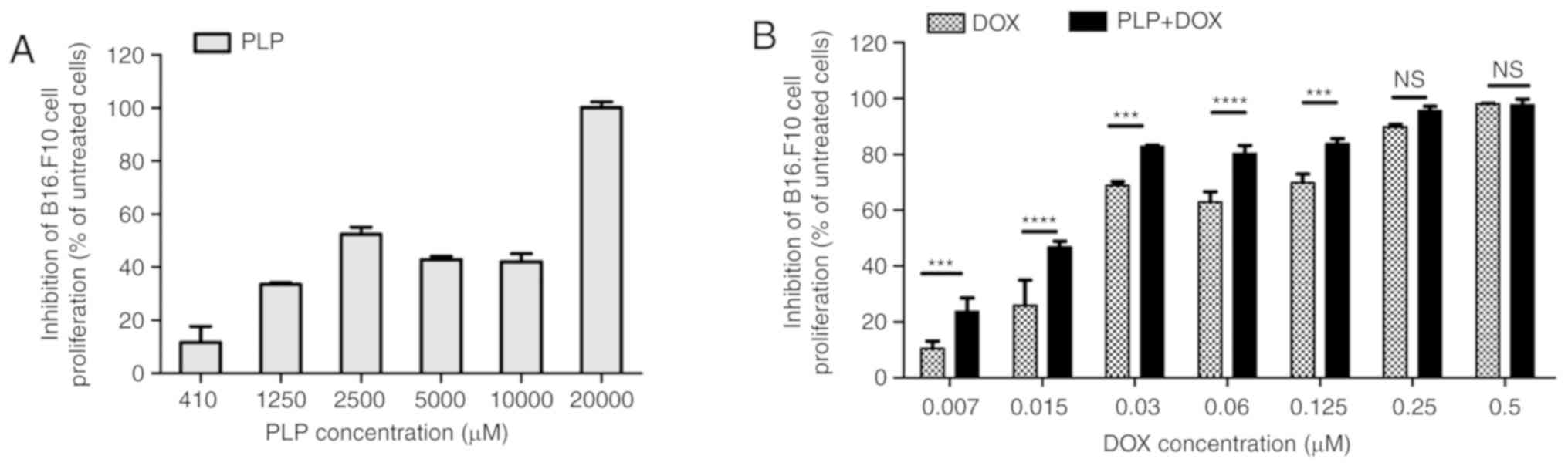
The effects of these treatments on cancer cell
proliferation were expressed as percentages of the inhibition of
cell proliferation compared to the proliferation of the B16.F10
cells used as controls (Fig. 1A and
B). The IC50 values for PLP and DOX administered as
single treatments, as well as in combination, on B16.F10 cell
proliferation are shown in Table
I.
In line with our previous study regarding the
cytotoxicity of PLP on cancer cells (28), 410 µM PLP was the first concentration
of glucocorticoid that exerted slight to moderate inhibitory
effects on melanoma cell proliferation. This concentration was
selected to be administered in combination with the cytotoxic drug,
DOX in the cell co-culture model. Apart from the highest
concentrations of DOX (0.25 µM and 0.5 µM), combined treatment with
410 µM PLP with each DOX concentration significantly enhanced the
anti-proliferative effects of the cytotoxic drug on the tumor cells
(Fig. 1B). As at the concentrations
of 0.25 µM and 0.5 µM DOX, the proliferation of B16.F10 cells was
completely compromised, the enhancing effects of PLP on the DOX
cytotoxicity on these cancer cells were overshadowed. Moreover, the
IC50 value of DOX decreased 2-fold when the cytotoxic
drug treatment was administered in combination with 410 µM PLP
(Table I). Thus, according to the
Chou-Talalay calculation method, the combination index (CI)
(42,43) indicated synergism between the
anti-proliferative effects of PLP and DOX on melanoma cells
(CI=0.668) (Table I). Since 0.06 µM
was the lowest concentration of DOX which, when used in combination
with 410 µM PLP exerted a potent inhibitory effect (by 80%
inhibition of B16.F10 cell proliferation) (Fig. 1B) on cancer cell proliferation, this
combination was used throughout the experiments to further
investigate the molecular mechanisms responsible for the
synergistic antitumor efficacy of both drugs on melanoma
microenvironment model in vitro.
Apoptotic and necrotic effects of the
combined treatment
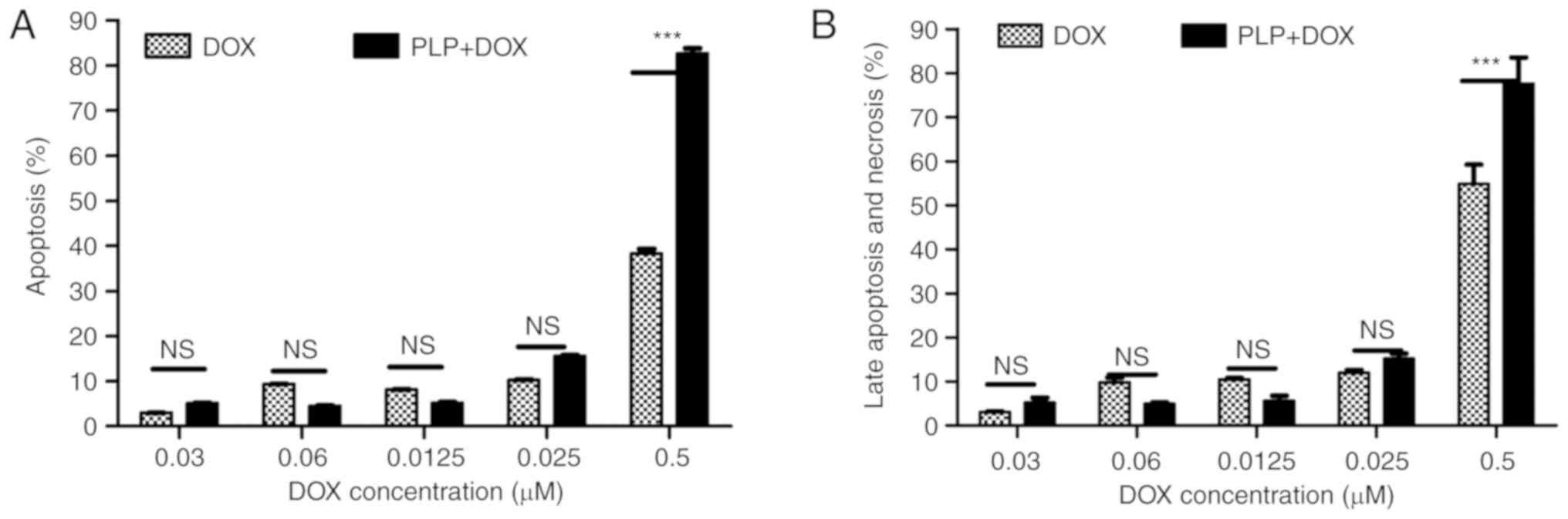
To gain insights into the mechanisms through which
the combination therapy inhibited tumor cell proliferation, we
assessed the ability of the treatment to induce the apoptosis of
B16.F10 murine melanoma cells co-cultured with TAMs. Thus, we used
Annexin V-FITC to stain the cells in early apoptosis and PI for
late apoptosis and necrosis. Relative fluorescence units measured
were normalized for the number of cells. Subsequently, all data
were compared with the positive control and shown as percentages of
apoptosis (Fig. 2A) and as
percentages of necrosis and late apoptosis (Fig. 2B).
