Introduction
Colorectal cancer (CRC) is one of the most highly
malignant cancers worldwide and is associated with high morbidity
and mortality rate (1,2). CRC evolves through multiple distinct
pathways, including the classical adenoma-carcinoma sequence and
the serrated pathways (3). These
pathways are defined based on molecular features and the pathology
of the precursor lesions. The classical adenoma-carcinoma sequence
originates in conventional adenomas (tubular adenomas and
tubulovillous adenomas), whereas the serrated pathway develops in
serrated polyps (SPs). Molecularly, the classical adenoma-carcinoma
sequence pathway is characterized by chromosomal instability (CIN)
and APC regulator Of WNT signaling pathway (APC) or KRAS
proto-oncogene, GTPase (KRAS) mutations (4), whereas in the serrated pathway the
genetic alterations include specific B-Raf proto-oncogene,
serine/threonine kinase (BRAF) and KRAS mutations, microsatellite
instability (MSI), and a CpG island methylator phenotype (CIMP)
(5,6).
KRAS mutations are found in 35–42% of CRCs and advanced adenomas
(7). Proteins expressed by KRAS genes
are involved in the RAF/MEK/ERK mitogen-activated protein kinase
(MAPK) signaling pathway, which is a downstream pathway of
epidermal growth factor receptor (EGFR). Mutations in KRAS and BRAF
genes lead to the persistent activation of this pathway and
accelerate the proliferation of tumor cells (8). It has been widely accepted that KRAS
mutations predict the poor efficacy of anti-EGFR therapy in
patients with metastatic CRC (9).
Previous studies indicate that KRAS mutations are linked with
statistically significant reductions in overall survival and
disease-free survival of CRC patients (10,11).
MicroRNAs (miRNAs or miRs) are endogenous RNA
molecules 19–25 nucleotides in length, that regulate the expression
of genes involved in cell differentiation, proliferation and
apoptosis (12–15). A number of studies have demonstrated
that altered expression of specific miRNAs contributes to the
initiation and progression of CRC (16–18). There
is also evidence that miRNAs can act as tumor suppressors or
oncogenes depending on the cellular environment in which they are
expressed (19,20). In an effort to uncover the
relationship between miRNA expression and carcinogenesis, it was
found that expression levels of miRNAs are correlated with
proto-oncogenes characteristically expressed in CRC. It was
demonstrated that miR-192 and miR-215 are both effectors and
regulators of p53 function to suppress colon carcinogenesis
(21). Another p53-related miRNA,
miR-34a, was shown to inhibit cell invasion in colon cancer cell
lines by targeting FRA1 (22).
miR-320b can target c-Myc in human CRC cells to suppress
proliferation (23). Another study
showed that c-Myc was able to regulate the miR-17 cluster, and
modulate E2F1 expression (24).
Johnson et al (25) reported
that the RAS oncogene is regulated by the let-7 miRNA family, and
decreased expression of let-7 miRNA in various types of human lung
tumors caused increased expression of RAS protein. miR-155 was
found to be positively regulated by CBX7 in colon carcinomas, and
targets the KRAS oncogene (26).
miR-143 was found to be tissue-specifically
expressed in mice (27). It was
observed that expression of mature miR-143 is frequently
downregulated in approximately 80% of human colorectal tumor
samples from cancer and adenoma patients compared with its level in
normal tissues (28). miR-143 can
regulate the cell growth and proliferation of CRC in vitro
by targeting different oncogenic protein-coding genes, and directly
repressing translation of extracellular signal-regulated kinase-5
(ERK5) (29) and DNA
(cytosine-5)-methyltransferase 3A (30), but the specific mechanism requires
further investigation.
In the present study, the miR-143 expression plasmid
was constructed and transfected into human colorectal carcinoma
cell lines (SW480, LoVo and HT-29). It was found that increased
accumulation of miR-143 suppressed expression of KRAS protein in
the transfected cells. Furthermore, luciferase reporter assay
demonstrated that miR-143 was likely to regulate translation of
target mRNAs by hybridization to complementary sites at the 3′
untranslated region (UTR).
Materials and methods
Tissue samples and cell lines
Colorectal tumors and the corresponding normal
mucosa were obtained from fresh surgical excision at the First
Hospital of Shanxi Medical University, Taiyuan, China from May 20
to July 26, 2018. This study was approved by the Ethics Commission
of the First Hospital of Shanxi Medical University [approval no.
2018(k012), Taiyuan, China)]. Informed consent was obtained from
all patients before surgery. A total of 8 patients, including 5
males and 3 females were recruited. The age range was 41–63 years,
with an average age of 51.8±6.72 years. Histopathological
examination of each sample was performed to confirm its diagnostic
yield. The tissues were placed in liquid nitrogen immediately after
resection and stored at −80°C. Human colorectal carcinoma cell
lines SW480 (KRAS mutant-type), LoVo (KRAS mutant-type), HT-29
(KRAS wild-type) and normal colon epithelial cell line NCM460 were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich; Merck
KGaA) at 37°C in a humidified atmosphere with 5%
CO2.
RNA isolation and quantitative reverse
transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cultured cells and
tissues using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The mature miR-143 was assessed using TaqMan miRNA Real-Time
PCR assays and the Rotor-Gene 3000 thermal cycler (Qiagen,
Shanghai, China). Briefly, 1 µg total RNA was reverse-transcribed
to cDNA using AWV reverse transcriptase (Takara, Dalian, China),
and the expression of miR-143 was measured by RT-qPCR. The
sequences of the primers (Sangon Biotech Co., Ltd., Shanghai,
China) were: miR-143 forward, 5′-GGGTGAGATGAAGCACTGTAGCTC-3′ and
reverse, 5′-GCTGTCAACATACGCTACGTAACG-3′; human U6 forward,
5′-CGCTTCACGAATTTGCGTGTCA-3′ and reverse,
5′-GCTTCGGCAGCACATATACTAAAAT-3′. The reaction conditions were as
follows: 95°C for 1 min, followed by 40 cycles at 95°C for 30 sec,
60°C for 30 sec, 95°C for 45 min, 55°C for 1 min and 55°C for 10
sec. U6 small nuclear (sn)RNA was used as an internal control, and
the relative expression levels of miR-143 were calculated by the
2−ΔΔCq method (31).
