Introduction
Colon cancer is the third most commonly diagnosed
cancer in the world (1) and the
second most commonly diagnosed cancer in Japan (2). The prognosis of advanced colon cancer
with metastasis remains poor, with the overall 5-year survival rate
being only 18.8% (3). Standard
treatment of unresectable advanced or recurrent colon cancer is
systemic chemotherapy. The recent development of several
chemotherapy regimens such as FOLFIRI and FOLFOX, which are
combined with bevacizumab, cetuximab, or panitumumab, has clearly
prolonged patient survival (3). In
Japan, fourth or fifth line continuous chemotherapy regimens are
administered for unresectable advanced colon cancer such that all
usable anticancer drugs are administered. Among them,
5-fluorouracil (5-FU) is a core cytotoxic drug that is included in
all first line regimens. 5-FU is an analogue of uracil, which is
one of the four bases found in RNA; it is also utilized more in
tumors than in normal tissue (4).
After entering the cell, 5-FU is converted to several metabolites
(5), which leads to misincorporation
into RNA and DNA, thereby inhibiting thymidine synthase and
initiating apoptosis. In general, cytotoxic anticancer drugs are
effective with respect to killing cancer cells at the beginning of
treatment. However, tumors gradually fail to respond to these drugs
after drug resistance sets in. Therefore, understanding the
mechanism of resistance to anticancer drugs is indispensable for
uncovering more efficacious treatments for refractory cancer.
In the present study, we established three colon
cancer cell lines with acquired resistance to continuous 5-FU
treatment and analyzed the mechanism of 5-FU resistance in terms of
anti-apoptosis using a well-designed functional apoptotic assay-BH3
profiling.
Materials and methods
Cell lines
Colorectal adenocarcinoma cell lines DLD-1 (ATCC
CCL-221TM), HCT-15 (ATCC CCL-225TM) and HT-29 (ATCC HTB-38TM) were
obtained from the American Type Culture Collection and cultured at
37°C in a humidified 5% CO2 incubator in either RPMI
(DLD-1, HCT-15) or DMEM (HT-29) medium supplemented with 10% FBS,
10 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin
(all media and supplements from Sigma-Aldrich; Merck KGaA).
Generation of the 5-FU-resistant cell
line
The 5-FU-resistant colon cancer cell line was
developed as previously reported (6).
Briefly, parental colon cancer cell lines were treated with
gradually increasing concentrations of 5-FU (Sigma-Aldrich; Merck
KGaA). The initial 5-FU concentration added to cells was 10% of the
IC50; this concentration was gradually increased. Colon
cancer cell lines resistant to 16 µM of continuous 5-FU
administered continuously were established according to a previous
study (7).
Cell viability assay
Each colon cancer cell line was treated for 72 h
with 5-FU at the indicated concentrations in 96-well plates (Thermo
Fisher Scientific, Inc.). Subsequently, cell proliferation was
determined using the Premix WST-1 Cell Proliferation Assay (Takara
Bio Inc., Kusatsu, Shiga, Japan) and an Infinite M1000 PRO
microplate reader (Tecan Japan, Kawasaki, Kanagawa, Japan). The
half-maximal inhibitory concentration (IC50) of 5-FU was
defined as the drug concentration resulting in 50% cell survival
relative to that of untreated cells. Triplicate wells were treated
with various drug concentrations and average IC50 values
were determined. As known antagonists of BCL2 and BCLXL in in
vitro studies, ABT-199 (Selleck Chemicals) and WEHI-539
hydrochloride (MedChem Express) were used and the IC50
values of each drug were obtained, respectively.
Apoptosis assays
Parental and 5-FU-resistant colon cancer cells were
allowed to adhere to 6-well plates for 24 h and cells were treated
with either 5-FU or WEHI-539 hydrochloride as indicated. Cells were
then stained with a phycoerythrin-conjugated Annexin V antibody and
7-AAD (BD Pharmingen; BD Biosciences). Apoptotic cells were
analyzed using a BD FACSCanto II flow cytometer (BD Biosciences)
with FACSDiva software (BD Biosciences). The percentage of
apoptotic cells was calculated by dividing the percentage of either
Annexin V-positive or 7-AAD positive cells by the total cells.
Apoptosis was also assessed using the Caspase-Glo® 3/7
Assay (Promega). Five thousand cells were plated in white-walled
96-well round plates (Thermo Fisher Scientific, Inc.) and treated
with the drugs as indicated. After incubation, 100 µl of
Caspase-Glo® reagent was added to each well and the
contents of the well were gently mixed with a plate shaker at 50 ×
g for 30 sec; this was followed by incubation at 20°C room
temperature for 1 h. The luminescence of each sample was measured
using an Infinite M1000 PRO microplate reader. The caspase
inhibitor Q-VD-OPH (Bay Bioscience, Kobe, Hyogo, Japan) was also
used.
Western blot analysis
Western blotting was performed as previously
described (8). Briefly, separated
proteins were transferred to polyvinylidene difluoride membranes
and blotted with specific antibodies to detect BCL2 (at dilution of
1:500; Thermo Fisher Scientific, Inc.; #13-8800), BCLW (1:1,000;
Cell Signaling Technology; cat. no. 2724), BCLXL (1:1,000; Cell
Signaling Technology; cat. no. 2764), MCL1 (1:1,000; Cell Signaling
Technology; cat. no. 5453), BAK (1:1,000; Cell Signaling
Technology; cat. no. 12105), BAX (1:1,000; Cell Signaling
Technology; cat. no. 5023), BIM (1:1,000; Cell Signaling
Technology; cat. no. 2933), BID (1:1,000; Cell Signaling
Technology; #2002), BAD (1:1,000; Cell Signaling Technology; cat.
no. 9292), NOXA (1:1,000; Cell Signaling Technology; cat. no.
14766), PUMA (1:1,000; Cell Signaling Technology; cat. no. 12450),
BMF [1:1,000; Abcam; cat. no. EPR10930 (2)], HRK (1:200; R&D Systems; cat. no.
AF851), and actin (1:3,000; Santa Cruz Biotechnology; cat. no.
sc1615). After incubation with either horseradish peroxidase-linked
anti-rabbit IgG (1:2,000; Cell Signaling Technology; cat. no.
7074S) or anti-mouse IgG (1:2,000; Cell Signaling Technology; cat.
no. 7076S), the membranes were stained with ECL Select Western
Blotting Detection Reagent (GE Healthcare UK Ltd.). Finally, the
bands were imaged either by exposing membranes to BIOMAX XAR film
(Sigma-Aldrich; Merck KGaA) and developing the images using a Kodak
X-OMAT 1000 Processor (Kodak via Thermo Fisher Scientific, Inc.) or
using an LAS-4000UV mini (GE Healthcare UK Ltd.) and MultiGauge
software (Fujifilm, Tokyo, Japan).
BCL2-homology domain 3 (BH3)
profiling
We conducted fluorescence activated cell sorting
(FACS)-based BH3 profiling as previously described (9,10). Nine
BH3 peptides were obtained as HPLC-purified products from
Sigma-Aldrich; Merck KGaA (Table I).