Following 24 h of incubation with DOX alone,
moderate apoptotic (approximately 40%) (Fig. 2A) and necrotic effects (approximately
50%) (Fig. 2B) were noted only at the
highest concentration tested. Notably, the same concentration of
DOX administered in combination with 410 µM PLP induced potent
apoptotic (approximately 80%) (Fig.
2A) and necrotic effects (approximately 80%) on the cell
co-culture (Fig. 2B).
Therefore, this combination treatment (410 µM PLP +
0.5 µM DOX) with potent apoptotic and necrotic effects on the cell
co-culture was also selected to be investigated with regard to the
underlying mechanisms of the cytotoxicity of both drugs on melanoma
microenvironment in vitro.
Effect of the combined treatments on
intracellular oxidative stress
As melanoma cells are under persistent oxidative
stress levels (44), we evaluated
whether each combined treatment could affect the physiological
production of ROS in murine melanoma cells cultured with TAMs.
Therefore, the levels of MDA, a general marker of oxidative stress,
in the cell co-culture lysates were assessed and are shown in
Fig. 3. Our data revealed that
treatment with 0.06 µM DOX either alone or in combination with PLP
markedly inhibited (by ~70%) the production of MDA in the B16.F10
melanoma cells co-cultured with murine macrophages (Fig. 3), while treatment with 410 µM PLP
alone had no effect on intracellular oxidative stress.
Nevertheless, the treatment with the pro-apoptotic concentration of
DOX (0.5 µM) alone, as well as in the presence of PLP, did not
induce any significant modification of the intracellular oxidative
stress in the cancer cells co-cultured with TAMs (Fig. 3).
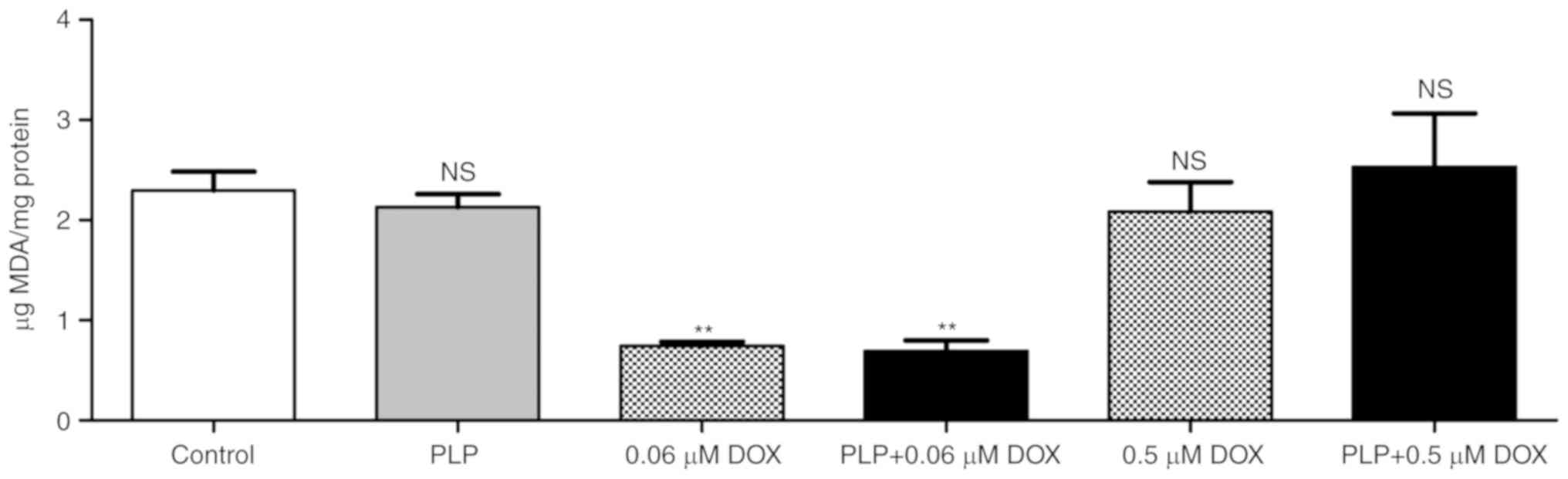 | Figure 3.Evaluation of the effect of PLP + DOX
on oxidative stress in B16.F10 melanoma cells co-cultured with
murine macrophages. The level of MDA was determined by HPLC
analysis. Control, MDA levels in untreated cells following 24 h of
incubation with culture media; 0.06 µM DOX, MDA levels in cells
afte following r 24 h of incubation with 0.06 µM DOX; PLP, MDA
levels in cells following 24 h of incubation with medium
supplemented with 410 µM PLP; PLP + 0.06 µM DOX, MDA levels in
cells following 24 h of treatment with 0.06 µM DOX + 410 µM PLP;
0.5 µM DOX, MDA levels in cells following 24 h of incubation with
0.5 µM DOX; PLP + 0.5 µM DOX, MDA levels in cells following 24 h of
treatment with 0.5 µM DOX + 410 µM PLP. The results are expressed
as the means ± SD of 2 independent measurements. One-way ANOVA with
the Bonferroni post hoc test was performed to analyze the
differences between the MDA levels in the control cells and treated
cells (NS, not significant, P>0.05; **P<0.01). MDA,
malondialdehyde; PLP, prednisolone disodium phosphate; DOX,
doxorubicin. |
Effects of the combined treatments on
the angiogenic/inflammatory capacity of B16.F10 cells co-cultured
with TAMs
The effects of the different treatments on the
expression levels of the angiogenic/inflammatory proteins in
B16.F10 cells co-cultured with murine macrophages were evaluated by
performing a screening for 24 angiogenic and inflammatory proteins
using a protein array (RayBiotech Inc.) and results are shown in
Fig. 4 and Table II. In accordance with our previously
published data (26), the overall
production of angiogenic/inflammatory proteins was notably
decreased by 40% (P<0.001) in the cells treated with 410 µM PLP
compared to their production in the untreated cell co-culture, as a
result of the well-known anti-inflammatory effects of this drug.