Cloning of pri-miR-143 and plasmid
construction
Genomic DNA was extracted using lysis buffer
containing 10 mM Tris-HCl, pH 8.0; 100 mM NaCl; 0.5% SDS; 25 mM
EDTA, treated with proteinase K (0.2 mg/ml), purified with
phenol/chloroform, precipitated with ethanol and dissolved in
DNase-free water. RT-PCR was performed using the following primers
(32):
5′-AGGTTTGGTCCTGGGTGCTCAAATGGCAGG-3′ (forward) and
5′-TGCCCAGACTCGTGAAGCAGATCGTGGCAC-3′ (reverse). pGEM-pri-miR-143
was constructed by T/A cloning of a 430-bp pri-miR-143 PCR product
into the pGEM-T-Easy vector (Promega). Mutant pri-miR-143 was
derived from pGEM-pri-miR-143 using the QuickChange method
(Stratagene). The reaction was performed in the presence of
pfu hotstart DNA polymerase (Stratagene) for 18 cycles at
95°C for 1 min, 60°C for 1 min and 68°C for 8 min using PTC-100
thermocycler (MJ-Research). The mutagenic primers were as follows:
5′-TGGTCAGTTGGGAGTCAGCACTGTAGCTCAGG-3′ (forward) and
5′-CCTGAGCTACAGTGCTGACTCCCAACTGACCA-3′ (reverse).
pcDNA3.1-pri-miR-143 and pcDNA3.1-pri-miR-143m were constructed by
subcloning pri-miR-143 and mutated pri-miR-143 into pcDNA3.1
eukaryotic expression vector (Invitrogen), respectively. The DNA
insert from the recombinant plasmid was sequenced by
chain-termination dideoxy sequencing to confirm the mutation.
Transfection in vitro and cell
proliferation assay
Transient transfections were performed using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
Human colorectal carcinoma cell lines SW480, LoVo or HT-29 cells
were plated in duplicate 1 day before transfection, so that the
cells were 90–95% confluent at the time of transfection. We used
1.6 µg plasmid DNA (expression plasmid pcDNA3.1-pri-miR-143,
pcDNA3.1-pri-miR-143 mutant or empty vector pcDNA3.1) for each well
of a 12-well plate. After 5 h of incubation, DNA-Lipofectamine 2000
complexes were removed and replaced by normal medium. Cells were
used for experiment 24 h after transfection.
Cell proliferation was evaluated by colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Transfected SW480, LoVo and HT-29 cells were respectively
seeded in 96-well plates in triplicate and incubated for 1–5 days.
A 1/10 volume of MTT solution (4 mg/ml in PBS) was added and
incubated for 4 h at 37°C. The supernatant was aspirated, and an
equal volume of dimethyl sulfoxide was added to the cells. MTT
formazan was dissolved by pipetting. The absorbance was measured on
an ELISA plate reader (Bio-Rad) at a wavelength of 570 nm. All the
experiments were repeated at least three times.
RT-qPCR analysis
To quantify the KRAS mRNA, total RNA was isolated 48
or 72 h after transfection using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). Total RNA (1 µg) was reverse-transcribed
to cDNA using oligo(dT) (Takara) and AMV reverse transcriptase
(Takara). The reaction conditions were: 16°C for 30 min, 42°C for
30 min and 85°C for 5 min. Real-time PCR was then processed using
the RT product, SYBR Green dye (Invitrogen; Thermo Fisher
Scientific, Inc.) and specific primers for KRAS and β-actin. The
relative amount of KRAS mRNA was normalized to β-actin. The
sequences of the primers were as follows: KRAS (sense),
5′-GACTCTGAAGATGTACCTATGGTCCTA-3′ and KRAS (antisense),
5′-CATCATCAACACCCTGTCTTGTC-3′; β-actin (sense),
5′-TCACCCACACTGTGCCCATCTACGA-3′ and β-actin (antisense),
5′-CAGCGGAACCGCTCATTGCCAATGG-3′. The reactions were incubated at
95°C for 5 min, followed by 40 cycles of 95°C for 30 sec, 60°C for
30 sec and 72°C for 30 sec. The results were confirmed by three
independent experiments.
Western blotting
Total protein was extracted from tissue samples and
cells with protein extraction reagent (Pierce; Thermo Fisher
Scientific, Inc.). The protein concentration was determined using a
BCA Protein Assay Reagent kit (Pierce; Thermo Fisher Scientific,
Inc.). Total protein (10 µg) was separated on 12% SDS-PAGE and
electrotransfered to polyvinylidene fluoride membranes. Primary
antibodies binding to KRAS (mouse monoclonal antibody, cat. no.
sc-30; 1:5,000 dilution; Santa Cruz Biotechnology), NRAS (mouse
monoclonal antibody, cat. no. sc-519; 1:5,000 dilution; Santa Cruz
Biotechnology), β-actin (mouse monoclonal antibody, cat. no. A5316;
1:1,000 dilution; Sigma-Aldrich; Merck KGaA) were detected with
horseradish-peroxidase-conjugated anti-mouse secondary antibodies
(cat. no. AP308P; 1:2,000 dilution; Sigma-Aldrich; Merck KGaA) and
the ECL Plus Western Blotting Detection kit (Amersham). Scanned
images were quantified using Quantity One software (version 4.6.9;
Bio-Rad Laboratories).
Luciferase assay
Two KRAS cDNA fragments, containing putative miR-143
complementary site (CS), were amplified by RT-PCR with the
following primer sets: KRAS-CS1, 5′-TTACAATCTCTAGGTTTGGCTAGTTCTC-3′
(forward) and 5′-GTCTAGAAGGTAGGGAGGCAAGATGAC-3′ (reverse);
KRAS-CS2, 5′-GCCTCTTGAATTTTTGATGTAGATG-3′ (forward) and
5′-GTCTAGACAAATGGAAATCTTCAGATA-3′ (reverse). pGL3-KRAS-CS was
constructed by insertion of XbaI-digested KRAS-CS in the
XbaI site of the pGL3 control vector (Promega). Various pGL3
reporters were constructed, and then cotransfected into the CRC
cells with pcDNA3 plasmids using Lipofectamine 2000 transfection
reagent. After 48 and 72 h of transfection, the luciferase and
β-galactosidase activities were determined.
Statistical analysis
All of the western blotting and proliferation assay
images are representative of at least three independent
experiments. RT-qPCR and luciferase reporter assays were performed
in triplicate. SPSS 20.0 software (IBM Corp., Armonk, NY, USA) was
used for statistical analysis, and the data are presented as the
mean ± standard error of the mean. Statistical analysis was
performed using Student's t-test or one-way ANOVA followed by the
Dunnett's t test. P<0.05 was considered indicative of a
statistically significant result.
