Introduction
Chronic myeloid leukemia (CML) is a malignant
myeloproliferative disease, originating from pluripotent
hematopoietic stem cells and comprising ~15% of leukemia cases
(1). CML is characterized by
increased proliferation of the granulocytic cell line and is
associated with erythroid cells and platelet hyperplasia (2). Chemotherapy remains the first line of
clinical treatment for CML. However, although chemotherapy improves
the survival of CML patients, and despite the development of novel
chemotherapeutic drugs and hematopoietic stem cell transplantation,
30% of CML patients relapse after remission. Tyrosine kinase
inhibitors (TKIs), the first-line treatment drug for CML at
present, are associated with a high rate of adverse effects and CML
recurrence (3). Thus, in order to
improve the outcome of CML patients, it is crucial to explore
alternative therapeutic approaches with enhanced efficacy, low
toxicity and multi-target combinations of anticancer drugs,
possibly with different mechanisms of action (4).
Emodin (1,3,8-trihydroxy-6-methyl anthraquinone) is
a traditional Chinese medicine isolated from rhubarb, with
effective antitumor, antibacterial and antiaging properties
(5,6).
Emodin has been reported to be effective in treating a number of
tumors that are insensitive to other chemotherapies, adding
exciting new prospects to cancer treatment (7). However, it has also been reported
(8) that emodin is potentially
nephrotoxic by damaging proximal tubules, which limits its
applicability in cancer therapy. However, it remains unknown
whether a combination of emodin and currently available
chemotherapeutic drugs can exert synergistic anticancer effects
while reducing side effects, such as nephrotoxicity.
Azidothymidine (AZT), a thymidine analogue and
telomerase inhibitor mainly used in anti-HIV therapy, has been
found to exert potent inhibitory effects on a variety of
malignancies, including leukemia (9–11).
However, the therapeutic dosage of AZT is usually associated with
severe toxic side effects, which restrict the antitumor application
of AZT in the clinical setting (12).
Recent studies indicate that AZT exerts marked therapeutic effects
acting synergistically with other antitumor drugs within a safe
dosage range (13,14). This observation prompts further
investigation to identify drugs optimally combined with AZT for the
treatment of leukemia.
The gene encoding early growth response protein-1
(EGR1), a member of the immediate early gene family, is
rapidly induced by extensive extracellular stimulation, and is a
converging point for a number of intracellular signaling cascades
that control tumor cell growth and proliferation, as well as other
signaling cascades associated with cell death mechanisms.
Accumulating evidence indicates that EGR1 acts as a tumor
suppressor (15).
The canonical Wnt/β-catenin pathway is characterized
by activation of transcriptional activity mediated by β-catenin.
Several components of this pathway are associated with a variety of
human diseases, particularly cancer (16–21). The
Wnt/β-catenin pathway has been proven to play an important role in
the malignant transformation of hematopoietic cells and the
development of hematological malignancies (22–24). In
CML, the majority of the cases are associated with BCR-ABL
oncoprotein mutations and BCR-ABL amplification. BCR-ABL fusion
gene regulates β-catenin, and it may be associated with the high
expression of β-catenin observed in CML (25). Therefore, the Wnt/β-catenin pathway
appears to play a key role in regulating leukemia resistance and
late recurrence, which provides a new approach to targeted
treatment of leukemia (26).
EGR1 has been demonstrated to induce an
increase in glycogen synthase kinase (GSK3) β activity. GSK3β
phosphorylates β-catenin and induces its proteasomal degradation,
which attenuates β-catenin expression and β-catenin-dependent
T-cell factor/lymphoid enhancer factor (TCF/LEF) transcriptional
activity (27). The combination of
bioinformatics, the NCBI database, Patch software, and KEGG
database were utilized to identify the EGR1 sequence,
EGR1 predicted target gene, and Wnt/β-catenin pathway genes.