All peptides were dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich; Merck KGaA) as 1 mM stock solutions and stored at
−80°C. As a control for mitochondrial depolarization,
p-trifluoromethoxy carbonyl cyanide phenyl hydrazine (FCCP) was
used. Two hundred thousand parental and 5-FU-resistant colon cancer
cells were suspended in TE-B buffer [300 mM trehalose, 10 mM
HEPES-KOH (pH 7.7), 80 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1% bovine
serum albumin (BSA), and 5 mM succinate; all from Sigma-Aldrich;
Merck KGaA] containing 0.001% digitonin (Sigma-Aldrich; Merck KGaA)
and 20 µg/ml oligomycin (Sigma-Aldrich; Merck KGaA), followed by
incubation with each BH3 peptide at a final concentration of 10 µM
for 30 min. After staining the cells with 25 nM
tetramethylrhodamine ethyl ester (Invitrogen/Thermo Fisher
Scientific, Inc.), fluorescence intensities were analyzed using a
BD FACSCanto II flow cytometer (BD Biosciences) with FACSDiva
software (BD Biosciences). The percentage of relative mitochondrial
depolarization was calculated using the following equation:
 | Table I.Sequences of the BH3 peptide. |
Table I.
Sequences of the BH3 peptide.
| Name | Sequence |
|---|
| BIM |
MRPEIWIAQELRRIGDEFNA |
| BID |
EDIIRNIARHLAQVGDSMDRY |
| BAD |
LWAAQRYGRELRRMSDEFEGSFKGL |
| NOXA |
AELPPEFAAQLRKIGDKVYC |
| PUMA |
EQWAREIGAQLRRMADDLNA |
| BMF |
HQAEVQIARKLQLIADQFHRY |
| HRK |
WSSAAQLTAARLKALGDELHQ |
| MS1 |
RPEIWMTQGLRRLGDEINAYYAR |
| XXA1 |
RPEIWYAQGLKRFGDEFNAYYAR |
%mitochondrialdepolarization=100×(DMSO(fv)-X(fv))(DMSO(fv)-FCCP(fv))
where DMSO(fv) is the mean fluorescence value as the
negative control, X(fv) indicates the fv as the tested BH3 peptide,
and FCCP(fv) indicates the fv as the positive control.
Inhibition of BCLXL expression via
small-interfering RNA (siRNA) transfection
One million parental and 5-FU-resistant HT-29 cells
in 6-well plates were transfected with either siRNAs targeting
human BCLXL (Dharmacon; siBCLXL#1: D-003458-03, siBCLXL#2:
D-003458-30) or a non-targeting siRNA (Dharmacon; siNT:
D-001210-03-05) using RNAi Max reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Immunoprecipitation
BCLXL immunoprecipitation experiments were conducted
using the ImmunoCruz™ IP/WB Optima F System (Santa Cruz
Biotechnology; #SC-45043). Separated total proteins (1,000 µg) were
incubated with a complex of BCLXL antibody and IP matrix overnight
at 4°C. After incubation and centrifugation, the eluted supernatant
was subjected to western blotting for BID, BIM, and BCLXL.
Tumor xenograft study
Fifteen female BALB/c nu/nu mice (six weeks of age,
18–20 g/each) were purchased from Sankyo Labo Service Corporation,
Inc. (Tokyo, Japan). Five million 5-FU-resistant HT-29 cells
suspended in 100 µl DMEM and 100 µl Matrigel (BD Biosciences) were
injected subcutaneously into the left flank. In place of WEHI-539,
which has a labile and potentially toxic hydrazone moiety, we used
another BCLXL inhibitor, A-1155463, which is known to be
efficacious on tumor growth suppression in vivo (11). Mice were divided into three
experimental groups: Control (vehicle-treated), 5-FU, and
A-1155463. When tumor volumes reached 100 mm3,
administration of either intravenous 5-FU (50 mg/kg) injections
once a week or intraperitoneal A-1155463 (5 mg/kg) injections every
day was conducted for four weeks as previously reported (11,12). Tumor
size and body weight were measured throughout the experimental
period. Tumor volumes were calculated with the following
formula:
Tumorvolume(mm3)=(Length(mm)xWidth(mm)xHeight(mm)×π)/6
Percent volume was calculated with the following
formula:
%volume=100x(Tumorvolume(mm3)ofday)/(Tumorvolume(mm3)ofday0)
Mice were sacrificed when the tumor diameter reached
20 mm or when the study period finished. Tumors and other organs
including the lung and liver were isolated, followed by hematoxylin
and eosin staining. Apoptotic cells were analyzed by TUNEL staining
using an in situ apoptosis detection kit (Takara Bio Inc.). The
percentage of TUNEL-positive cells was calculated by counting cells
in 5 different fields. The animal study was conducted according to
protocols approved by the Animal Ethics Committee of the Sapporo
Medical University School of Medicine (Protocol no. 17-142).
Euthanasia was conducted by high concentration carbon dioxide and
all efforts were made to minimize suffering.
Statistical analysis
All data represent the mean ± SD. For comparisons
between two groups, two-tailed unpaired Student's t-tests were
performed. For multiple comparisons of in vitro experiments,
one way analysis of variance followed by Tukey test was conducted.
IC50 values for each drug tested and dose-response
curves were analyzed using Graph-Pad PRISM 5 (GraphPad Software,
Inc., La Jolla, CA, USA). For in vivo experiments, one way
analysis of variance followed by Dunnett's post hoc test was
adopted for comparison with the control group. All statistical
analyses were performed using SPSS software version 21 (IBM,
Armonk, NY, USA).
Results
Enhanced survival in three
5-FU-resistant colon cancer cell lines
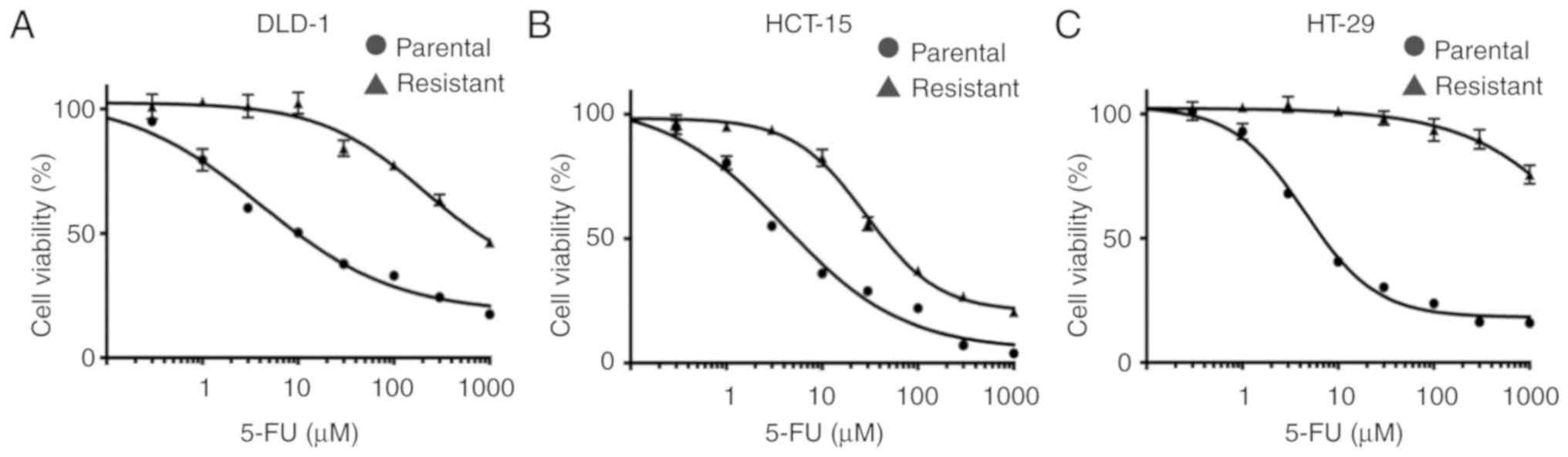
We established three colon cancer cell lines which
were resistant to 16 µM continuous 5-FU treatment. The 5-FU
IC50 values of resistant colon cancer cells were
markedly higher than those of the parental cells (DLD-1:
5-FU-resistant 776.0 µM, parental 10.3 µM; HCT-15: 5-FU-resistant
44.2 µM, parental 4.1 µM; HT-29: 5-FU-resistant >1,000 µM,
parental 7.2 µM, respectively) (Fig.