Results
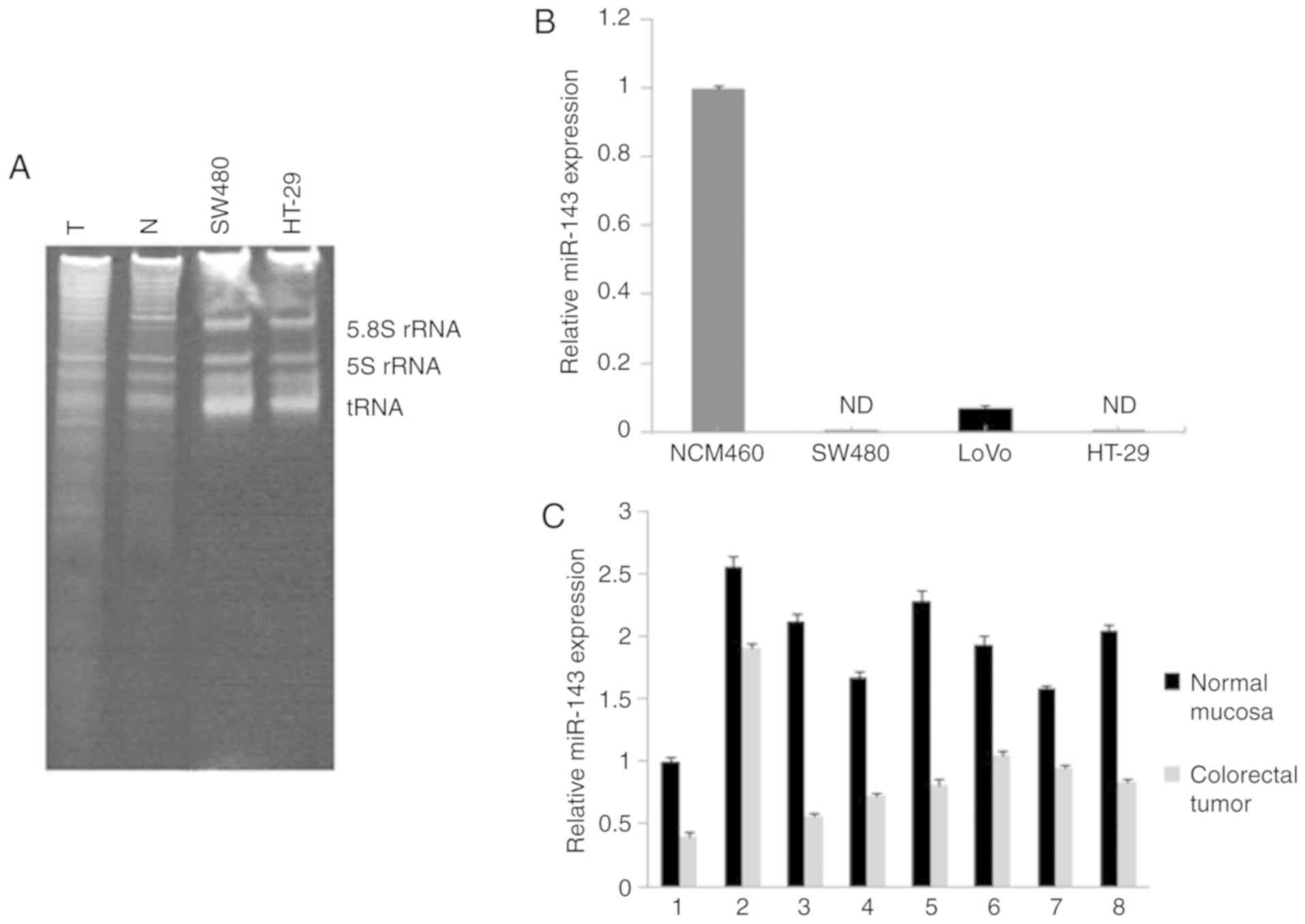
Reduced expression of miR-143 in
colorectal neoplasia
Total RNA was isolated from colorectal carcinoma
tissues, normal adjacent mucosa and cell lines. snRNA was examined
to confirm its high quality by staining with ethidium bromide
(Fig. 1A). RT-qPCR assay was
performed to validate the expression levels of mature miR-143 in
cell lines. The results showed a lack of miR-143 in human
colorectal carcinoma cells SW480, LoVo and HT-29, compared to high
expression in normal colon epithelial cell line NCM460 (Fig. 1B). The same method revealed an ~53.2%
in reduction of the miR-143 level in tumors compared with that
noted in the normal adjacent tissues (Fig. 1C). This observation was consistent
with the findings of a previous study reporting reduction of
miR-143 in colorectal neoplasia (28).
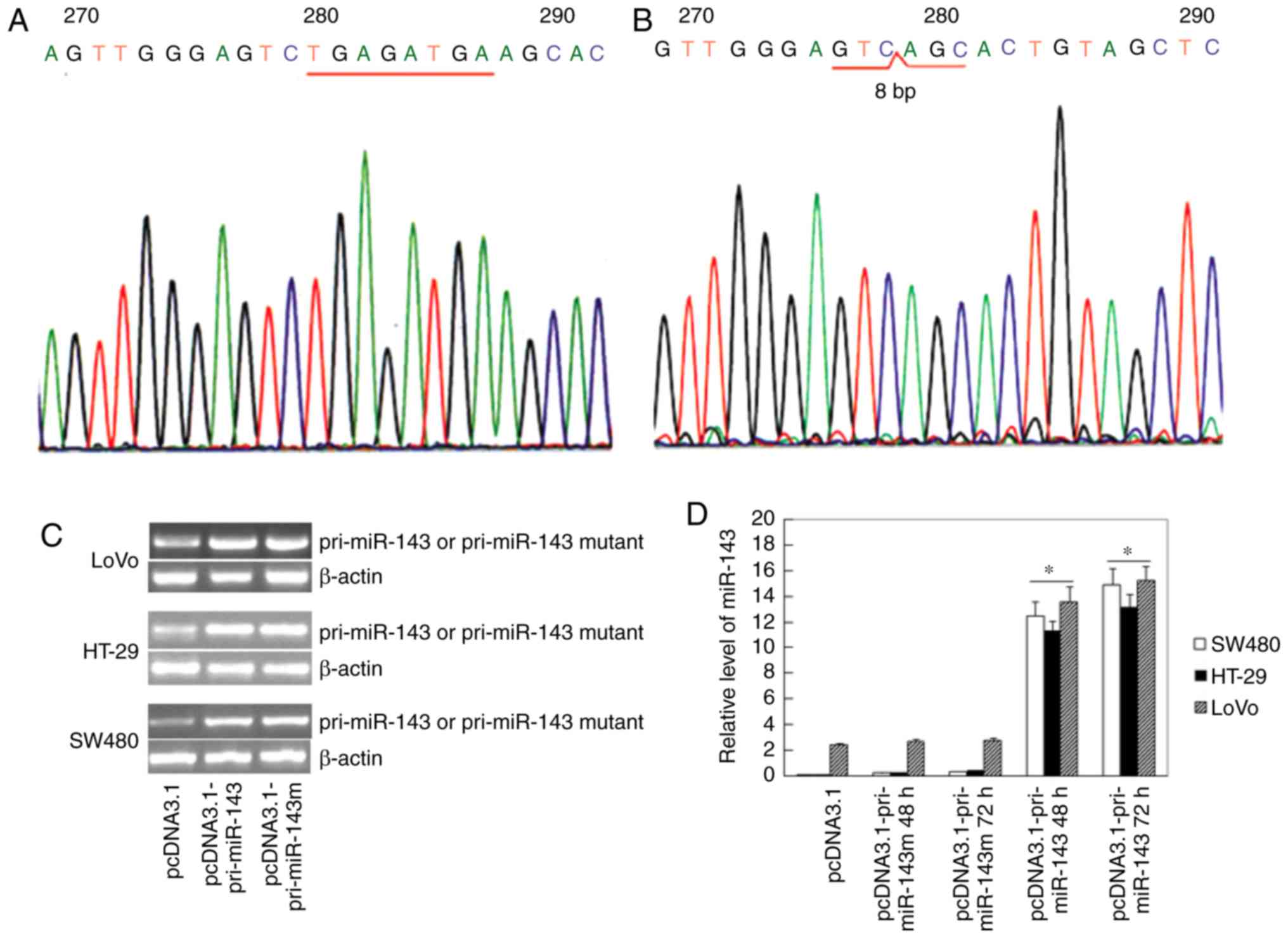
Accumulation of miR-143 in the
transfected cells
Expression plasmid pcDNA3.1-pri-miR-143 was
constructed. The 430-bp DNA insert was sequenced (Fig. 2A), and compared with the GenBank
database (NT_029289). A pcDNA3.1-pri-miR-143 mutant was derived by
knockdown of 8 nucleotides (TGAGATGA) at the 5′end of miR-143,
which was a putative binding site (Fig.