It was revealed that TCF, an important gene in the Wnt/β-catenin
pathway, is also a target gene of EGR1. However, it remains
unclear whether the Wnt/β-catenin signaling pathway is regulated by
the Egr-1 gene in CML.
Based on the abovementioned data, it is reasonable
to hypothesize that enhancing the antitumor effect of drugs at a
similar or lower dosage would be of great value. To the best of our
knowledge, the effects of the combination of emodin and AZT on the
inhibition of proliferation and induction of apoptosis in the human
CML cell line K562 has not been determined. The present study was
designed to investigate the effects of a combination of emodin and
AZT on CML cell proliferation inhibition and apoptosis induction
in vitro, explore the possible underlying mechanisms, and
provide an experimental basis for further development and clinical
application of emodin and AZT.
Materials and methods
Cell culture
The human CML K562 cell line was purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). K562 cells were cultured in RPMI-1640 (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal calf
serum, penicillin (100 U/ml), streptomycin (100 µg/ml) and
glutamine (0.292 mg/ml). These cells were incubated at 37°C in a
humidified atmosphere with 5% CO2. Cells in the
logarithmic growth phase were harvested for experiments.
Chemicals and reagents
Emodin (purity >98%) was purchased from
Biological Technology Development Co. Ltd. AZT, MTT and dimethyl
sulfoxide were purchased from Sigma-Aldrich; Merck KGaA. RPMI-1640
was purchased from Gibco; Thermo Fisher Scientific, Inc. Fetal calf
serum was purchased from Hangzhou Evergreen Co., Ltd. Gene primers
and the reverse transcription kit were purchased from Beijing
Tiangen Biotech Co., Ltd. The Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit was purchased
from Nanjing KGI Biological Ltd. The Nucleofector™ nuclear
transfection instrument was purchased from Germany LONZA Group,
Ltd.
Cell proliferation assay
K562 cells were plated at a concentration of
1×105/ml in 96-well plates in 100 µl of culture medium
per well. The cells were treated with emodin (8, 16 and 32 µM), AZT
(80, 160 and 320 µM) and a combination of emodin and AZT (8/80,
16/160 and 32/320 µM) for 24, 48 and 72 h. The experimental cells
were incubated with 10 ml MTT (5 mg/ml in serum-free medium) for 4
h, followed by addition of 100 µl lysis solution (10% SDS in 0.01 M
HCl) per well and incubation for a further 24 h. The cell survival
rate was measured using optical density (OD) at 490 nm on a
microplate reader (Ultramark™ Microplate system; Bio-Rad
Laboratories, Inc.) in triplicate. The experiment was repeated
three times. The inhibition rate (IR) was calculated as follows: IR
(%)=[1-Mean OD of experiment)/Mean OD of control] ×100%.
Synergy determination
The synergistic effects of emodin and AZT at a fixed
concentration ratio (emodin: AZT=1:10) and at different
concentrations were analyzed using the CalcuSyn 2.0 software
(Biosoft). The drug half maximal inhibitory concentration
(IC50) and combination index (CI) were calculated. A CI
value of 1 indicates an additive effect, values <1 indicate
synergistic action, and values >1 indicate antagonism.
Detection of apoptotic cells
Using flow cytometry, the apoptosis analysis was
performed as described in the Annexin V-FITC/PI apoptosis detection
kit manual. The harvested K562 cells were resuspended in fresh
medium, adjusting cell density to 1×105/ml. A total of
2.5 ml cell suspension per well were added into 6-well plates. The
cells were treated with emodin at a final concentration of 32 µM,
AZT at a final concentration of 320 µM, and a combination of the
two at a final concentration of 32 µM (emodin)/320 µM (AZT) for 24,
48 and 72 h. Equal volumes of RPMI-1640 were used as control. Cells
were collected by centrifugation (111.8 × g, 5 min, 37°C) and
resuspended in 500 µl of 1X binding buffer in tubes. Annexin V-FITC
(5 µl) and PI (5 µl) were added, the tubes were incubated at room
temperature for 5 min in the dark, and the samples were then
examined by flow cytometry. The experiment was repeated three
times.