1A-C).
Resistance to apoptosis in
5-FU-resistant colon cancer cells and BCL2 protein induction in
5-FU-resistant HT-29 cells
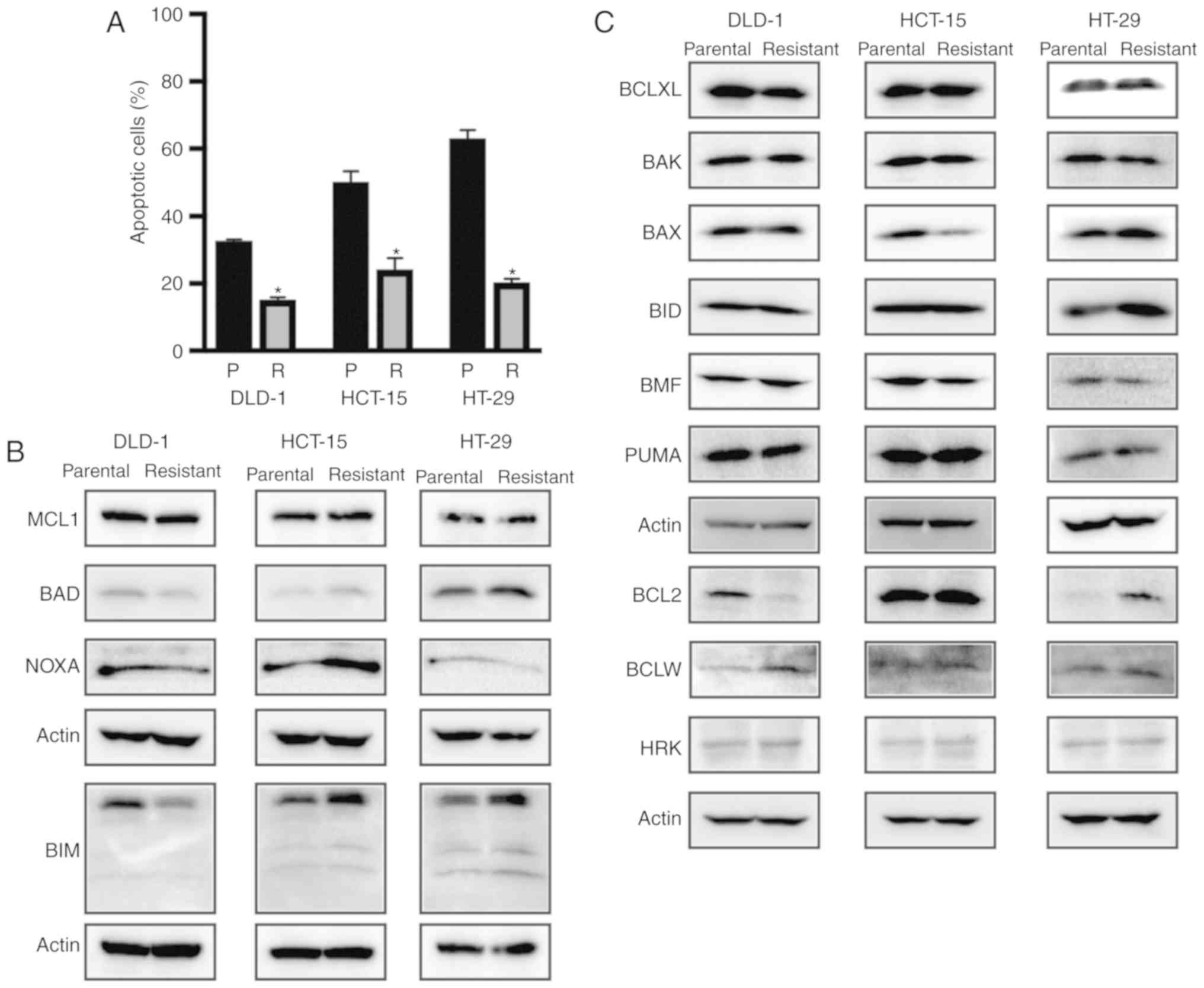
To evaluate the mechanisms of 5-FU resistance, we
first verified the occurrence of apoptosis in parental cells and
three 5-FU-resistant colon cancer cell lines. Each 5-FU-resistant
colon cancer cell line contained significantly fewer
Annexin-positive apoptotic cells compared with the parental cells,
suggesting that apoptotic resistance had been acquired by
5-FU-resistant colon cancer cell lines (Figs. 2A and S1). We then examined expression alterations
in both anti-apoptotic and apoptotic proteins between parental and
5-FU-resistant colon cancer cells (Fig.
2B and C). Western blot analysis showed that BCLXL and MCL1
protein expression levels were similar between the parental and
5-FU-resistant cells in the three colon cancer cell lines. Whereas
BCL2 protein expression in 5-FU-resistant DLD-1 cells was
decreased, both BCLW expression in DLD-1 and BCL2 protein
expression in HT-29 was increased in the 5-FU-resistant cells. The
expression of apoptosis-related proteins, e.g., BAX was decreased
whereas BIM, BAD, and NOXA expression was increased in the
5-FU-resistant HCT-15 cells. Whereas NOXA expression was decreased
in the 5-FU-resistant HT-29 cells, BIM expression was increased,
suggesting that the expression patterns of both anti-apoptotic and
apoptotic proteins were altered during the acquisition of 5-FU
resistance in the three colon cancer cell lines. From this analysis
of apoptosis-related protein expression, it may be difficult to
identify specific apoptotic responses to 5-FU resistance solely by
quantifying protein expression.