2B). The mutated plasmid pcDNA3.1-pri-miR-143m was utilized as
a control in research of miR-143 function. To investigate whether
changes in miR-143 expression levels were associated with cell
proliferation and tumorigenesis, we transfected pcDNA3.1,
pcDNA3.1-pri-miR-143 or pcDNA3.1-pri-miR-143m into SW480, LoVo and
HT-29 cells respectively. The mRNA levels of pri-miR-143 or
pri-miR-143 mutant in the transfected cells were detected by
RT-PCR, Subsequently the product was sequenced to confirm the
efficiency of gene transfection. The results showed the
significantly increased expression of pri-miR-143 in the
transfected cells with pcDNA3.1-pri-miR-143, related to the empty
vector, and transfection of pcDNA3.1-pri-miR-143m was confirmed by
gene sequencing (Fig. 2C). RT-qPCR
was used to detect the level of mature miR-143. Increased
accumulation of mature miR-143 was found in the
pcDNA3.1-pri-miR-143-transfected cells, while transfection of
pcDNA3.1-pri-miR-143m had no effects on the level of miR-143
(Fig. 2D). It should be emphasized
that expression of the miR-143 mutant was not measured by RT-qPCR,
due to its poor specificity (14 base-fragment,
3′-ACTCGATGTCACGA-5′).
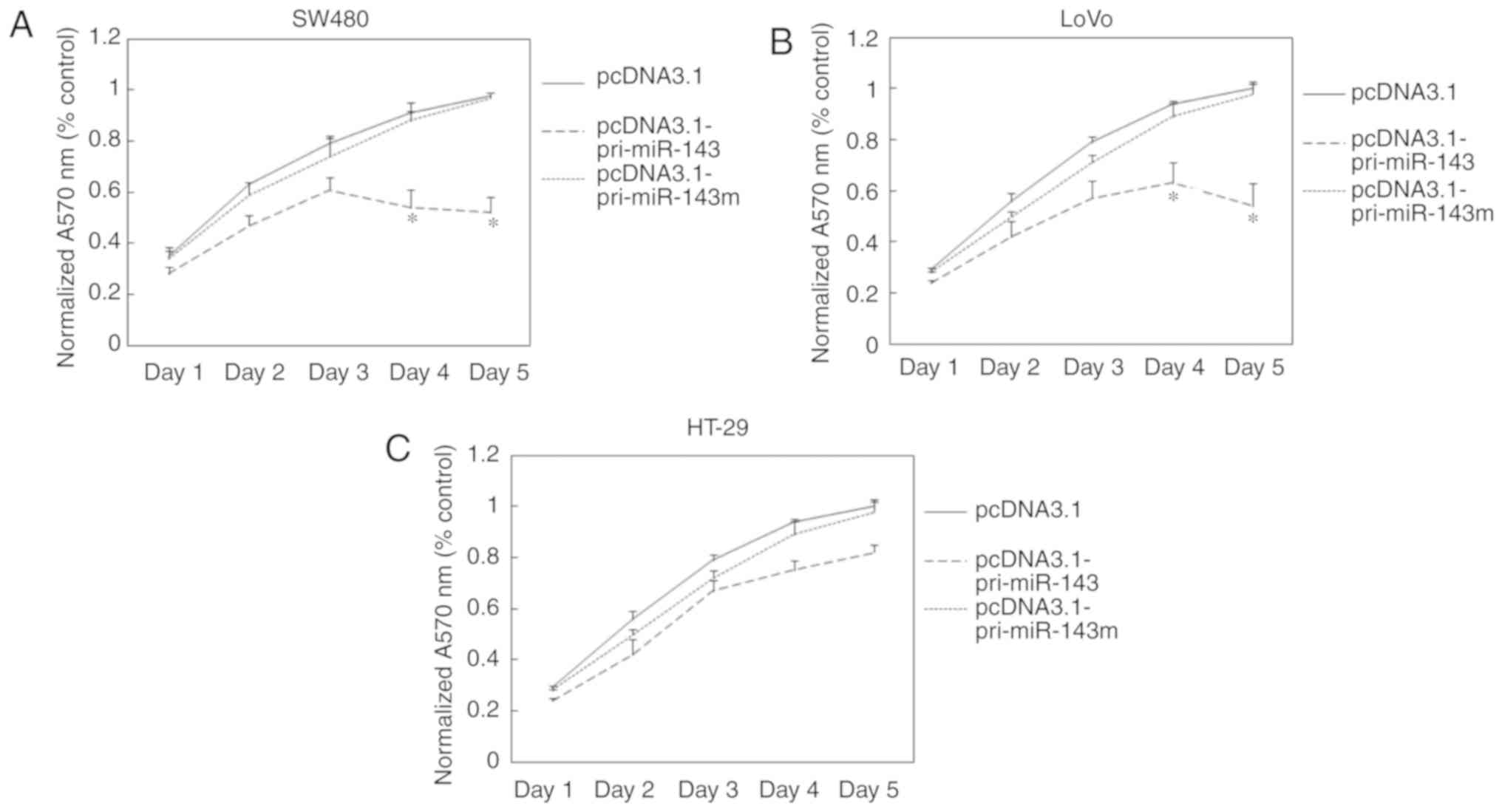
Effects of miR-143 on cell
proliferation
miRNAs are involved in cell proliferation and
differentiation. Therefore, we examined the effects of miR-143
transfection on the proliferation of CRC cells. The cells
transfected with pcDNA3.1-pri-miR-143, pcDNA3.1-pri-miR-143 mutant
or pcDNA3.1 were incubated for 1–5 days before measuring absorbance
by colorimetric MTT assay. In SW480 cells, transfection of
pcDNA3.1-pri-miR-143 resulted in 35 and 47% reduction in cell
growth after incubation for 4 and 5 days, respectively, compared
with transfection of pcDNA3.1-pri-miR-143m (Fig. 3A), while in LoVo cells, transfection
of pcDNA3.1-pri-miR-143 resulted in 33 and 46% reduction in cell
growth after incubation for 4 and 5 days, respectively (Fig. 3B). In contrast, transfection of
pcDNA3.1-pri-miR-143 had no significantly effects on HT-29 cell
growth (Fig. 3C). This demonstrated
that increased accumulation of miR-143 is capable of inhibiting the
proliferation of KRAS-mutant cell lines, but had no effects on KRAS
wild-type cell lines.