The addition of 0.06 µM DOX to the co-culture did not lead to a
significant decrease in the mean production of the
angiogenic/inflammatory proteins compared with cells treated with
PLP alone. More specifically, 0.06 µM DOX stimulated the production
of two proteins with an important role in tumor progression:
Insulin-like growth factor (IGF)-II (by 45%) and IL-1 β (by 94%),
while the levels of the antitumor protein, tissue inhibitor of
metalloproteinases (TIMP)-1, were markedly decreased (by 80%). Only
the production of tumorigenic proteins, such as granulocyte-colony
stimulating factor (G-CSF), M-CSF, IL-6, basic fibroblast growth
factor (bFGF) and IL-13 was moderately decreased (by 35–55%)
(Fig. 4).
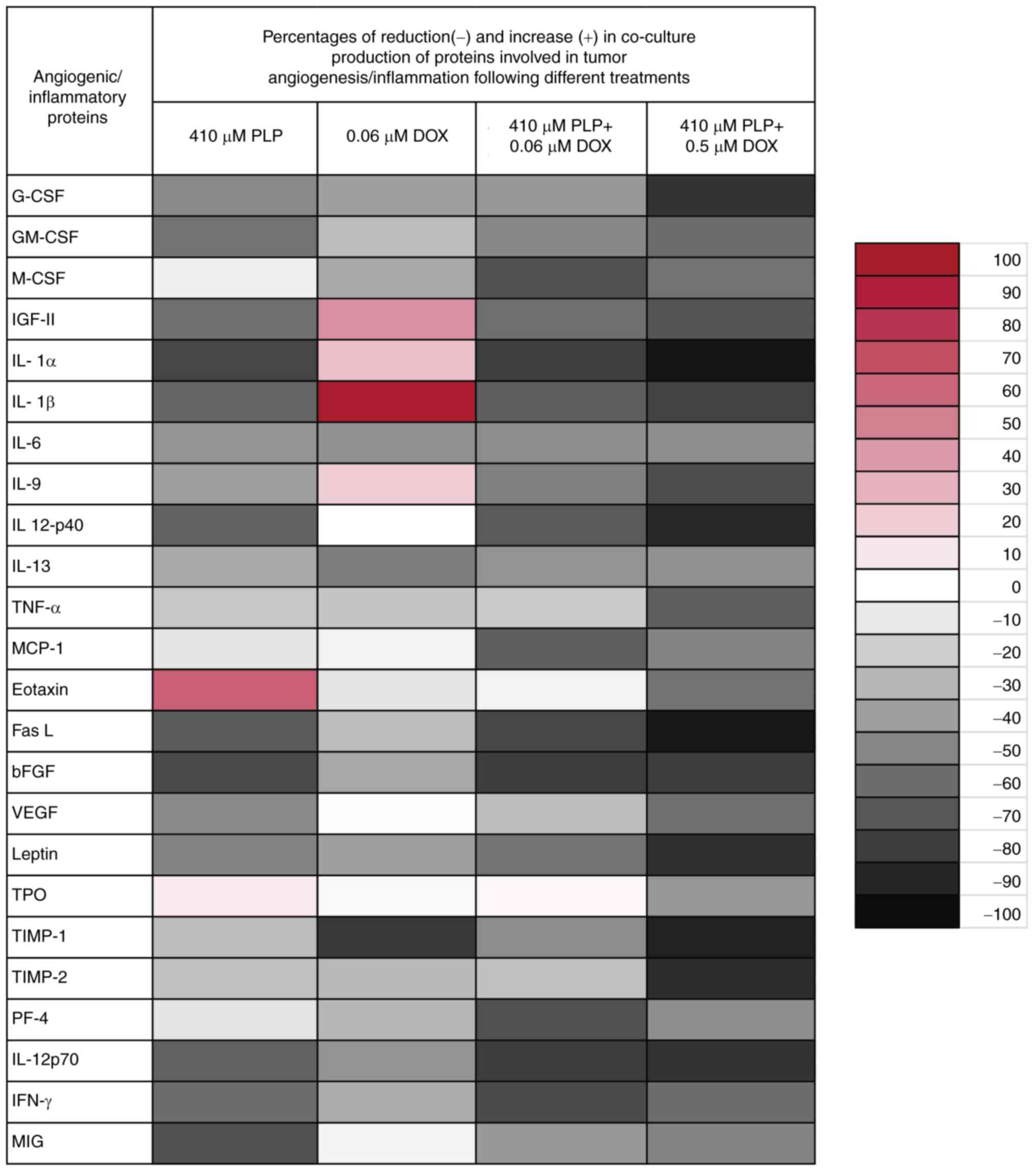 | Figure 4.Effects of PLP, DOX and PLP + DOX on
the production of angiogenic/inflammatory proteins in the
co-culture of B16F10 melanoma cells and murine macrophages. Results
are presented either as % of reduction (−) of tumor protein levels
ranging from 0% (white) to 100% (black with vertical line pattern)
or as % of stimulation (+) of production of proteins ranging from
0% (white) to 100% (red) in cells after different treatments
compared to levels of the same proteins in untreated cells. G-CSF,
granulocyte-colony stimulating factor; GM-CSF,
granulocyte-macrophage-colony stimulating factor; M-CSF,
monocyte-colony stimulating factor; IGF-II, insulin growth factor
II; IL, interleukin; TNF-α, tumor necrosis factor α; MCP-1,
monocyte chemoattractant protein-1; FasL, Fas ligand; bFGF, basic
fibroblast growth factor; VEGF, vascular endothelial growth factor;
TPO, thrombopoietin; TIMP, tissue inhibitor of matrix
metalloproteinase; PF-4, platelet factor 4; IFN-γ, interferon γ;
MIG, monokine induced by IFN-γ. |
| Table II.Effects of PLP + DOX on the
production of angiogenic/inflammatory proteins in B16.F10 murine
melanoma cells co-cultured with murine macrophages for 24 h. |
Table II.
Effects of PLP + DOX on the
production of angiogenic/inflammatory proteins in B16.F10 murine
melanoma cells co-cultured with murine macrophages for 24 h.