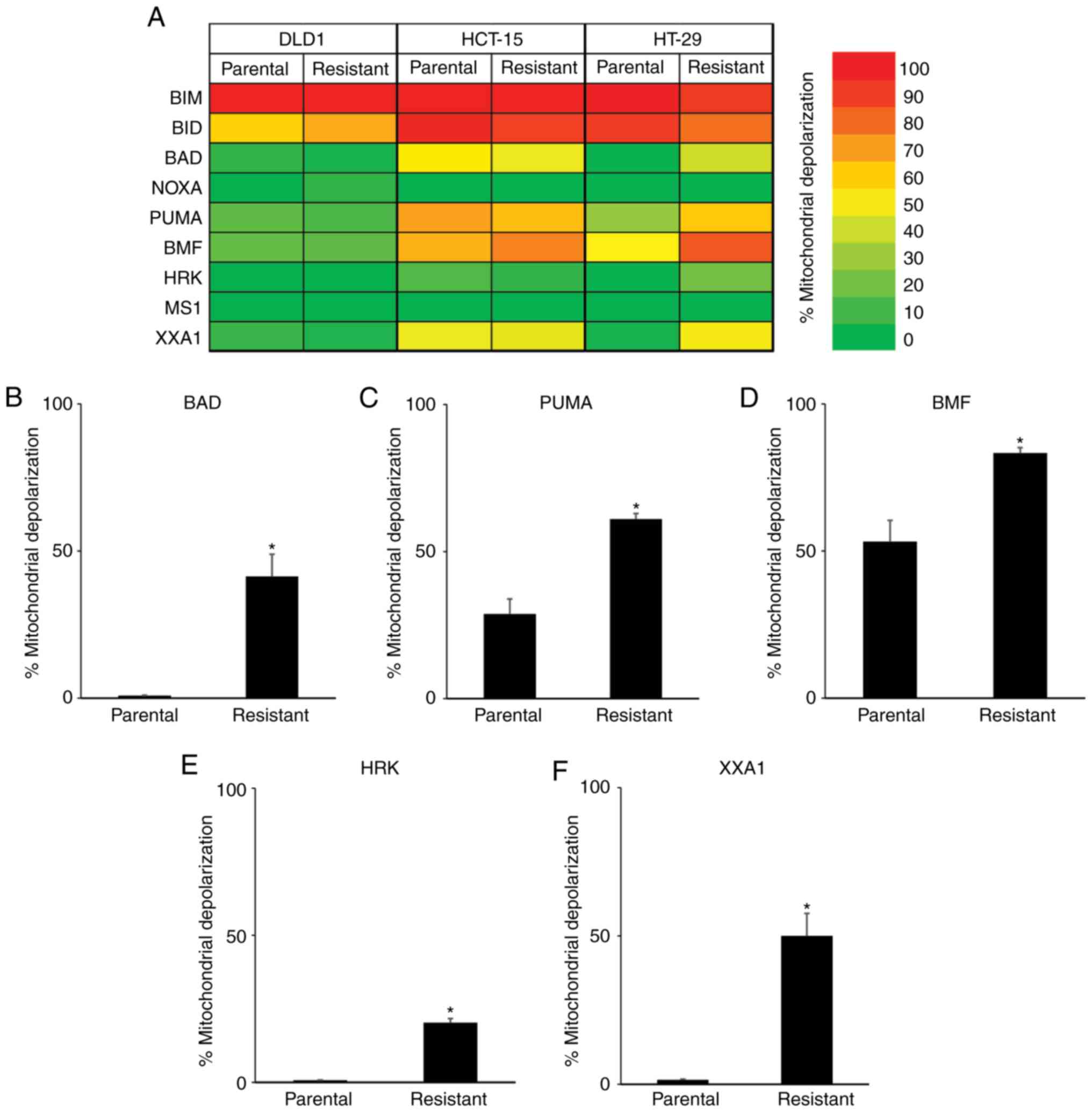
BH3 profiling reveals that
5-FU-resistant HT-29 cells are dependent on BCLXL for survival
To further clarify the relationship between 5-FU
resistance and apoptosis-related proteins, we conducted a
functional analysis via BH3 profiling (Fig. 3A). When treated with BIM and BID BH3
peptides, which interact directly with BAK and BAX proteins
resulting in the release of cytochrome c into the cytoplasm,
strong mitochondrial depolarization was observed in the parental
and all 5-FU-resistant cells. We then treated cells with BAD, NOXA,
PUMA, BMF, and HRK BH3 peptides, which have variable affinities for
anti-apoptotic proteins, and compared the mitochondrial
depolarization between parental and 5-FU-resistant colon cancer
cells. In DLD-1 and HCT-15 cells, no significant differences were
observed between parental and 5-FU-resistant cells when treated
with these BH3 peptides. However, greater mitochondrial
depolarization was observed in 5-FU-resistant HT-29 cells when
treated with BAD (Fig. 3B), PUMA
(Fig. 3C), and BMF (Fig. 3D) BH3 peptides, which was correlated
with dependency on BCL2, BCLXL or BCLW. Furthermore, treatment with
HRK peptide, which binds specifically to BCLXL, caused a small
increase in mitochondrial depolarization in the 5-FU-resistant
HT-29 cells (Fig. 3E mitochondrial
depolarization: Parental, 0.7%; 5-FU-resistant, 20.3%). To further
clarify the dependency on BCLXL protein, HT-29 cells were treated
with the XXA1 peptide, which has been structurally identified
(13) as a specific inhibitor of
BCLXL. Significant differences in mitochondrial depolarization were
observed between the parental and 5-FU-resistant HT-29 cells
(Fig. 3F mitochondrial
depolarization: Parental, 1.5%; 5-FU-resistant, 50.0%). As for
DLD-1 and HCT-15, there were no differences in mitochondrial
depolarization between parental and 5-FU-resistant cells when
treated with XXA1 peptide. We also treated cells with MS1 peptide
as a specific MCL inhibitor (14); no
changes in mitochondrial depolarization were observed in both
parental and 5-FU-resistant colon cancer cells. Taken together,
results of the BH3 profiling of 5-FU-resistant HT-29 cells revealed
strong dependency on BCLXL for cell survival compared with parental
cells, while no relationship to apoptosis-related protein
expression profiles was determined, especially BCLXL protein
expression. We then focused on the BCLXL dependence of
5-FU-resistant HT-29 colon cancer cells.
Inhibition of BCLXL selectively
induces apoptosis in 5-FU-resistant HT-29 cells
To confirm the BCLXL dependency in 5-FU-resistant
HT-29 cells, we first treated parental and 5-FU-resistant colon
cancer cells with the BCLXL-specific inhibitor WEHI-539 and
assessed the effect of this inhibition on cell survival. As
expected, the IC50 of WEHI-539 in 5-FU-resistant HT-29
cells was markedly lower compared to that in parental cells
(Fig. 4A: 5-FU-resistant, 0.66 µM;
parental, >100 µM), whereas no difference in IC50
values between parental and 5-FU-resistant cells was observed in
DLD-1 (Fig. 4B: 5-FU-resistant, 9.94
µM; parental, 7.12 µM) and HCT-15 (Fig.
4C: 5-FU-resistant, 9.83 µM; parental, 6.31 µM) cells. To rule
out any possibility of the contribution of induced BCL2 expression
to 5-FU resistance in HT-29 cells as shown in Fig. 2B, we also treated HT-29 cells with the
BCL2-specific inhibitor ABT-199 (Fig.
4D). No marked differences in IC50 values were
observed between the parental and 5-FU-resistant HT-29 cells
(IC50: Parental, 16.5 µM; 5-FU-resistant, 13.5 µM),
suggesting that these inhibitory data support the result of the BH3
profiling rather than that of the anti-apoptotic protein expression
profiles. We then examined the effect of WEHI-539 on apoptosis in
5-FU-resistant HT-29 cells to examine how BCLXL inhibition could
kill these cells. An significantly increased percentage of
apoptotic cells was observed in the 5-FU-resistant HT-29 cells,
whereas no additional Annexin V-positive cells were noted in the
parental cells (Fig. 4E).
Furthermore, the activities of caspase-3 and caspase-7, both of
which are key effectors of apoptosis, were upregulated in the
5-FU-resistant HT-29 cells when treated with WEHI-539 and were
completely inhibited through co-treatment with the caspase
inhibitor Q-VD-OPH (Fig. 4F). These
results indicate that BCLXL, but not BCL2, exerts substantial
anti-apoptotic functions in 5-FU-resistant HT-29 cells.