miR-143 partially suppresses KRAS
protein expression
There have been several efforts to use
bioinformatics techniques to identify miRNA target mRNAs. KRAS
transcript was one of the predicted miR-143 targets (33,34). When
expression plasmids were transfected into SW480, LoVo and HT-29
cells, respectively, increased accumulation of miR-143 was achieved
only in cells transfected with pcDNA3.1-pri-miR-143 (Fig. 2D). We observed an ~58% decrease in
KRAS protein levels in the pcDNA3.1-pri-miR-143-transfected SW480
cells, but not in the pcDNA3.1-pri-miR-143m-transfected cells
(Fig. 4B and C). Similarly,
transfection with pcDNA3.1-pri-miR-143 resulted in approximately 54
and 43% KRAS protein reduction in the LoVo and HT-29 cells,
respectively, compared with pcDNA3.1-pri-miR-143m (Fig. 4D-G). As a control for target
specificity, expression of NRAS protein was not affected (Fig. 4B-G). We also found that levels of KRAS
mRNA were not affected by transfection of miR-143 (Fig. 4A). These results suggest that
increased accumulation of miR-143 specifically suppresses KRAS
protein expression at the post-transcriptional level.
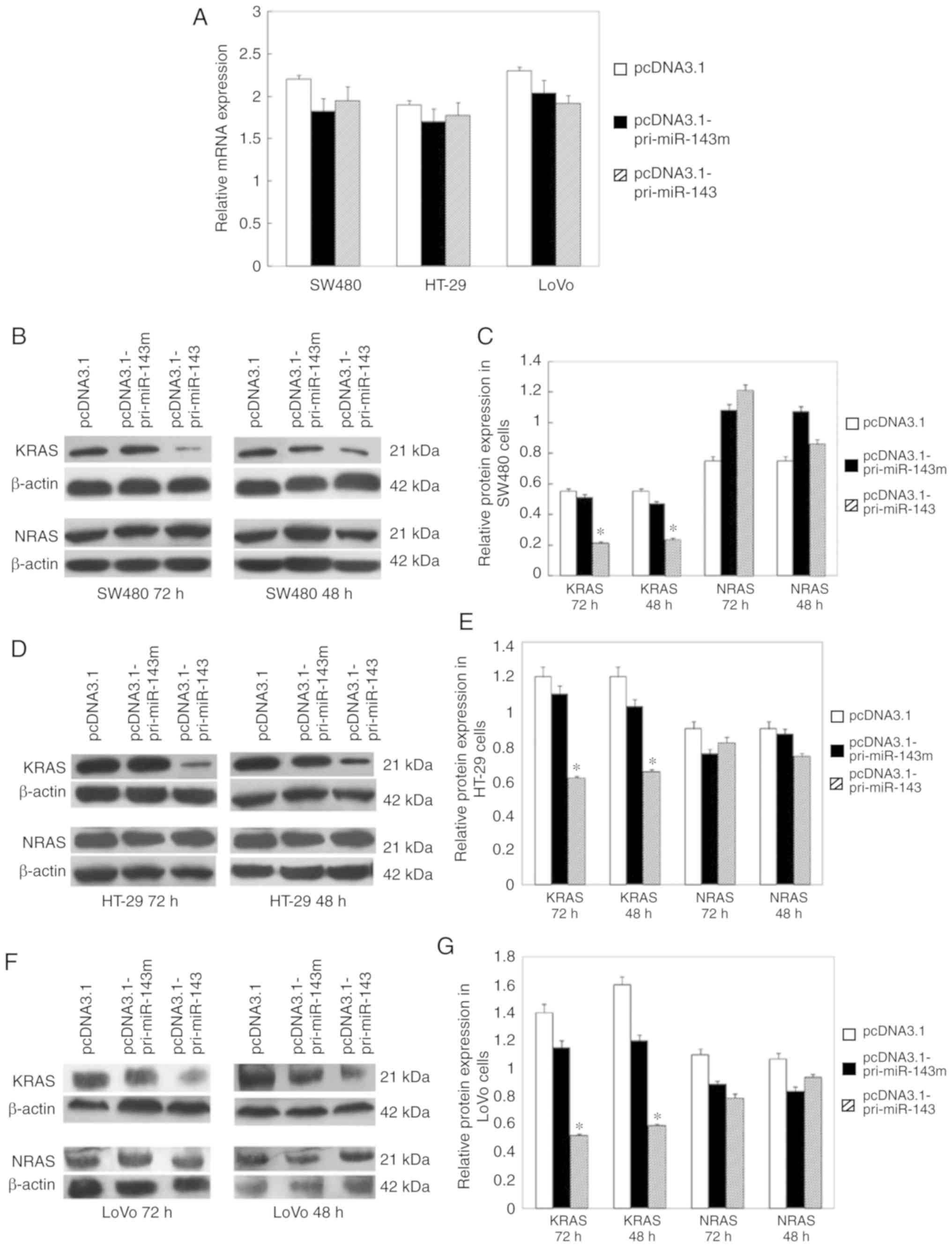 | Figure 4.miR-143 partially suppresses KRAS
protein expression without altering mRNA abundance. (A) RT-qPCR
analysis of KRAS mRNA levels in transfected CRC cells 72 h after
transfection. β-actin served as an endogenous control.
Normalization to the control, allowing comparison of mRNA levels.