Detection of cell cycle
distribution
The harvested K562 cells were resuspended in fresh
medium, adjusting cell density to 3×105/ml. A total of
2.5 ml cell suspension per well were added into 6-well plates. The
cells were treated with emodin at a final concentration of 32 µM,
AZT at a final concentration of 320 µM, and a combination of the
two at a final concentration of 32 µM (emodin)/320 µM (AZT) for 24,
48 and 72 h. The cells were collected by centrifugation (111.8 × g,
5 min, 37°C), washed twice with PBS, fixed with 70% iced ethanol
overnight, stained in the dark for 30 min with 0.6 ml PI, and
examined by flow cytometry. The experiment was repeated three
times.
Detection of EGR1 mRNA expression
The harvested K562 cells were resuspended in fresh
medium, adjusting cell density to 1×106/ml. A total of 5
ml cell suspension per well was added into 6-well plates. The cells
were treated with emodin at a final concentration of 32 µM, AZT at
a final concentration of 320 µM, and a combination of the two at a
final concentration of 32 µM (emodin)/320 µM (AZT) for 0, 0.5, 1,
2, 4 or 8 h; subsequently, the K562 cells were subjected to reverse
transcription-polymerase chain reaction (RT-PCR) analysis. Total
cellular RNA at different time points was obtained using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Subsequently, RNA concentration was
measured by UV spectrophotometry. RNA was reverse-transcribed using
random primers and AMV reverse transcriptase (Tiangen) for 5 min at
70°C, 5 min on ice, and 60 min at 37°C. The single-stranded cDNA
was amplified by PCR using GoTaq DNA polymerase (Tiangen). PCR of
the EGR1 gene was performed under the following conditions:
45 sec at 94°C, 45 sec at 55°C and 45 sec at 72°C, for a total of
30 cycles. After PCR of the EGR1 gene, equal amounts of
RT-PCR products were loaded on 1.0% agarose gels. The results were
analyzed using Quantity One software (V4.6.6; Premier Biosoft, Palo
Alto, CA, USA). The primers for PCR are shown in Table I.
 | Table I.Primer sequences, and length of
RT-PCR. |
Table I.
Primer sequences, and length of
RT-PCR.
| Gene | Primer sequences
(5→3) | Length |
|---|
| EGR1 | Upstream:
TTCGCTAACCCCTCTGTCTACTACTATT |
|
|
| Downstream:
GACTCCACTGGGCAAGCGTAA | 180 bp |
| β-actin | Upstream:
GACTCCACTGGGCAAGCGTAA |
|
|
| Downstream:
GTGGGGCGCCCCAGGCACCA | 548 bp |
Cell transfection
For the EGR1 gene mRNA, a specific base
sequence was selected as the interference target, and the siRNA
fragment was used as follows: si-EGR1 sense,
5′-CCCGGUUACUACCUCUUAUTT-3′ and antisense,
5′-AUAAGAGGUAGUAACCGGGTT-3′. Target-free siRNAs with fluorescently
labeled homologous sequences were used as negative controls (NC).
K562 cells were transfected by electroporation. Nucleofector I
nuclear transfection apparatus and Amaxa® Cell Line
Nucleofector® (Lonza, Cologne, Germany) were used. K562
cells in the logarithmic growth phase were collected, the cell
density was adjusted to 1–2×106/ml, and the cells were
divided into a blank control group (K562, only subjected to
electroporation), a non-specific control group (K562/NC,
transfected with the FAM-labeled universal random negative control
NC-siRNA), and the target siRNA group (K562/siRNA, transfected with
Egr-1 siRNA). The transfection methods and procedures were
performed in accordance with the kit instructions. Transfected
cells emitting green fluorescence were observed under a
fluorescence microscope, and RT-PCR was used to determine the
integration of EGR1 mRNA expression in the transfected K562
cells. Each set of experiments was repeated 3 times.