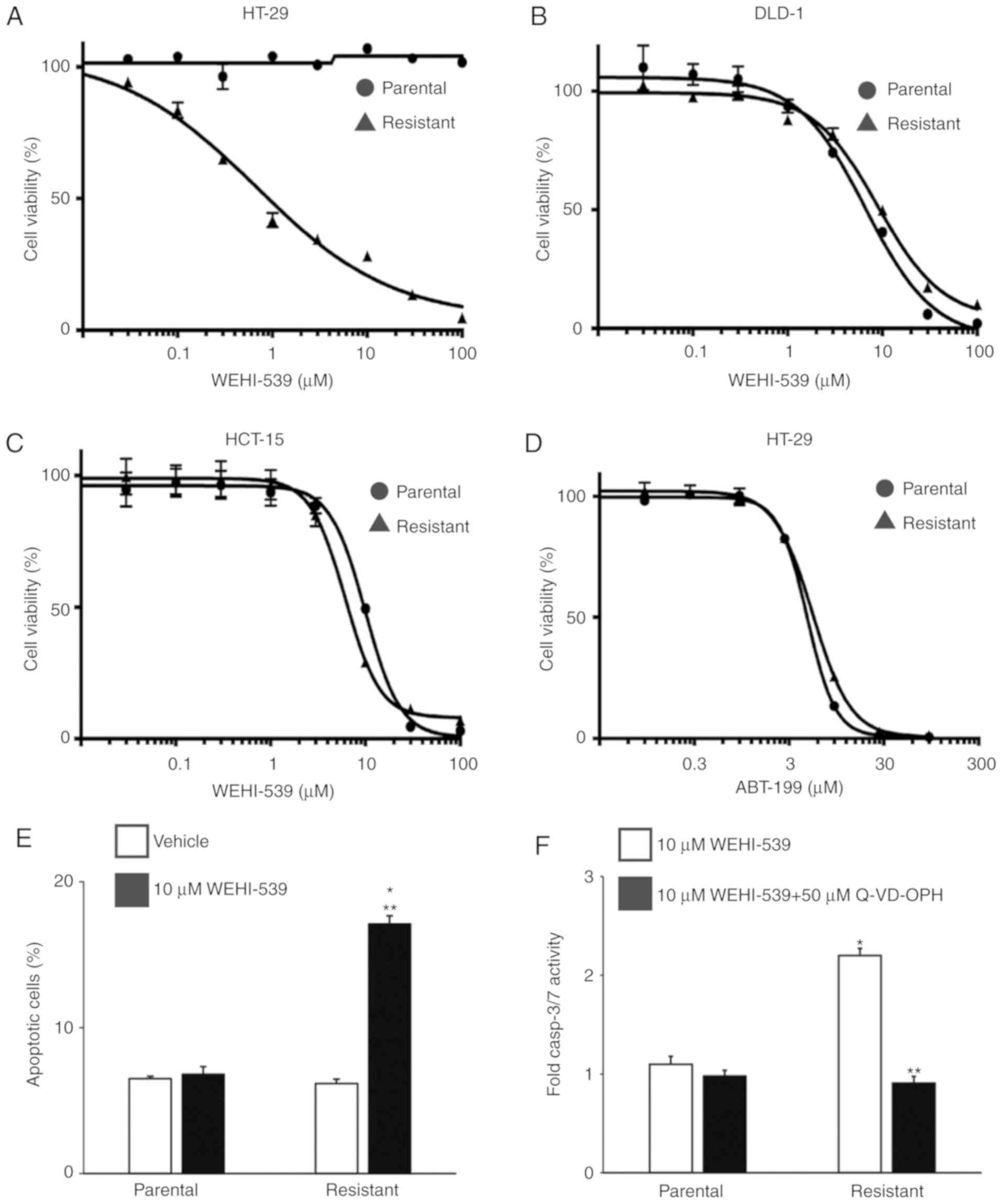 | Figure 4.A BCLXL inhibitor not only inhibits
proliferation but also promotes apoptosis in 5-FU-resistant HT-29
cells. Both parental and 5-FU-resistant HT-29 (A), DLD-1 (B) and
HCT-15 (C) cells were treated with different concentrations of the
BCLXL inhibitor, WEHI-539, for 72 h, followed by measurement of
cell viability. (D) Parental and 5-FU-resistant HT-29 cells were
treated with different concentrations of the BCL2 inhibitor ABT-199
for 72 h, followed by measurement of cell viability. Each point
indicates the mean ± SD (n=3). (E) Parental and 5-FU-resistant
HT-29 cells were treated with or without 10 µM WEHI-539 for 48 h,
followed by the assessment of apoptosis by flow cytometry.
*P<0.01, statistical significance when compared to parental
HT-29 cells; **P<0.01, statistical significance when compared to
vehicle-treated cells. (F) Caspase3/7 activity in parental and
5-FU-resistant HT-29 cells when treated with 10 µM WEHI-539, with
or without the caspase inhibitor (50 µM Q-VD-OPH) for 24 h. Values
are fold-changes in caspase3/7 activity compared to those with
medium treatment only. *P<0.01, statistical significance when
compared to parental HT-29 cells; **P<0.01, statistical
significance when compared to 5-FU-resistant HT-29 cells without
the caspase inhibitor (vehicle). All data represent the mean ± SD
(n=3). 5-FU, 5-fluorouracil. |
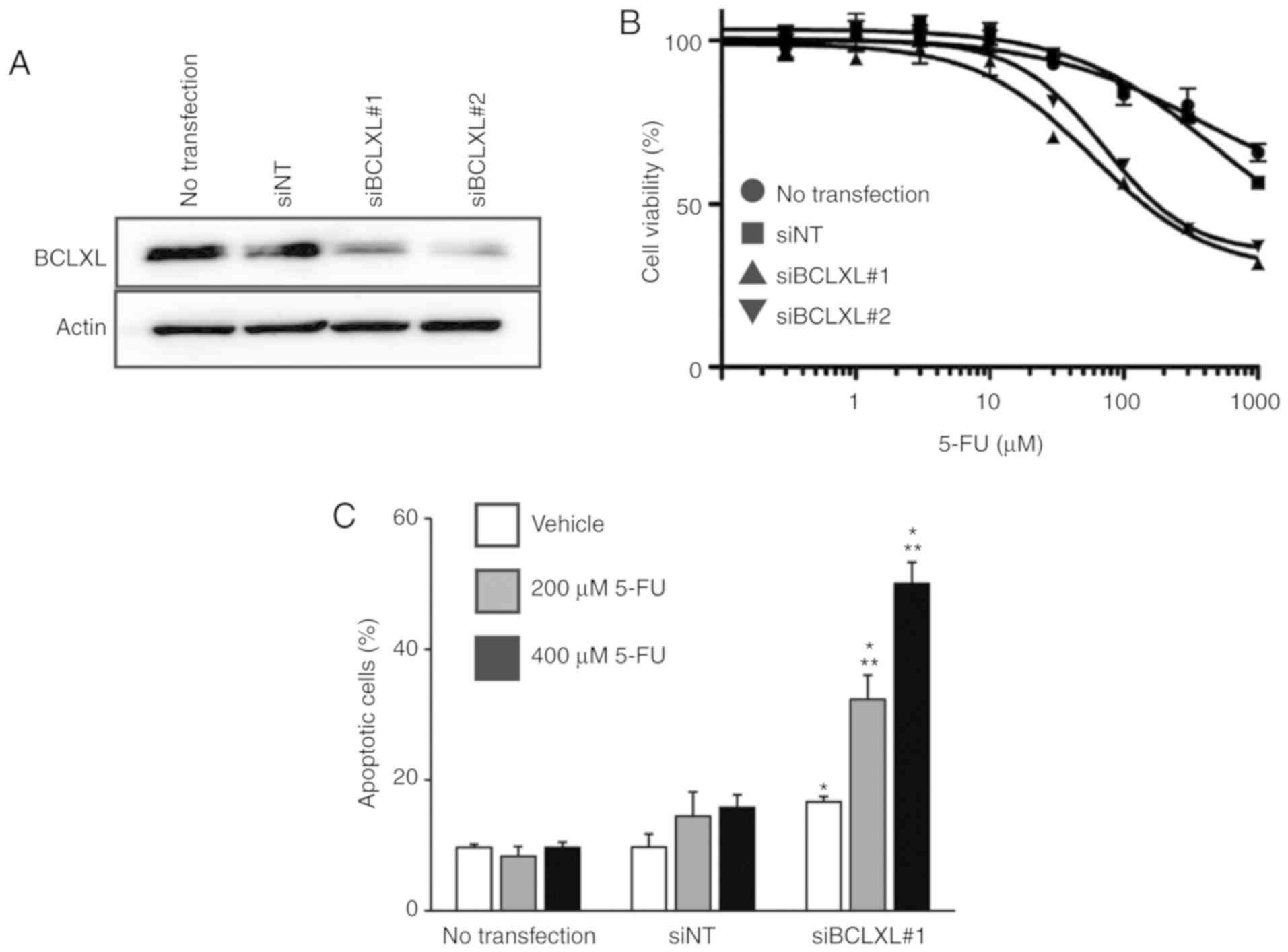
Restoration of 5-FU sensitivity by
inhibiting BCLXL in 5-FU-resistant HT-29 cells
We then inhibited BCLXL protein expression in
5-FU-resistant HT-29 cells through siRNA transfection (Fig. 5A), followed by treatment with 5-FU.