(B) Western blotting of KRAS and NRAS proteins in SW480 cells
transfected with pcDNA3.1-pri-miR-143, pcDNA3.1-pri-miR-143m and
pcDNA3.1 48 and 72 h after transfection. β-actin was detected as a
loading control. (C) Normalization to the control, allowing
comparison of protein expression in transfected SW480 cells. (D)
Western blotting of KRAS and NRAS proteins in HT-29 cells
transfected with pcDNA3.1-pri-miR-143, pcDNA3.1-pri-miR-143m and
pcDNA3.1 48 and 72 h after transfection. (E) Normalization to the
control, allowing comparison of protein expression in transfected
HT-29 cells. (F) Western blotting of KRAS and NRAS proteins in LoVo
cells transfected with pcDNA3.1-pri-miR-143, pcDNA3.1-pri-miR-143m
and pcDNA3.1 48 and 72 h after transfectinon. (G) Normalization to
the control, allowing comparison of protein expression in
transfected LoVo cells. Error bars for all panels represent SDs
derived from at least three independent measurements. *P<0.05.
CRC, colorectal cancer; KRAS, KRAS proto-oncogene, GTPase; NRAS,
NRAS proto-oncogene, GTPase. |
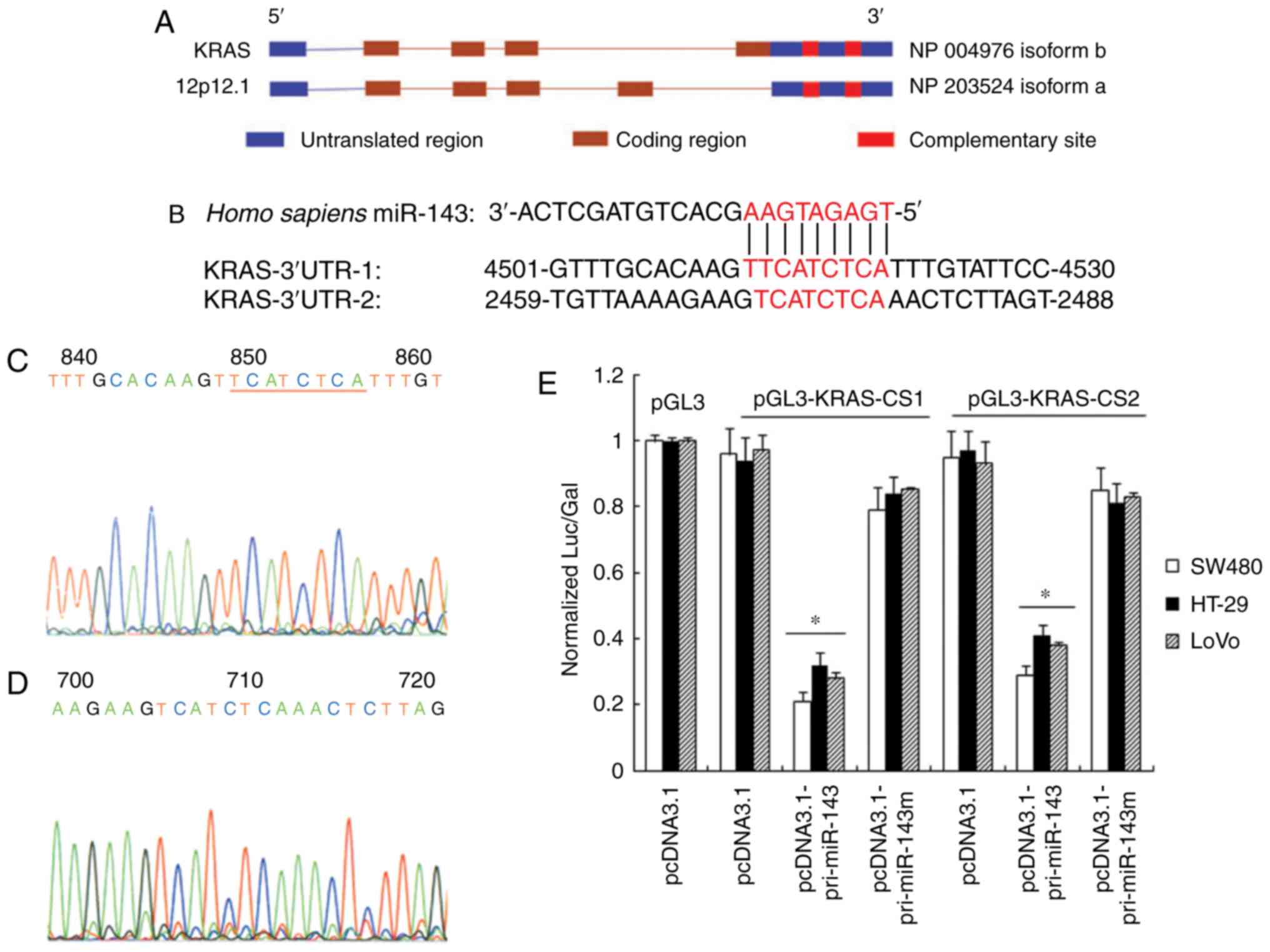
Putative miR-143 complementary sites
in the 3′UTRs of the KRAS gene
It is believed that miRNAs inhibit expression of
target mRNAa by interacting with imperfect target sites in the
3′UTR (Fig. 5B). A putative binding
site was located at the 5′end of miR-143 (TGAGATGA). We found that
the human KRAS 3′UTR contained two miR-143 complementary sites,
locating at nucleotides 2471–2478 and 4513–4521 (Fig. 5A). Two fragments containing the
complementary sites were fused to the XbaI site of the pGL3
control vector, constructing the luciferase reporter pGL3-KRAS-CS1
and pGL3-KRAS-CS2, respectively (Fig. 5C
and D). After transfection, pri-miRNA-143 and pri-miRNA-143m
were detected by RT-PCR and gene sequencing, which confirmed the
efficiency and reliability of gene transfection. The expression of
miR-143 can be measured by RT-qPCR, while the miR-143 mutant is
difficult to detect because of its poor specificity. Cotransfection
of pcDNA3.1-pri-miR-143 and pGL3-KRAS-CS1 into SW480 cells resulted
in 4.6-fold inhibition of luciferase activity compared with
pGL3-KRAS-CS1 transfection alone, while cotransfection of
pcDNA3.1-pri-miR-143 and pGL3-KRAS-CS2 resulted in 3.3-fold
inhibition compared with pGL3-KRAS-CS2 transfection alone. We also
found a 4.0- and 3.2-fold inhibition of luciferase activity in LoVo
cells, while a 3.7- and 3.1-fold inhibition in HT-29 cells, when
pcDNA3.1-pri-miR-143 was cotransfected with pGL3-KRAS-CS1 and
pGL3-KRAS-CS2, respectively. Differences in pGL3-KRAS-CS1 and
pGL3-KRAS-CS2 activity were not significant. As a control,
transfection of pcDNA3.1-pri-miR-143m had no effects on luciferase
activity (Fig. 5E). This supported
specific miR-143 interaction with complementary sites of KRAS
3′UTR.