Detection of β-catenin protein
expression in the Wnt/β-catenin pathway by western blotting
K562 cells in the logarithmic growth phase and K562
cells transfected with siRNA were resuspended in fresh medium to
adjust the cell density to 1×106/ml. A total of 5 ml
cell suspension per well was added to a 6-well plate. The final
concentration of emodin was 32 µM, the final concentration of AZT
was 320 µM, and the final concentration of emodin/AZT was 32/320 µM
for 72 h. The total protein concentration was determined by the BCA
method. Electrophoresis was performed using a 10% polyacrylamide
gel, and western blotting was conducted in a conventional manner.
Finally, color development was performed by ECL chemiluminescence,
and imaging was performed using a gel imager. Each set of
experiments was repeated 3 times.
Bioinformatics analysis
The EGR1 transcription factor sequence was
downloaded from the National Center for Biotechnology Information
(NCBI) (https://www.ncbi.nlm.nih.gov), and
the EGR1 target gene was predicted by Patch software
(http://gene-regulation.com/cgi-bin/pub/program/patch/bin/patch.cgi).
The KEGG database (https://www.genome.jp/kegg/) was used to search for
all genes of the WNT/β-catenin pathway. The intersection of the
target genes predicted by EGR1 and the genes in the
WNT/β-catenin pathway was selected.
Statistical analysis
The dose inhibition effect of each drug alone and
the effects of the combination of the two drugs were calculated
using CalcuSyn 2.0 statistical software (Premier Biosoft).
Experimental data are shown as means ± standard deviation.
Comparisons were performed using Student's-test and one-way ANOVA,
and the multiple test were performed with post hoc test. All data
were processed using SPSS 13.0 (SPSS, Inc.).
Results
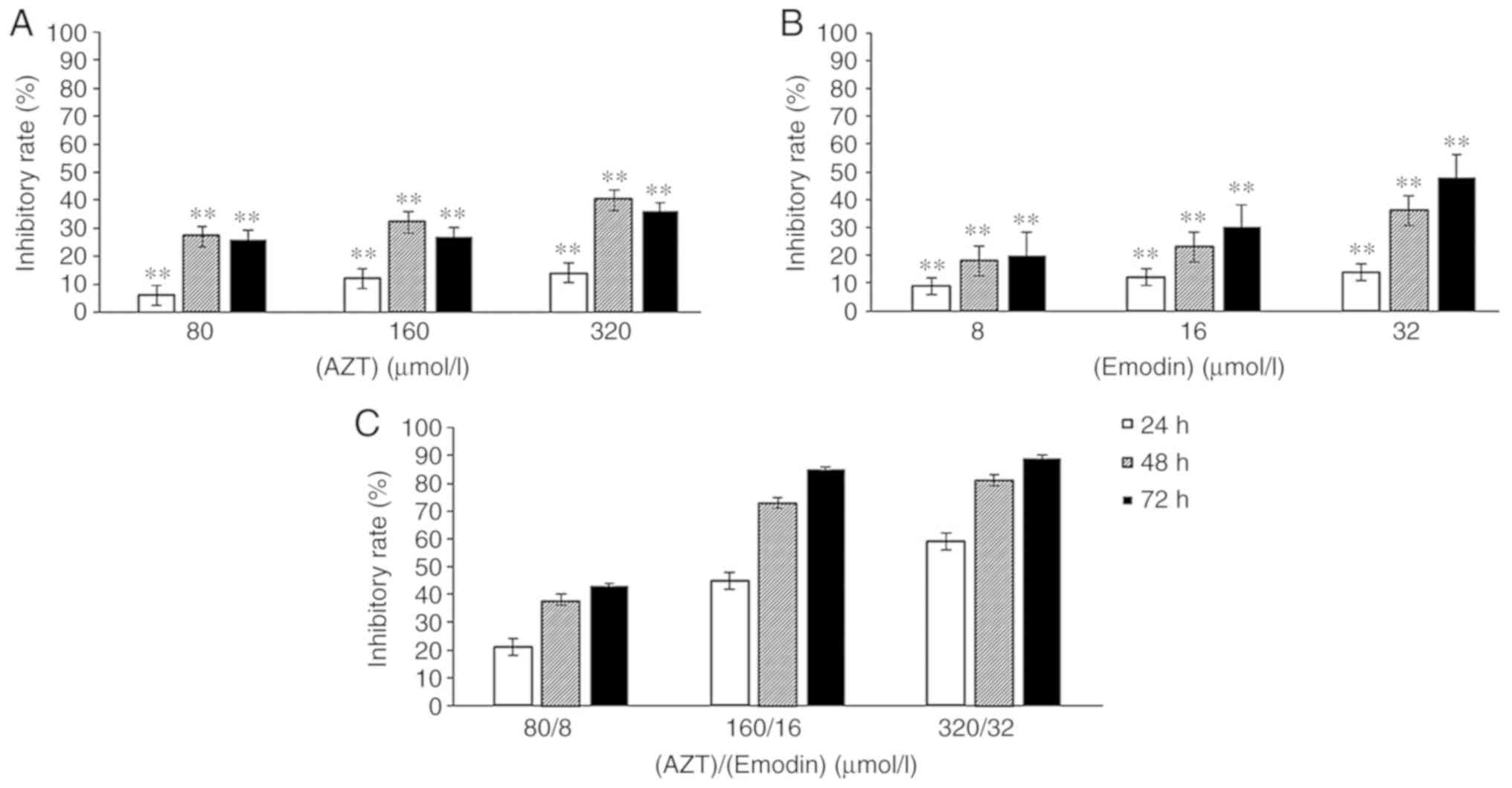
Inhibitory effects of emodin, AZT and