|
| Percentage of
reduction(−)/increase (+) in the cell co-culture production of
proteins involved in tumor angiogenesis/inflammation following
different combined treatments compared to untreated cells |
|---|
|
|
|
|---|
|
Angiogenic/inflammatory proteins | 410 µM PLP | 410 µM PLP + 0.06
µM DOX | 410 µM PLP + 0.5 µM
DOX |
|---|
| G-CSF |
−49.11±9.01c |
−42.45±4.76c |
−79.72±0.88c,f |
| GM-CSF |
−58.61±0.40c |
−49.31±8.34c |
−57.40±4.61c,d |
| M-CSF |
−6.69±32.26a |
−71.58±2.87c |
−55.53±4.90c,d |
| IGF-II |
−59.21±2.12c |
−59.56±0.20c |
−66.83±2.02c,d |
| IL-1α |
−77.01±9.54c |
−79.39±3.95c |
−91.38±0.09c,d |
| IL-1β |
−63.39±1.52c |
−66.30±0.61c |
−74.37±0.54c,d |
| IL-6 |
−44.62±1.79c |
−48.03±4.21c |
−45.05±12.33c,d |
| IL-9 |
−39.75±3.19b |
−52.90±2.24c |
−69.65±3.09c,e |
| IL 12-p40 |
−64.75±1.24c |
−68.24±1.48c |
−84.69±3.30c,d |
| IL-13 |
−35.42±3.14b |
−44.66±0.27c |
−43.26±10.9c,d |
| TNF-α |
−23.34±1.04a |
−21.19±0.71a |
−63.20±1.70a,f |
| MCP-1 |
11.16±10.13a |
−66.69±0.67c |
−48.53±1.50c,e |
| Eotaxin |
62.79±24.13c |
−6.31±4.64a |
−55.66±15.11c,f |
| FasL |
−67.41±1.89c |
−75.96±0.85c |
−90.21±1.09c,d |
| bFGF |
−75.46±0.48c |
−80.16±1.60c |
−76.77±1.22c,d |
| VEGF |
−48.35±1.93c |
−26.86±16.64a |
−56.74±0.91c,f |
| Leptin |
−50.62±2.14c |
−57.91±5.01c |
−81.72±2.40c,f |
| TPO |
9.69±3.79a |
+4.51±5.67a |
−41.10±0.02c,f |
| TIMP-1 |
−27.53±7.18a |
−47.98±0.72c |
−87.82±1.52c,f |
| TIMP-2 |
25.23±13.87a |
−25.50±5.15a |
−82.97±0.27c,f |
| PF4 |
−11.36±6.98a |
−72.45±4.64c |
−44.83±0.14c,f |
| IL-12p70 |
−65.28±2.69c |
−80.26±0.96c |
−80.09±0.96c,d |
| IFN-γ |
−60.24±2.70c |
−74.73±2.69c |
−57.13±0.17c,e |
| MIG |
−72.60±2.29c |
−41.83±7.01c |
−48.43±8.03c,d |
Notably, a marked overall reduction by 52%
(P<0.001) of angiogenic protein production in the cell
co-culture lysates was observed following combined treatment (410
µM PLP + 0.06 µM DOX) with anti-proliferative effects, while the
reduction following co-treatment with both drugs at concentrations
with pro-apoptotic, as well as anti-proliferative effects (410 µM
PLP + 0.5 µM DOX) was slightly higher (by 66%, P<0.001). More
specifically, the production of the majority of the pro-angiogenic
proteins was reduced almost completely (70–100% reduction in G-CSF,
IL-1α, IL-1β, IL-9, IL-12p40, FasL, bFGF, leptin, TIMP-1 and
TIMP-2) following treatment with 410 µM PLP + 0.5 µM DOX.
Nevertheless, the levels of antitumor proteins [platelet factor
(PF)-4, IL-12p70, IFN-γ and monokine induced by IFN-γ (MIG)] were
also moderately to strongly decreased following treatment with each
of the combination treatments tested (Table II).
Combined treatments affect the
pro-angiogenic functions of TAMs
As TAMs are key cell players in the tumor
angiogenesis, we investigated whether the anti-angiogenic effects
of the combined treatments on the cell co-culture model could be
linked to their effects on TAMs angiogenic capacity. Thus, IL-4
polarized macrophages were treated with 410 µM PLP + 0.06 µM DOX,
as well as 410 µM PLP + 0.5 µM DOX. Following 24 h of incubation
with 410 µM PLP + 0.06 µM DOX, the average angiogenic protein
production in the polarized macrophages was not affected. Only the
levels of specific pro-angiogenic proteins (GM-CSF, IL-1β, TNF-α,
eotaxin, FasL, bFGF and VEGF) were moderately (by approximately
40–50%) decreased by this combined treatment. Notably, combined
treatment with both drugs at concentrations demonstrating
pro-apoptotic, as well as anti-proliferative effects (410 µM PLP +
0.5 µM DOX) markedly inhibited the overall expression of the
angiogenic proteins (by approximately 70%, P<0.001). In
particular, treatment with 410 µM PLP + 0.5 µM DOX decreased the
production of the majority of the tumorigenic proteins (55–100%
reduction in G-CSF, GM-CSF, M-CSF, IGF-II, IL-1α, IL-1β, IL-6,
IL-9, IL-12p40, IL-13, MCP-1, eotaxin, FasL, bFGF, leptin and
TIMP-2) (Table III), as well as
that of the anti-angiogenic proteins (55–95% reduction in PF-4,
IL-12p70, IFN-γ and MIG) (Table
III).
| Table III.Effects of the combined treatments on
the production of angiogenic/inflammatory proteins in IL-4
polarized murine macrophages. |
Table III.
Effects of the combined treatments on
the production of angiogenic/inflammatory proteins in IL-4
polarized murine macrophages.
|
| Percentage of
reduction (−)/increase (+) in IL-4 polarized macrophages production
of proteins involved in tumor angiogenesis/inflammation following
the treatment with DOX and PLP compared to untreated
macrophages |
|---|
|
|
|
|---|
|
Angiogenic/inflammatory proteins | 410 µM PLP + 0.06
µM DOX | 410 µM PLP + 0.5 µM
DOX |
|---|
| G-CSF |
−16.58±4.38a |
−69.06±3.33d,h |
| GM-CSF |
−40.67±3.52c |
−87.93±0.54d,h |
| M-CSF |
17.62±7.15a |
−75.82±3.80d,h |
| IGF-II |
65.93±29.00d |
−80.63±0.9d,h |
| IL-1α |
38.64±2.20b |
−84.63±2.03d,h |
| IL-1β |
−44.84±7.51c |
−60.25±5.71d,e |
| IL-6 |
−28.59±2.57a |
−80.58±9.13d,h |
| IL-9 |
−1.98±7.55a |
−84.80±3.72d,h |
| IL 12-p40 |
−13.26±0.94a |
−92.62±0.09d,h |
| IL-13 |
−10.11±4.65a |
−70.03±3.07d,h |
| TNF-α |
−45.07±9.47c |
−55.91±1.91d,g |
| MCP-1 |
+26.15±13.48a |
−81.71±0.34d,f |
| Eotaxin |
−42.71±8.93c |
−88.89±1.36d,h |
| FasL |
−47.74±2.21d |
−94.03±0.82d,h |
| bFGF |
−50.00±1.71d |
−77.00±5.04b,e |
| VEGF |
−51.06±0.58d |
−34.30±5.23b,e |
| Leptin |
−32.70±11.94a |
−98.77±0d,h |
| TPO |