The IC50 of 5-FU in the resistant HT-29 cells was
markedly decreased following BCLXL inhibition compared to that in
both cells transfected with non-targeting siRNA (siNT) and
untransfected HT-29 cells (Fig. 5B:
siBCLXL#1 transfected, 174.9 µM; siBCLXL#2 transfected, 236.5 µM;
siNT transfected, >1,000 µM; untransfected, >1,000 µM).
Furthermore, a significantly increased percentage of apoptotic
cells was observed following BCLXL inhibition during 5-FU treatment
(Fig. 5C), suggesting that BCLXL
downregulation restored 5-FU sensitivity in the resistant HT-29
cells by enhancing their vulnerability to apoptosis.
Sequestration of BIM by BCLXL mediates
sensitization to apoptosis in 5-FU-resistant HT-29 cells
To examine whether an interaction between BCLXL and
an apoptotic protein would account for functional BCLXL dependency,
we performed immunoprecipitation on cell extracts of parental and
5-FU-resistant HT-29 cells using a BCLXL antibody and examined the
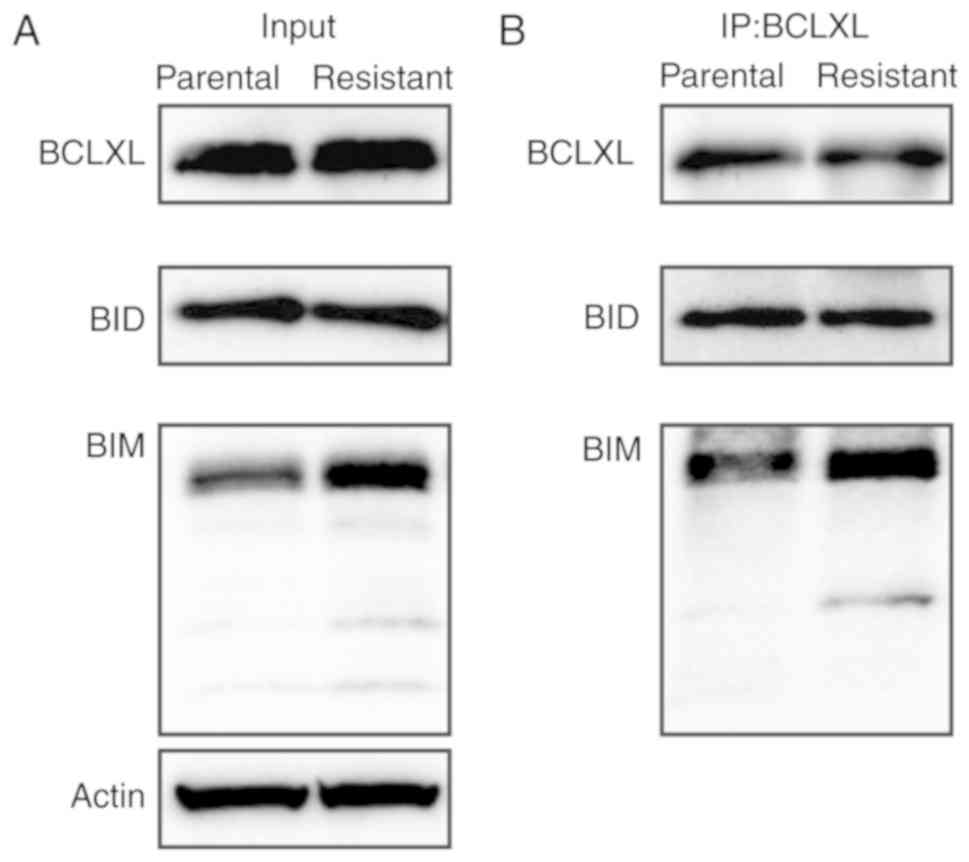
binding of apoptotic BIM and BID proteins. As shown in Fig. 6A and B, BIM protein preferentially
bound to BCLXL in the 5-FU-resistant HT-29 cells compared to the
parental cells. In summary, BCLXL dependence in 5-FU-resistant
HT-29 cells was mediated through the sequestration of BIM by
BCLXL.
Inhibition of BCLXL controls tumor
growth in the 5-FU-resistant HT-29 cells
We further tested the effect of BCLXL inhibition on
the growth of 5-FU-resistant HT-29 cells using a tumor xenograft
model. Prior to our in vivo study, we found that the
A-1155643 IC50 values of 5-FU-resistant HT-29 cells were
markedly lower compared to those in parental cells (5-FU-resistant,
1.2 µM; parental, >100 µM). After transplantation of these cells
into the left flank, mice were treated for four weeks with either
5-FU or BCLXL inhibitor A-1155643, respectively. Whereas tumor
volume gradually increased in the mice treated with 5-FU, similar
to vehicle-treated mice, BCLXL inhibitor-treated mice showed stable
tumor size as well as a significantly different size compared with
vehicle-treated mice (Figs. 7A and B
and S2). A significant increase in
TUNEL-positive apoptotic tumor cells was also observed in mice
treated with A-1155643 (Fig. 7C and
D). A-1155643 treatment was well tolerated, without
significantly decreasing body weight (day 28 body weight: Vehicle,
17.9±1.8 g; 5-FU treatment, 18.3±1.7 g; A-1155643 treatment,
17.7±1.9 g, respectively; Fig. S3).
Also, no severe bleeding in the lung and liver (Fig. S4), which usually occurs due to
thrombocytopenia, was observed in the BCLXL inhibitor-treated
mice.