their combination on the proliferation of the K562 cells
The proliferation of K562 cells treated with AZT
(Fig. 1A), emodin (Fig. 1B) and the combination of emodin/AZT
(Fig. 1C) at three different
concentration levels for 24, 48 or 72 h was decreased in a dose-and
time-dependent manner. The inhibition rates of the combination of
emodin and AZT in K562 cells (Fig.
1C) were significantly higher compared with those of either AZT
or emodin alone at the same concentrations and time
(P<0.01).
Analysis of the effects of emodin, AZT
and their combination Associations between inhibitory effects and
the dose of emodin, AZT and their combination
K562 cells were treated with emodin and/or AZT for
24, 48 and 72 h. The IC50 values for emodin in K562
cells at 24, 48 and 72 h were all >32 µmol/l, and the
IC50 values for AZT in K562 cells at 24, 48 and 72 h
were all >320 µmol/l; however, the IC50 values of the
emodin/AZT combination in K562 cells at 24, 48 and 72 h were
22.43/224.31, 10.53/105.26, and 7.31/73.07 µmol/l, respectively
(Table II).
| Table II.IC50 of emodin, AZT and
the combination in regards to the growth of K562 cells. |
Table II.
IC50 of emodin, AZT and
the combination in regards to the growth of K562 cells.
|
| IC50
(µM) at: |
|---|
|
|
|
|---|
| Treatment | 24 h | 48 h | 72 h |
|---|
| Emodin | >32 | >32 | >32 |
| AZT | >320 | >320 | >320 |
| Emodin + AZT | 22.43/224.31 | 10.53/105.26 | 7.31/73.07 |
CI of emodin and AZT in the K562
cells
As calculated by CalcuSyn 2.0 statistical software,
the CI values of the combination of emodin and AZT at a fixed
concentration ratio in K562 cells treated for 24, 48 and 72 h were
all <1 (Table III). These
results indicate a strongly synergistic inhibitory effect of the
emodin + AZT combination on the CML cell line K562.
| Table III.CI values at different treatment
times. |
Table III.