−25.24±11.43a |
−40.95±4.81c,e |
| TIMP-1 |
−1.12±3.71a |
22.93±2.18a,e |
| TIMP-2 |
−12.87±2.54a |
−89.81±5.81d,h |
| PF-4 |
11.18±27.46a |
−54.42±4.40d,g |
| IL-12p70 |
−3.16±0.72a |
−79.50±1.42d,h |
| IFN-γ |
−8.50±11.00a |
−92.25±2.22d,h |
| MIG |
−29.17±16.36a |
−93.32±1.69d,h |
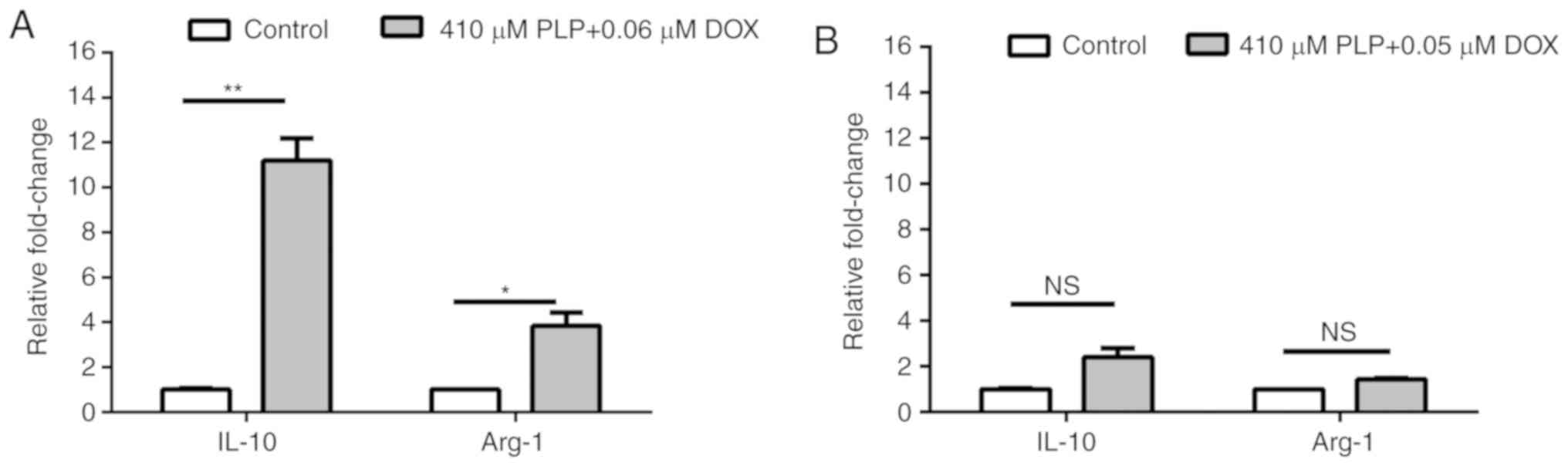
Combined treatment does not affect the
polarization of TAMs
To assess whether the applied treatments can affect
the polarization of TAMs, the mRNA relative expression of two
markers for this protumor phenotype of macrophages (IL-10 and
Arg-1) (45) was quantified by
RT-qPCR. The results revealed that administration of each combined
treatment increased the expression of both markers, albeit with
lower and not statistically significant degrees for pro-apoptotic
combined treatment (Fig. 5).
Previously reported data have demonstrated that TAMs
that induce chemoresistance to DOX are characterized by a high
expression of CD68, CD206, CD163, programmed death-ligand 1 (PD-L1)
and release immunosuppressive cytokines, such as IL-10 and TGF-β
(46). Moreover, in mouse models for
breast and lung cancer, the administration of cyclophosphamide,
paclitaxel and DOX, was shown to promote the protumor phenotype of
TAMs (46). Thus, the strong
overexpression of both M2 macrophage markers induced by the
anti-proliferative dose (0.06 µM) of DOX suggest that a low dose of
the cytotoxic drug favors the pro-tumor and immunosuppressive
function of TAMs. Thus, it is suggested that the combined
treatments tested did not affect the M2 phenotype of macrophages
and only had the ability to suppress the pro-angiogenic functions
of this cell type.
Discussion
Regardless of recent advances in melanoma treatment,
the development of acquired drug resistance remains a major issue,
limiting the effectiveness of these therapies (9,47). One of
the principal causes for melanoma cell resistance to different
treatments is the involvement of the tumor microenvironment cell
types (49,50). Among these supportive cell types, TAMs
are the most abundant immune cells infiltrated in the stroma of
solid cancers, their elevated number being an indicate of a poor
prognosis for >80% of tumor cases. Intratumor macrophages
exhibit a high plasticity in response to microenvironment stimuli,
promoting immune suppression, tumor cell proliferation,
angiogenesis, invasion and metastasis (51). In particular, it has been demonstrated
that TAMs may interfere via different means with the response of
melanoma cells to various drugs (52–55).
Nevertheless, our previous studies demonstrated that melanoma
growth can be markedly inhibited following the administration of
the water-soluble salt of prednisolone (PLP; as a liposomal
formulation), as a result of its inhibitory effect on TAM-mediated
tumor angiogenesis (26,56). Based on these findings, the aim of the
present study was to investigate whether the combination of the
TAM-targeting drug, PLP (26), with a
conventional cytotoxic drug, DOX, could lead to an improved
therapeutic outcome on the melanoma microenvironment. To the best
of our knowledge, the cytotoxicity of this combined therapeutic
approach on the melanoma microenvironment model has not been
described to date. The results of this study provided confirmatory
evidence for the synergistic inhibitory effects of PLP and DOX on
the proliferation of B16.F10 melanoma cells (Fig. 1 and Table
I). Moreover, the pro-apoptotic effects of DOX (noted at the
highest concentration tested) on the melanoma microenvironment were
also significantly potentiated by glucocorticoid administration
(Fig. 2). To elucidate the molecular
mechanisms responsible for the potent cytotoxicity of the combined
therapeutic approach on B16.F10 melanoma cells, we explored the
effects of two different combined treatments based on 410 µM PLP
administration with either the anti-proliferative DOX concentration
(0.06 µM) or the pro-apoptotic concentration of DOX (0.5 µM), on
TAM-mediated protumor processes, such as oxidative stress and
angiogenesis.
As several lines of evidence have demonstrated the
pro-oxidant capacity of DOX in both normal and cancer cells
(57,58) the role of the modulation of oxidative
stress in the cytotoxicity of the combined administration of PLP
and DOX on B16.F10 cells co-cultured with TAMs was assessed. The
results suggested that only the administration of 0.06 µM DOX
exerted potent antioxidant effects on the melanoma
microenvironment, irrespective of the presence of PLP (Fig. 3). Although the majority of studies
have indicated the stimulatory effects of DOX on oxidative stress
in endothelial and myocardial cells (59,60), the
suppressive effects of a lower concentration of DOX on ROS levels
in the melanoma microenvironment may be explained by its potential
to activate the antioxidant enzymes, as previously suggested
(61,62). Taken together, these data suggested
that the modulation of intratumor oxidative stress by this
therapeutic approach may be responsible for the anti-proliferative
activity rather than the inducing action of the combined
administration of DOX and PLP on cell apoptosis. Moreover, it has
been demonstrated that DOX-induced oxidative stress is mainly
responsible for apoptosis in endothelial and myocardial cells,
while tumor cell apoptosis is mediated via a different mechanism
(59,60).