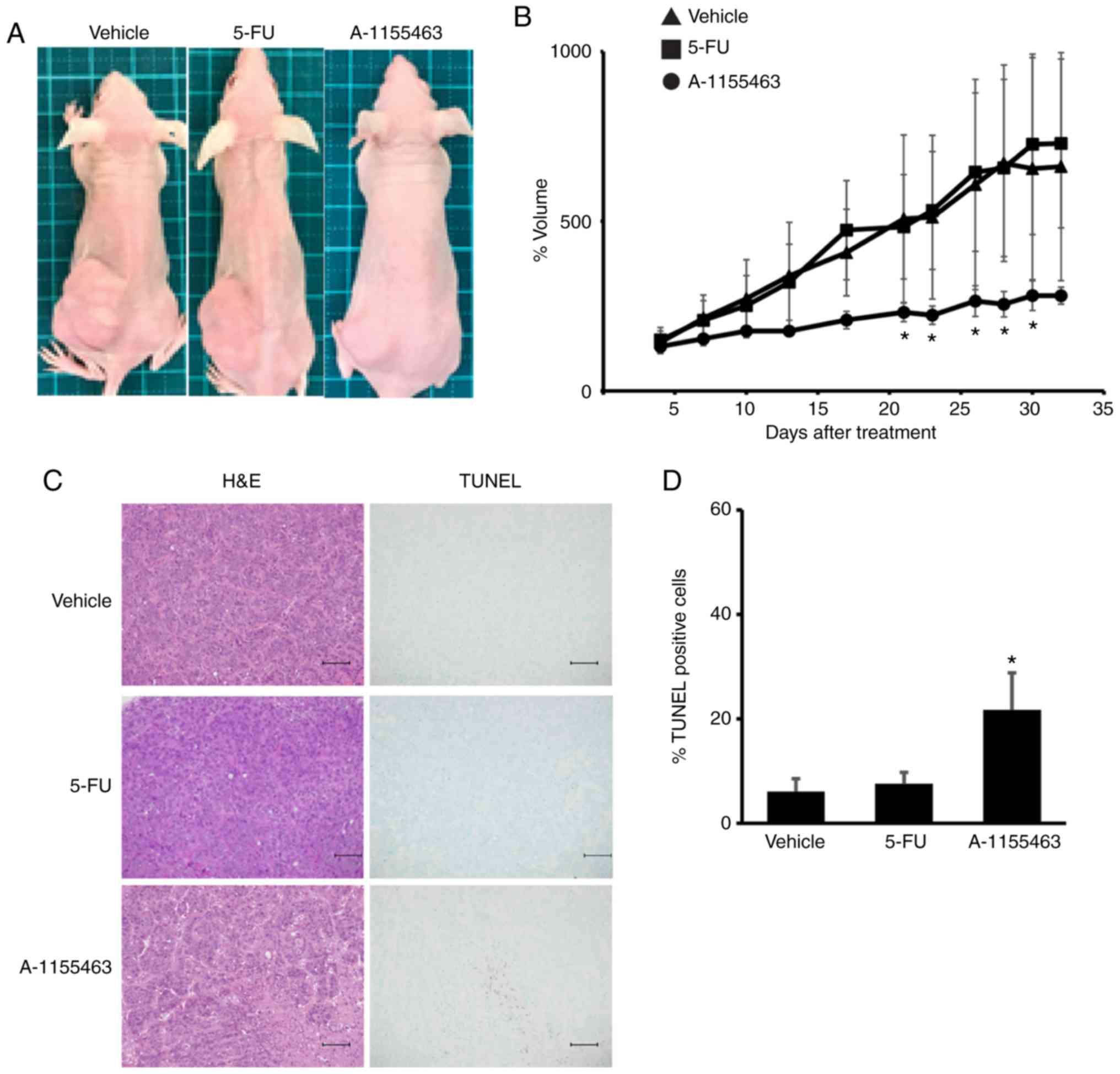 | Figure 7.BCLXL inhibitor controls the growth
of 5-fluorouracil (5-FU)-resistant HT-29 cells in vivo.
After engraftment with tumors of 5-FU-resistant HT-29 cells, BALB/c
nu/nu mice were divided into three experimental groups; Vehicle
(vehicle-treated/control), 5-FU, and A-1155463 groups. (A)
Representative images of subcutaneous tumors at day 32 after
treatment. (B) Tumor volumes were calculated by measuring the
length, height, and width. Data represent the mean ± SD (n=5).
*P<0.05, statistical significance when compared to
vehicle-treated mice, based on one way analysis of variance
followed by Dunnett's post hoc test. (C) Histologic analysis of
tumor sections stained with hematoxylin and eosin (H&E) and
TUNEL. Scale bars, 100 µm. (D) Statistical analysis of TUNEL
staining. *P<0.05, statistical significance when compared to
vehicle-treated mice, based on one way analysis of variance
followed by Dunnett's post hoc test. |
Discussion
In the present study, we presented a new strategy to
identify the mechanisms of 5-FU resistance, namely, BH3 profiling.
Results of this profiling identified strong dependence on BCLXL
regardless of induced BCL2 protein expression in 5-FU-resistant
HT-29 cells among three 5-FU-resistant colon cancer cell lines.
Furthermore, this BCLXL dependence may facilitate the treatment of
5-FU-resistant colon cancer cells by BCLXL inhibitors in
vitro and in vivo. As reported in a previous study
(15), our results suggest that
analysis of the expression patterns of apoptosis-related proteins
alone is insufficient to explain how cancer cells survive.
In clinical settings, anticancer drugs are continued
until they no longer elicit a response by the cancer cells or cause
severe adverse effects in the treated patient. During prolonged
drug treatment, certain cancer cell clones acquire drug resistance;
these cells gradually become predominant in the population, while
other cancer cells that are sensitive to the drug die. To elucidate
the mechanism of 5-FU resistance, we established three colon cancer
cell lines which were resistant to 16 µM 5-FU administered
continuously. Expression analysis of apoptosis-related proteins
indicated a marked induction of anti-apoptotic BCL2 expression in
the 5-FU-resistant HT-29 cells, consistent with previous studies
that showed little to no expression of BCL2 in wild-type HT-29
colon cancer cells (16,17) and BCL2 stabilization in 5-FU-resistant
HT-29 cells (18). However, our
results showed that ABT-199, a BCL2 inhibitor, had no
anti-apoptotic effect on 5-FU-resistant HT-29 cells, suggesting
that BCL2 induction did not contribute to cell survival.
In general, a complex network comprising many
apoptosis-related proteins makes it difficult to predict apoptotic
responses merely by quantifying protein expression. Recently, a new
functional assay, namely, BH3 profiling, was reported (15); this technique was shown to predict
sensitivity to a single drug in patients with acute myelogenous
leukemia (19), multiple myeloma
(20) and gastric cancer (21). Based on our BH3 profiling results,
5-FU-resistant HT-29 colon cancer cells have specific BCLXL
dependence, irrespective of the induction and expression of several
apoptosis-related proteins. Disparate results between the
quantification of protein expression and BH3 profiling provides
great insight in clinical settings, as expression analysis
including immunohistochemistry and real-time PCR has been conducted
previously for assessing cancer cells. Therefore, to accurately
assess protein dependence for cancer cell survival, BH3 profiling
stands to become an auxiliary tool for targeted therapy, even after
tumors acquire drug resistance.
BIM was induced in 5-FU-resistant HT-29 cells
(Fig. 2A) and this protein enhanced
sequestration by BCLXL (Fig. 6B).
BIM, which is usually bound to microtubules under physiological
conditions and recruited to mitochondria after treatment with
cytotoxic drugs (22), functions as a
pro-apoptotic activator of BAK and BAX (15). In our study, sequestration of induced
BIM by BCLXL in 5-FU-resistant HT-29 cells resulted in the evasion
of apoptosis caused by 5-FU; consequently, this BIM sequestration
resulted in dependence on BCLXL even in the absence of 5-FU
treatment, whereas parental HT-29 cells without BIM induction were
unprimed for apoptosis to BCLXL-related BH3 peptide (Fig. 3A and D-F).
Similar to HT-29 cells, increased mitochondrial
depolarization in 5-FU-resistant HCT-15 cells was also observed
upon treatment with the XXA1 peptide (Fig. 3A). However, this BCLXL dependence is
not associated with 5-FU resistance, as parental HCT-15 cells
already tended to be primed by this XXA1 peptide. These results
prompted us to consider trying a combination of 5-FU and BCLXL
inhibitor prior to the acquisition of 5-FU resistance. However, as
BCLXL plays a pivotal role in determining platelet life span
(23), the combination of BCLXL
inhibitor with a cytotoxic drug may cause severe thrombocytopenia.
Indeed, clinical studies targeting BCLXL in glioblastoma multiforme
(NCT00540722; http://clinicaltrials.gov/ct2/show/NCT00540722) and
small cell lung cancer (NCT03080311; http://clinicaltrials.gov/ct2/show/NCT03080311) are
ongoing; monotherapy has been conducted in these studies.