CI values at different treatment
times.
| Treatment time
(h) | AZT (µmol/l) | Emodin
(µmol/l) | Fa | CI |
|---|
| 24 | 80 | 8 | 0.221224 | 0.221 |
|
| 160 | 16 | 0.435789 | 0.102 |
|
| 320 | 32 | 0.592201 | 0.087 |
| 48 | 80 | 8 | 0.338468 | 0.789 |
|
| 160 | 16 | 0.741626 | 0.107 |
|
| 320 | 32 | 0.813899 | 0.111 |
| 72 | 80 | 8 | 0.424044 | 0.302 |
|
| 160 | 16 | 0.853403 | 0.047 |
|
| 320 | 32 | 0.872798 | 0.077 |
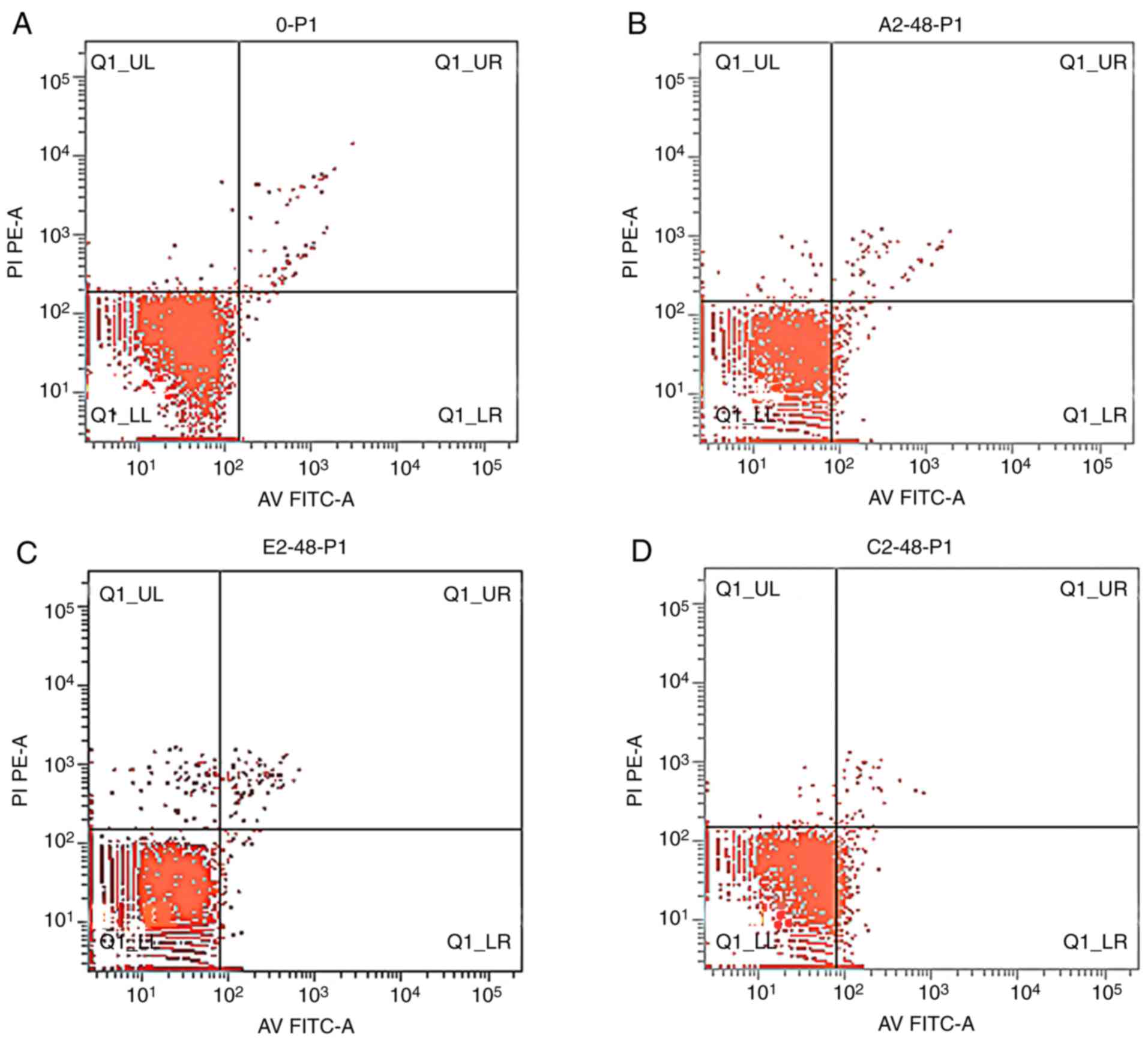
Apoptosis and cell cycle
Apoptosis rates of the K562 cells
The apoptosis rates of K562 cells treated with
emodin and/or AZT were increased when compared with the control
(Fig. 2) in an overall time-dependent
manner; however, the increase in apoptosis rates at 48 h was the
most significant. The apoptosis rates in the combination treatment
group were higher compared with those in the control and
single-agent groups, and the differences were statistically
significant (P<0.05) (Table
IV).
| Table IV.Apoptosis rates in the different
treatment groups (n=3). |
Table IV.
Apoptosis rates in the different
treatment groups (n=3).
|
| Apoptosis rate (%)
at: |
|---|
|
|
|
|---|
| Group (µmol/l) | 24 h | 48 h | 72 h |
|---|
| Control | 1.04±0.22 | 1.19±0.25 | 1.31±0.37 |
| Emodin 32 | 1.47±0.23 | 2.1±0.57 | 1.58±0.41 |
| AZT 320 | 1.11±0.32 | 7.1±1.42 | 5.98±1.45 |
| Emodin 32 + AZT
320 |
1.75±0.29b,c,f |
9.59±2.16b,d,e |
7.03±1.27b,d,e |