To link the cytotoxicity of the therapeutic approach
to its ability to inhibit melanoma angiogenesis, we investigated
whether the combined treatments affected the production of proteins
involved in angiogenesis, as well as in inflammation-associated
angiogenesis (Table II and Fig. 4). The data revealed that both combined
treatments considerably decreased the levels of the majority of the
pro-angiogenic/ pro-inflammatory proteins in the cell co-culture
(Table II and Fig. 4). These antitumor effects of the
combined therapeutic approach may be linked to the anti-angiogenic
activity of PLP on the melanoma microenvironment, as a single
administration of the glucocorticoid also exerted marked inhibitory
effects on the angiogenic capacity of the cell co-culture (Fig. 4). Notably, treatment with DOX alone
did not markedly influence tumor angiogenesis (Fig. 4). These findings are consistent with
the results of a previous study by our group, showing that the
administration of DOX as a liposomal form, to melanoma
tumor-bearing mice generated only a slight inhibitory effect on the
production of angiogenic proteins (27). Taken together, these data suggest that
the anti-angiogenic activity of PLP on the melanoma
microenvironment may be potentiated by its combination with DOX in
a cytotoxic drug concentration-dependent manner (Table II). Moreover, the higher amplitude of
the anti-angiogenic effects of co-administration of PLP with 0.5 µM
DOX than that induced by the administration of the same
concentration of PLP in the presence of 0.06 µM DOX may also
explain the pro-apoptotic effects of the first treatment. Thus, the
production of several tumorigenic proteins, such as TIMP-1, TIMP-2,
TNF-α and thrombopoietin (TPO), also known for their anti-apoptotic
activity (63–67), were markedly inhibited by the
co-administration of PLP with 0.5 µM DOX.
Furthermore, to gain deeper insight into the role of
TAMs in modulating melanoma angiogenesis and finally, in the
response of melanoma cells to the combination therapy, the effects
of the simultaneous administration of both drugs on the angiogenic
capacity of TAMs were also evaluated. The results demonstrated
differences in the underlying mechanisms of the anti-angiogenic
action of the combined treatments on TAMs (Table III). Thus, the anti-proliferative
DOX concentration (0.06 µM) administered in combination with PLP,
exerted moderate suppressive effects on the TAM levels of specific
pro-angiogenic proteins (GM-CSF, IL-1β, TNF-α, eotaxin, FasL, bFGF
and VEGF), while the anti-angiogenic and anti-inflammatory proteins
(PF-4, IL-12p70, IFN-γ and MIG) were not affected by this
treatment. When the same concentration of PLP was administered in
the presence of the pro-apoptotic concentration of DOX (0.5 µM),
the majority of the pro-angiogenic, as well as anti-angiogenic
protein levels in TAMs were almost completely decreased (Table III). Nevertheless, the M2 phenotype
of TAMs was not altered by any treatment, as the expression of
IL-10 and Arg-1 was increased following combined treatment with PLP
and 0.06 µM DOX, and was not affected by pro-apoptotic combined
treatment (Fig. 5).
Collectively, the data of the present study
suggested that both combined treatments inhibited the
pro-angiogenic function of TAMs in the melanoma microenvironment,
while the immunosuppressive phenotype of these macrophages
(45,68,69) was
not affected by these treatments. Consequently, the combined
therapeutic approach developed in the present study could be
improved by supplementation with IL-12 or IFN-γ as re-polarizing
agents of M2-like TAMs toward M1-like antitumor macrophages
(70).
In conclusion, the results of this study
demonstrated that PLP enhanced the antitumor effects of DOX on
B16.F10 murine melanoma cells compared with the effects induced by
the cytotoxic drug administered alone. The cytotoxicity of DOX was
potentiated mainly via the anti-angiogenic activity of PLP in the
melanoma microenvironment. Moreover, the amplitude of the
cytotoxicity of the combined treatments might be linked to the
degree of the suppression of the pro-angiogenic function of TAMs.
Nevertheless, the immunosuppressive phenotype of TAMs was still
preserved after co-administration of PLP with DOX. Therefore,
further investigations with regard to the re-activation of TAMs to
combat melanoma cells are required.
Acknowledgements
Not applicable.
Funding
The authors alone are responsible for the content
and writing of this article. This study was supported by grants of
the Romanian Ministry of Research and Innovation, CNCS-UEFISCDI,
project no. PN-II-RU-TE-2014-4-1191, contract no. 235/2015 within
PNCDI–II and project no. PN-III-P4-ID-PCE-2016-0342, contract no.
91/2017 within PNCDI–III.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
EL and MB conceived and designed the study. EL, VFR,
LL, LP and AS acquired the data, and performed the experiments and
statistical analysis. EL and MB analyzed and interpreted the data.
EL and MB drafted and edited the manuscript. All authors have given
final approval of the version to be published.
Ethics approval and consent to
participate
Experiments using laboratory mice were performed
according to the European and national regulations and were
approved by the Committee on the Ethics of Animal Experiments of
the Babes-Bolyai University (registration no.
31444/27.03.2017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hsueh EC and Gorantla KC: Novel melanoma
therapy. Exp Hematol Oncol. 5:232016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
La Porta CA: Mechanism of drug sensitivity
and resistance in melanoma. Curr Cancer Drug Targets. 9:391–397.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuphal S and Bosserhoff A: Recent progress
in understanding the pathology of malignant melanoma. J Pathol.