Therefore, when treating cancers with a BH3 profiling pattern
similar to parental and 5-FU-resistant HCT-15 cells,
BCLXL-inhibitor treatment may be better after acquiring 5-FU
resistance.
There are certain limitations to our study. Although
BCLXL inhibition in 5-FU-resistant HT-29 cells may reverse
sensitivity (Fig. 5B and C), the
detailed mechanism by which BCLXL dependence affects cell survival
exclusively in 5-FU-resistant HT-29 cells remains unknown.
Furthermore, though BH3 profiling may distinguish a cell line
sensitive to WEHI-539 out of three colon cancer cell lines in
vitro, the efficacy of this profiling in vivo remains to
be determined. Therefore, BH3 profiling of colon cancer samples
before and after chemotherapy in clinical settings will be
required.
In conclusion, our research involving BH3 profiling
demonstrated a clear dependence on BCLXL in the acquisition of 5-FU
drug resistance in HT-29 colon cancer cells among three colon
cancer cell lines. In addition, we showed that the sequestration of
the apoptosis-related BIM protein by BCLXL results in dependence on
the latter protein. Clinical studies targeting BCLXL in laryngeal
(NCT01633541; http://clinicaltrials.gov/ct2/show/NCT01633541) and
small cell lung cancer (NCT03080311) are ongoing. Assessing BCLXL
dependence in colon cancer cells through BH3 profiling will enable
a more detailed stratification of individual sensitivities to this
class of BCLXL selective inhibitors, thereby increasing the
efficacy of precision medicine.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Yukie Nakamura and Yumiko
Kaneko for research assistance.
Funding
The present study was supported by JSPS KAKENHI
(grant no. JP17K07200).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
KI and YK conducted the experiments. YA, TK, KT, KMu
and KMi participated in the data collection and analysis. YK and MK
participated in the design of the study. YK and JK participated in
the writing of the manuscript and data interpretation. All authors
read and approved the final manuscript and agree to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
All experiments on animals were performed following
approval from the Animal Ethics Committee of the Sapporo Medical
University School of Medicine (Sapporo, Hokkaido, Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hori M, Matsuda T, Shibata A, Katanoda K,
Sobue T and Nishimoto H; Japan Cancer Surveillance Research Group,
: Cancer incidence and incidence rates in Japan in 2009: A study of
32 population-based cancer registries for the Monitoring of Cancer
Incidence in Japan (MCIJ) project. Jpn J Clin Oncol. 45:884–891.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watanabe T, Muro K, Ajioka Y, Hashiguchi
Y, Ito Y, Saito Y, Hamaguchi T, Ishida H, Ishiguro M, Ishihara S,
et al: Japanese society for cancer of the colon and rectum (JSCCR)
guidelines 2016 for the treatment of colorectal cancer. Int J Clin
Oncol. 23:1–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rutman RJ, Cantarow A and Paschkis KE:
Studies in 2-acetylaminofluorene carcinogenesis. III. The
utilization of uracil-2-C14 by preneoplastic rat liver and rat
hepatoma. Cancer Res. 14:119–123. 1954.PubMed/NCBI
|
|
5
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu W, Fang Y, Wang XT, Liu J, Dan X and
Sun LL: Overcoming 5-Fu resistance of colon cells through
inhibition of Glut1 by the specific inhibitor WZB117. Asian Pac J
Cancer Prev. 15:7037–7041. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P,
van Buren G II, Samuel S, Kim MP, Lim SJ and Ellis LM:
Chemoresistant colorectal cancer cells, the cancer stem cell
phenotype, and increased sensitivity to insulin-like growth
factor-I receptor inhibition. Cancer Res. 69:1951–1957. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshida M, Horiguchi H, Kikuchi S, Iyama
S, Ikeda H, Goto A, Kawano Y, Murase K, Takada K, Miyanishi K, et
al: miR-7977 inhibits the Hippo-YAP signaling pathway in the bone
marrow mesenchymal stromal cells. PLoS One. 14:e02132202019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ryan J and Letai A: BH3 profiling in whole
cells by fluorimeter or FACS. Methods. 61:156–164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fraser C, Ryan J and Sarosiek K: BH3
profiling: A functional assay to measure apoptotic priming and
dependencies. Methods Mol Biol. 1877:61–76. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tao ZF, Hasvold L, Wang L, Wang X, Petros
AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, et al:
Discovery of a potent and selective BCL-XL inhibitor with in vivo
activity. ACS Med Chem Lett. 5:1088–1093. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jian YS, Chen CW, Lin CA, Yu HP, Lin HY,
Liao MY, Wu SH, Lin YF and Lai PS: Hyaluronic acid-nimesulide
conjugates as anticancer drugs against CD44-overexpressing HT-29
colorectal cancer in vitro and in vivo. Int J Nanomedicine.
12:2315–2333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dutta S, Ryan J, Chen TS, Kougentakis C,
Letai A and Keating AE: Potent and specific peptide inhibitors of
human pro-survival protein Bcl-xL. J Mol Biol. 427:1241–1253. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Foight GW, Ryan JA, Gullá SV, Leta A and
Keating AE: Designed BH3 peptides with high affinity and
specificity for targeting Mcl-1 in cells. ACS Chem Biol.
9:1962–1968. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montero J and Letai A: Why do BCL-2
inhibitors work and where should we use them in the clinic? Cell
Death Differ. 25:56–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nita ME, Nagawa H, Tominaga O, Tsuno N,
Fujii S, Sasaki S, Fu CG, Takenoue T, Tsuruo T and Muto T:
5-Fluorouracil induces apoptosis in human colon cancer cell lines
with modulation of Bcl-2 family proteins. Br J Cancer. 78:986–992.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao DP, Ding XW, Peng JP, Zheng YX and
Zhang SZ: Prognostic significance of bcl-2 and p53 expression in
colorectal carcinoma. J Zhejiang Univ Sci B. 6:1163–1169. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu DW, Huang CC, Chang SW, Chen TH and Lee
H: Bcl-2 stabilization by paxillin confers 5-fluorouracil
resistance in colorectal cancer. Cell Death Differ. 22:779–789.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vo TT, Ryan J, Carrasco R, Neuberg D,
Rossi DJ, Stone RM, Deangelo DJ, Frattini MG and Letai A: Relative
mitochondrial priming of myeloblasts and normal HSCs determines
chemotherapeutic success in AML. Cell. 151:344–355. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan
JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P,
Tai YT, et al: Pretreatment mitochondrial priming correlates with
clinical response to cytotoxic chemotherapy. Science.
334:1129–1133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kubo T, Kawano Y, Himuro N, Sugita S, Sato
Y, Ishikawa K, Takada K, Murase K, Miyanishi K, Sato T, et al: BAK
is a predictive and prognostic biomarker for the therapeutic effect
of docetaxel treatment in patients with advanced gastric cancer.
Gastric Cancer. 19:827–838. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klotz DM, Nelson SA, Kroboth K, Newton IP,
Radulescu S, Ridgway RA, Sansom OJ, Appleton PL and Näthke IS: The
microtubule poison vinorelbine kills cells independently of mitotic
arrest and targets cells lacking the APC tumour suppressor more
effectively. J Cell Sci. 125:887–895. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan Y, Xie R, Zhang Q, Zhu X, Han J and
Xia R: Bcl-xL/Bak interaction and regulation by miRNA let-7b in the
intrinsic apoptotic pathway of stored platelets. Platelets.
30:75–80. 2019. View Article : Google Scholar : PubMed/NCBI
|