Discussion
We investigated the expression levels of miR-143 in
colorectal carcinoma tissue specimens. The results showed a
reduction in mature miR-143 in tumor tissues compared with that
noted in normal adjacent tissues, which are consistent with
previous reports (28). miRNA
downregulation in tumor tissue may be a consequence of the
undifferentiated state. Several studies have revealed the
prognostic significance of miRNA profiles in colorectal cancer
(CRC) (35,36). Expression levels of miR-143 are
frequently altered in tumors (29,37–39). There
is also evidence that miR-143 is related to expression of KRAS in
CRC. A novel synthetic miR-143 can strongly silence KRAS, and
increase efficacy of epidermal growth factor receptor (EGFR)
inhibitors. Downregulation of KRAS-interacting miRNA-143 predicts
poor prognosis but not response to EGFR-targeted agents in CRC
(40).
The RAS protein family plays a key role in
regulating diverse cellular pathways important for cell growth,
differentiation and survival (41).
Constitutive RAS signaling as a result of RAS mutations or
overexpression can induce oncogenesis. Activated KRAS protein was
observed in 40–50% of human colorectal adenomas and carcinomas
(42). To explore the effects of
miR-143 on KRAS, we constructed the pri-miR-143 expression plasmid
and mutation was introduced into the predicted binding site.
Colorectal carcinoma cell lines SW480 and LoVo were used in
experiments in vitro. SW480 cells carry two mutated alleles
at codon 12 of the KRAS oncogene, and LoVo cells harbor a KRAS
mutation at codon 13 (43). As a
control cell line, HT-29 cells with KRAS-wild-type and BRAF mutant
were selected. We detected the expression levels of miR-143 in cell
lines and found a lack of miR-143 in human colorectal carcinoma
cells SW480, LoVo and HT-29, compared to high expression in normal
colon epithelial cell line NCM460. Then the plasmids were
transfected into colorectal carcinoma cells respectively, and
achieved accumulation of mature miR-143. The results showed that
KRAS protein expression was partially inhibited in
miR-143-transfected cells, compared with that in the
mutated-miR-143-transfected cells. However, the protein expression
levels of NRAS were unaffected by the increased miR-143, indicating
that miR-143 regulation is specific to KRAS. Similar effect of
miR-143 on KRAS expression in KRAS-wild-type and KRAS-mutant cell
lines indicates that miR-143 interacts with the KRAS untranslated
region, but not the translated region. By cell proliferation assay
we found that the cell growth of SW480 and LoVo cells was obviously
inhibited after miR-143 transfection, probably due to the
downregulation of mutant KRAS protein. In contrast, transfection of
miR-143 had no significant effects on HT-29 cell growth. There is
evidence that blocking KRAS activity in tumor cell lines can result
in cell death or reversion to a nonmalignant phenotype (44,45).
Another possible explanation is that increased accumulation of
miR143 suppresses cell proliferation by regulating ERK5 (32) or IGF1R (46).
miRNAs have been shown to regulate gene expression
by inducing mRNA cleavage or translational repression. In animals,
most miRNAs bind with imperfect complementarity to multiple sites
in the 3′UTR of their target mRNAs and cause translational
repression (12). The potential
targets of miRNA were predicted by bioinformatic approaches with an
emphasis on the critical pairing at the 5′end of the miRNA
(47). To demonstrate that miR-143
directly regulates KRAS expression, we identified two putative
binding sites at the 3′UTR of KRAS mRNA, which are complementary
with the base sequence at the 5′end of miR-143. The two
complementary sites were cloned into the pGL3 control vector,
constructing the luciferase reporter. Then, the reporter plasmids
were cotransfected into CRC cells, respectively. We found that
miR-143 negatively regulated luciferase activity compared with
mutated miR-143, which indicates that miR-143 can interact
specifically with complementary sites of KRAS 3′UTR. The results
suggested that miRNA-mediated gene regulation at the
post-transcriptional level is involved in tumorigenesis and tumor
progression in colorectal neoplasia, which provides a clue for
research of the function of miRNAs and their targets.
Acknowledgements
The authors would like to thank Dr Chenghong Peng
(Department of General Surgery, Ruijin Hospital Shanghai Jiao Tong
University School of Medicine, Shanghai, China) for his great
assistance in the writing of the manuscipt.
Funding
The present study was supported by the Natural
Science Foundation of Shanxi Province in China (grant no.
2010011047-4).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL, JH, KaiLi, HG and YY performed the experiments.
HL and KaihuaLi analyzed the data. JL guided the experiments and
designed the study. HL wrote the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Commission of
First Hospital of Shanxi Medical University [approval no.
2018(k012), Taiyuan, China]. Written informed consent was obtained
from all participants included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jass JR: Classification of colorectal
canfcer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leggett B and Whitehall V: Role of the
serrated pathway in colorectal cancer pathogenesis.
Gastroenterology. 138:2088–2100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rex DK, Ahnen DJ, Baron JA, Batts KP,
Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, et
al: Serrated lesions of the colorectum: Review and recommendations
from an expert panel. Am J Gastroenterol. 107:1315–1329. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
8
|
McCubrey JA, Steelman LS, Abrams SL, Lee
JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA,
D'Assoro AB, et al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT
pathways in malignant transformation and drug resistance. Adv
Enzyme Regul. 46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Modest DP, Ricard I, Heinemann V,
Hegewisch-Becker S, Schmiegel W, Porschen R, Stintzing S, Graeven
U, Arnold D, von Weikersthal LF, et al: Outcome according to KRAS-,
NRAS- and BRAF-mutation as well as KRAS mutation variants: Pooled
analysis of five randomized trials in metastatic colorectal cancer
by the AIO colorectal cancer study group. Ann Oncol. 27:1746–1753.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andreatos N, Ronnekleiv-Kelly S, Margonis
GA, Sasaki K, Gani F, Amini N, Wilson A and Pawlik TM: From bench
to bedside: Clinical implications of KRAS status in patients with
colorectal liver metastasis. Surg Oncol. 25:332–338. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rusek AM, Abba M, Eljaszewicz A, Moniuszko
M, Niklinski J and Allgayer H: MicroRNA modulators of epigenetic
regulation, the tumor microenvironment and the immune system in
lung cancer. Mol Cancer. 14:342015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee SC, Tan HT and Chung MC: Prognostic
biomarkers for prediction of recurrence of hepatocellular
carcinoma: Current status and future prospects. World J
Gastroenterol. 20:3112–3124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Chen X, Cheng R, Yang F, Yu M, Wang
C, Cui S, Hong Y, Liang H, Liu M, et al: The Jun/miR-22/HuR
regulatory axis contributes to tumourigenesis in colorectal cancer.