219:400–409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zaretsky JM, Garcia-Diaz A, Shin DS,
Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY,
Abril-Rodriguez G, Sandoval S, Barthly L, et al: Mutations
associated with acquired resistance to PD-1 blockade in melanoma. N
Engl J Med. 375:819–829. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Larkin J, Chmielowski B, Lao CD, Hodi FS,
Sharfman W, Weber J, Suijkerbuijk KPM, Azevedo S, Li H, Reshef D,
et al: Neurologic serious adverse events associated with nivolumab
plus ipilimumab or nivolumab alone in advanced melanoma, including
a case series of encephalitis. Oncologist. 22:709–718. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Long GV, Atkinson V, Cebon JS, Jameson MB,
Fitzharris BM, McNeil CM, Hill AG, Ribas A, Atkins MB, Thompson JA,
et al: Standard-dose pembrolizumab in combination with reduced-dose
ipilimumab for patients with advanced melanoma (KEYNOTE-029): An
open-label, phase 1b trial. Lancet Oncol. 18:1202–1210. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wolchok JD, Kluger H, Callahan MK, Postow
MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K,
et al: Nivolumab plus ipilimumab in advanced melanoma. N Engl J
Med. 369:122–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Melis C, Rogiers A, Bechter O and van den
Oord JJ: Molecular genetic and immunotherapeutic targets in
metastatic melanoma. Virchows Arch. 471:281–293. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Son B, Lee S, Youn H, Kim E, Kim W and
Youn B: The role of tumor microenvironment in therapeutic
resistance. Oncotarget. 8:3933–3945. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang T, Xiao M, Ge Y, Krepler C, Belser E,
Lopez-Coral A, Xu X, Zhang G, Azuma R, Liu Q, et al: BRAF
inhibition stimulates melanoma-associated macrophages to drive
tumor growth. Clin Cancer Res. 21:1652–1664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith BD, Kaufman MD, Leary CB, Turner BA,
Wise SC, Ahn YM, Booth RJ, Caldwell TM, Ensinger CL, Hood MM, et
al: Altiratinib inhibits tumor growth, invasion, angiogenesis, and
microenvironment-mediated drug resistance via balanced inhibition
of MET, TIE2, and VEGFR2. Mol Cancer Ther. 14:2023–2034. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Castells M, Thibault B, Delord JP and
Couderc B: Implication of tumor microenvironment in
chemoresistance: Tumor-associated stromal cells protect tumor cells
from cell death. Int J Mol Sci. 13:9545–9571. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Affara NI, Ruffell B, Medler TR, Gunderson
AJ, Johansson M, Bornstein S, Bergsland E, Steinhoff M, Li Y, Gong
Q, et al: B cells regulate macrophage phenotype and response to
chemotherapy in squamous carcinomas. Cancer Cell. 25:809–821. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ries CH, Cannarile MA, Hoves S, Benz J,
Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I,
et al: Targeting tumor-associated macrophages with anti-CSF-1R
antibody reveals a strategy for cancer therapy. Cancer Cell.
25:846–859. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Solinas G, Germano G, Mantovani A and
Allavena P: Tumor-associated macrophages (TAM) as major players of
the cancer-related inflammation. J Leukoc Biol. 86:1065–1073. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: Tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tham M, Tan KW, Keeble J, Wang X, Hubert
S, Barron L, Tan NS, Kato M, Prevost-Blondel A, Angeli V and
Abastado JP: Melanoma-initiating cells exploit M2 macrophage
TGFbeta and arginase pathway for survival and proliferation.
Oncotarget. 5:12027–12042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yaddanapudi K, Putty K, Rendon BE, Lamont
GJ, Faughn JD, Satoskar A, Lasnik A, Eaton JW and Mitchell RA:
Control of tumor-associated macrophage alternative activation by
macrophage migration inhibitory factor. J Immunol. 190:2984–2993.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clear AJ, Lee AM, Calaminici M, Ramsay AG,
Morris KJ, Hallam S, Kelly G, Macdougall F, Lister TA and Gribben
JG: Increased angiogenic sprouting in poor prognosis FL is
associated with elevated numbers of CD163+ macrophages
within the immediate sprouting microenvironment. Blood.
115:5053–5056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leek RD, Lewis CE, Whitehouse R, Greenall
M, Clarke J and Harris AL: Association of macrophage infiltration
with angiogenesis and prognosis in invasive breast carcinoma.
Cancer Res. 56:4625–4629. 1996.PubMed/NCBI
|
|
22
|
Liu T, Larionova I, Litviakov N, Riabov V,
Zavyalova M, Tsyganov M, Buldakov M, Song B, Moganti K, Kazantseva
P, et al: Tumor-associated macrophages in human breast cancer
produce new monocyte attracting and pro-angiogenic factor YKL-39
indicative for increased metastasis after neoadjuvant chemotherapy.
Oncoimmunology. 7:e14369222018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shieh YS, Hung YJ, Hsieh CB, Chen JS, Chou
KC and Liu SY: Tumor-associated macrophage correlated with
angiogenesis and progression of mucoepidermoid carcinoma of
salivary glands. Ann Surg Oncol. 16:751–760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marech I, Ammendola M, Sacco R, Sammarco
G, Zuccalà V, Zizzo N, Leporini C, Luposella M, Patruno R,
Filippelli G, et al: Tumour-associated macrophages correlate with
microvascular bed extension in colorectal cancer patients. J Cell
Mol Med. 20:1373–1380. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alupei MC, Licarete E, Patras L and Banciu
M: Liposomal simvastatin inhibits tumor growth via targeting
tumor-associated macrophages-mediated oxidative stress. Cancer
Lett. 356:946–952. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banciu M, Metselaar JM, Schiffelers RM and
Storm G: Antitumor activity of liposomal prednisolone phosphate
depends on the presence of functional tumor-associated macrophages
in tumor tissue. Neoplasia. 10:108–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Banciu M, Schiffelers RM and Storm G:
Investigation into the role of tumor-associated macrophages in the
antitumor activity of Doxil. Pharm Res. 25:1948–1955. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sylvester B, Porfire A, Muntean DM, Vlase
L, Lupuţ L, Licarete E, Sesarman A, Alupei MC, Banciu M, Achim M
and Tomuţă I: Optimization of prednisolone-loaded long-circulating
liposomes via application of quality by design (QbD) approach. J
Liposome Res. 28:49–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
De Palma M and Lewis CE: Macrophage
regulation of tumor responses to anticancer therapies. Cancer Cell.
23:277–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cortes-Funes H and Coronado C: Role of
anthracyclines in the era of targeted therapy. Cardiovasc Toxicol.
7:56–60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fink W, Zimpfer-Rechner C, Thoelke A, Figl
R, Kaatz M, Ugurel S and Schadendorf D: Clinical phase II study of
pegylated liposomal doxorubicin as second-line treatment in
disseminated melanoma. Onkologie. 27:540–544. 2004.PubMed/NCBI
|
|
32
|
Schadendorf D, Worm M, Algermissen B,
Kohlmus CM and Czarnetzki BM: Chemosensitivity testing of human
malignant melanoma. A retrospective analysis of clinical response
and in vitro drug sensitivity. Cancer. 73:103–108. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vorobiof DA, Rapoport BL, Mahomed R and
Karime M: Phase II study of pegylated liposomal doxorubicin in
patients with metastatic malignant melanoma failing standard
chemotherapy treatment. Melanoma Res. 13:201–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Haase-Kohn C, Wolf S, Herwig N, Mosch B
and Pietzsch J: Metastatic potential of B16-F10 melanoma cells is
enhanced by extracellular S100A4 derived from RAW264.7 macrophages.
Biochem Biophys Res Commun. 446:143–148. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X, Goncalves R and Mosser DM: The
isolation and characterization of murine macrophages. Curr Protoc
Immunol. 14:Unit 14 11. 2008. View Article : Google Scholar
|
|
36
|
Rauca VF, Licarete E, Luput L, Sesarman A,
Patras L, Bulzu P, Rakosy-Tican E and Banciu M: Combination therapy
of simvastatin and 5,6-dimethylxanthenone-4-acetic acid
synergistically suppresses the aggressiveness of B16.F10 melanoma
cells. PLoS One. 13:e02028272018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Banciu M, Fens MH, Storm G and Schiffelers
RM: Antitumor activity and tumor localization of liposomal
glucocorticoids in B16 melanoma-bearing mice. J Control Release.
127:131–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Licarete E, Sesarman A, Rauca VF, Luput L,
Patras L and Banciu M: HIF-1α acts as a molecular target for
simvastatin cytotoxicity in B16.F10 melanoma cells cultured under
chemically induced hypoxia. Oncol Lett. 13:3942–3950. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alupei MC, Licarete E, Cristian FB and
Banciu M: Cytotoxicity of lipophilic statins depends on their
combined actions on HIF-1α expression and redox status in B16.F10
melanoma cells. Anticancer Drugs. 25:393–405. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Karatas F, Karatepe M and Baysar A:
Determination of free malondialdehyde in human serum by
high-performance liquid chromatography. Anal Biochem. 311:76–79.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Venza M, Visalli M, Beninati C, De Gaetano
GV, Teti D and Venza I: Cellular mechanisms of oxidative stress and
action in melanoma. Oxid Med Cell Longev. 2015:4817822015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rőszer T: Understanding the mysterious M2
macrophage through activation markers and effector mechanisms.
Mediators Inflamm. 2015:8164602015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Larionova I, Cherdyntseva N, Liu T,
Patysheva M, Rakina M and Kzhyshkowska J: Interaction of
tumor-associated macrophages and cancer chemotherapy.
Oncoimmunology. 8:15960042019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Manzano JL, Layos L, Bugés C, de Los
Llanos Gil M, Vila L, Martínez-Balibrea E and Martínez-Cardús A:
Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl
Med. 4:2372016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Winder M and Viros A: Mechanisms of drug
resistance in melanoma. Handb Exp Pharmacol. 249:91–108. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vera RE, Lamberti MJ, Rivarola VA and
Rumie Vittar NB: Developing strategies to predict photodynamic
therapy outcome: The role of melanoma microenvironment. Tumour
Biol. 36:9127–9136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Klemm F and Joyce JA: Microenvironmental
regulation of therapeutic response in cancer. Trends Cell Biol.
25:198–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Szebeni GJ, Vizler C, Kitajka K and Puskas
LG: Inflammation and cancer: Extra- and intracellular determinants
of tumor-associated macrophages as tumor promoters. Mediators
Inflamm. 2017:92940182017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gajewski TF: Identifying and overcoming
immune resistance mechanisms in the melanoma tumor
microenvironment. Clin Cancer Res. 12:2326s–2330s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Varney ML, Olsen KJ, Mosley RL and Singh
RK: Paracrine regulation of vascular endothelial growth factor-a
expression during macrophage-melanoma cell interaction: Role of
monocyte chemotactic protein-1 and macrophage colony-stimulating
factor. J Interferon Cytokine Res. 25:674–683. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang T, Ge Y, Xiao M, Lopez-Coral A, Azuma
R, Somasundaram R, Zhang G, Wei Z, Xu X, Rauscher FJ III, et al:
Melanoma-derived conditioned media efficiently induce the
differentiation of monocytes to macrophages that display a highly
invasive gene signature. Pigment Cell Melanoma Res. 25:493–505.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ruffell B and Coussens LM: Macrophages and
therapeutic resistance in cancer. Cancer Cell. 27:462–472. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Banciu M, Metselaar JM, Schiffelers RM and
Storm G: Liposomal glucocorticoids as tumor-targeted
anti-angiogenic nanomedicine in B16 melanoma-bearing mice. J
Steroid Biochem Mol Biol. 111:101–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gouaze V, Mirault ME, Carpentier S,
Salvayre R, Levade T and Andrieu-Abadie N: Glutathione peroxidase-1
overexpression prevents ceramide production and partially inhibits
apoptosis in doxorubicin-treated human breast carcinoma cells. Mol
Pharmacol. 60:488–496. 2001.PubMed/NCBI
|
|
58
|
Ubezio P and Civoli F: Flow cytometric
detection of hydrogen peroxide production induced by doxorubicin in
cancer cells. Free Radic Biol Med. 16:509–516. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Berthiaume JM and Wallace KB:
Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell
Biol Toxicol. 23:15–25. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang S, Konorev EA, Kotamraju S, Joseph J,
Kalivendi S and Kalyanaraman B: Doxorubicin induces apoptosis in
normal and tumor cells via distinctly different mechanisms.
Intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem.
279:25535–25543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Verma N and Vinayak M: A low dose of
doxorubicin improves antioxidant defence system and modulates
anaerobic metabolism during the development of lymphoma. Indian J
Pharmacol. 44:308–313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pritsos CA and Ma J: Basal and
drug-induced antioxidant enzyme activities correlate with
age-dependent doxorubicin oxidative toxicity. Chem Biol Interact.
127:1–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dong QM, Ling C, Chen X and Zhao LI:
Inhibition of tumor necrosis factor-α enhances apoptosis induced by
nuclear factor-kB inhibition in leukemia cells. Oncol Lett.
10:3793–3798. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kirito K, Watanabe T, Sawada K, Endo H,
Ozawa K and Komatsu N: Thrombopoietin regulates Bcl-xL gene
expression through Stat5 and phosphatidylinositol 3-kinase
activation pathways. J Biol Chem. 277:8329–8337. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nalluri S, Ghoshal-Gupta S, Kutiyanawalla
A, Gayatri S, Lee BR, Jiwani S, Rojiani AM and Rojiani MV: TIMP-1
inhibits apoptosis in lung adenocarcinoma cells via interaction
with Bcl-2. PLoS One. 10:e01376732015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Valacca C, Tassone E and Mignatti P:
TIMP-2 interaction with MT1-MMP activates the AKT pathway and
protects tumor cells from apoptosis. PLoS One. 10:e01367972015.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Valente P, Fassina G, Melchiori A,
Masiello L, Cilli M, Vacca A, Onisto M, Santi L, Stetler-Stevenson
WG and Albini A: TIMP-2 over-expression reduces invasion and
angiogenesis and protects B16F10 melanoma cells from apoptosis. Int
J Cancer. 75:246–253. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Georgoudaki AM, Prokopec KE, Boura VF,
Hellqvist E, Sohn S, Östling J, Dahan R, Harris RA, Rantalainen M,
Klevebring D, et al: Reprogramming tumor-associated macrophages by
antibody targeting inhibits cancer progression and metastasis. Cell
Rep. 15:2000–2011. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sica A and Mantovani A: Macrophage
plasticity and polarization: In vivo veritas. J Clin Invest.
122:787–795. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zheng X, Turkowski K, Mora J, Brüne B,
Seeger W, Weigert A and Savai R: Redirecting tumor-associated
macrophages to become tumoricidal effectors as a novel strategy for
cancer therapy. Oncotarget. 8:48436–48452. 2017.PubMed/NCBI
|