Mol Cancer. 17:112018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chai J, Guo D, Ma W, Han D, Dong W, Guo H
and Zhang Y: A feedback loop consisting of
RUNX2/LncRNA-PVT1/miR-455 is involved in the progression of
colorectal cancer. Am J Cancer Res. 8:538–550. 2018.PubMed/NCBI
|
|
18
|
Huang L, Cai JL, Huang PZ, Kang L, Huang
MJ, Wang L and Wang JP: miR19b-3p promotes the growth and
metastasis of colorectal cancer via directly targeting ITGB8. Am J
Cancer Res. 7:1996–2008. 2017.PubMed/NCBI
|
|
19
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schetter AJ, Okayama H and Harris CC: The
role of microRNAs in colorectal cancer. Cancer J. 18:244–252. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baraniskin A, Birkenkamp-Demtroder K,
Maghnouj A, Zöllner H, Munding J, Klein-Scory S, Reinacher-Schick
A, Schwarte-Waldhoff I, Schmiegel W and Hahn SA: miR-30a-5p
suppresses tumor growth in colon carcinoma by targeting DTL.
Carcinogenesis. 33:732–739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Braun CJ, Zhang X, Savelyeva I, Wolff S,
Moll UM, Schepeler T, Ørntoft TF, Andersen CL and Dobbelstein M:
p53-responsive micrornas 192 and 215 are capable of inducing cell
cycle arrest. Cancer Res. 68:10094–10104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang H, Cao F, Li X, Miao H, E J, Xing J
and Fu CG: miR-320b suppresses cell proliferation by targeting
c-Myc in human colorectal cancer cells. BMC Cancer. 15:7482015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Forzati F, De Martino M, Esposito F, Sepe
R, Pellecchia S, Malapelle U, Pellino G, Arra C and Fusco A:
miR-155 is positively regulated by CBX7 in mouse embryonic
fibroblasts and colon carcinomas, and targets the KRAS oncogene.
BMC Cancer. 17:1702017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lagos-Quintana M, Rauhut R, Yalcin A,
Meyer J, Lendeckel W and Tuschl T: Identification of
tissue-specific microRNAs from mouse. Curr Biol. 12:735–739. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Michael MZ, O'Connor SM, van Holst
Pellekaan NG, Young GP and James RJ: Reduced accumulation of
specific microRNAs in colorectal neoplasia. Mol Cancer Res.
1:882–891. 2003.PubMed/NCBI
|
|
29
|
Akao Y, Nakagawa Y and Naoe T:
MicroRNA-143 and −145 in colon cancer. DNA Cell Biol. 26:311–320.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li
JJ, Röcken C, Ebert MP, Kwok TT and Sung JJ: MicroRNA-143 targets
DNA methyltransferases 3A in colorectal cancer. Br J Cancer.
101:699–706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Esau C, Kang X, Peralta E, Hanson E,
Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, et
al: MicroRNA-143 regulates adipocyte differentiation. J Biol Chem.
279:52361–52365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lewis BP, Shih IH, Jones-Rhoades MW,
Bartel DP and Burge CB: Prediction of mammalian microRNA targets.
Cell. 115:787–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Griffiths-Jones S, Grocock RJ, van Dongen
S, Bateman A and Enright AJ: miRBase: microRNA sequences, targets
and gene nomenclature. Nucleic Acids Res 34 (Database Issue).
D140–D144. 2006. View Article : Google Scholar
|
|
35
|
Eslamizadeh S, Heidari M, Agah S,
Faghihloo E, Ghazi H, Mirzaei A and Akbari A: The role of microRNA
signature as diagnostic biomarkers in different clinical stages of
colorectal cancer. Cell J. 20:220–230. 2018.PubMed/NCBI
|
|
36
|
Wang CJ, Zhou ZG, Wang L, Yang L, Zhou B,
Gu J, Chen HY and Sun XF: Clinicopathological significance of
microRNA-31, −143 and −145 expression in colorectal cancer. Dis
Markers. 26:27–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahmad I, Singh LB, Yang ZH, Kalna G,
Fleming J, Fisher G, Cooper C, Cuzick J, Berney DM, Møller H, et
al: Mir143 expression inversely correlates with nuclear ERK5
immunoreactivity in clinical prostate cancer. Br J Cancer.
108:149–154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shen JZ, Zhang YY, Fu HY, Wu DS and Zhou
HR: Overexpression of microRNA-143 inhibits growth and induces
apoptosis in human leukemia cells. Oncol Rep. 31:2035–2042. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Johannessen C, Moi L, Kiselev Y, Pedersen
MI, Dalen SM, Braaten T and Busund LT: Expression and function of
the miR-143/145 cluster in vitro and in vivo in human breast
cancer. PLoS One. 12:e01866582017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pichler M, Winter E, Stotz M, Eberhard K,
Samonigg H, Lax S and Hoefler G: Down-regulation of
KRAS-interacting miRNA-143 predicts poor prognosis but not response
to EGFR-targeted agents in colorectal cancer. Br J Cancer.
106:1826–1832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Friday BB and Adjei AA: K-ras as a target
for cancer therapy. Biochim Biophys Acta. 1756:127–144.
2005.PubMed/NCBI
|
|
42
|
Forrester K, Almoguera C, Han K, Grizzle
WE and Perucho M: Detection of high incidence of K-ras oncogenes
during human colon tumorigenesis. Nature. 327:298–303. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haliassos A, Chomel JC, Grandjouan S, Kruh
J, Kaplan JC and Kitzis A: Detection of minority point mutations by
modified PCR technique: A new approach for a sensitive diagnosis of
tumor-progression markers. Nucleic Acids Res. 17:8093–8099. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brummelkamp TR, Bernards R and Agami R:
Stable suppression of tumorigenicity by virus-mediated RNA
interference. Cancer Cell. 2:243–247. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Smakman N, Veenendaal LM, van Diest P, Bos
R, Offringa R, Borel Rinkes IH and Kranenburg O: Dual effect of
Kras(D12) knockdown on tumorigenesis: Increased immune-mediated
tumor clearance and abrogation of tumor malignancy. Oncogene.
24:8338–8342. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Su J, Liang H, Yao W, Wang N, Zhang S, Yan
X, Feng H, Pang W, Wang Y, Wang X, et al: miR-143 and miR-145
regulate IGF1R to suppress cell proliferation in colorectal cancer.
PLoS One. 9:e1144202014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res 39 (Database issue). D163–D169.
2011. View Article : Google Scholar
|