K562 cell cycle distribution
Emodin, AZT and their combination exerted inhibitory
effects on cell cycle progression in K562 cells, as determined by
flow cytometry. After K562 cells were treated with the combination
of emodin and AZT for 48 h, the percentage of cells in the
G0/G1 phase was increased, and that of cells
in the G2/M and S phases was decreased. Therefore,
emodin/AZT combination treatment arrested cells in the
G0/G1 phase (Table
V).
| Table V.Effect of the different treatments on
the cell cycle distribution of K562 cells (n=3). |
Table V.
Effect of the different treatments on
the cell cycle distribution of K562 cells (n=3).
|
|
| Percentage of cells
(%) |
|---|
|
|
|
|
|---|
| Treatment
(µmol/l) | Time (h) |
G0/G1 phase | S phase | G2/M
phase |
|---|
| AZT 320 | 24 | 65.63±2.66 | 24.79±1.22 | 0.54±0.04 |
|
| 48 | 65.35±2.58 | 28.92±0.98 | 2.74±0.12 |
|
| 72 | 75.86±4.04 | 18.43±1.78 | 0.29±0.03 |
| Emodin 32 | 24 | 39.12±3.12 | 29.97±2.03 | 26.28±1.36 |
|
| 48 | 43.19±1.85 | 35.04±0.97 | 17.15±0.96 |
|
| 72 | 41.26±1.35 | 36.06±1.14 | 15.28±0.88 |
| Emodin 32 + AZT
320 | 24 |
70.54±2.44b,d |
22.94±0.95a,d |
1.85±0.08a,d |
|
| 48 |
77.5±2.98b,d |
18.87±0.88b,d |
0.07±0.01b,d |
|
| 72 |
77.38±3.01d |
16.24±0.86a,d |
0.5±0.04a,d |
Effects of the combination of emodin
and AZT on EGR1 mRNA expression
It was demonstrated by RT-PCR (Fig. 3) that the transcriptional levels of
EGR1 in K562 cells treated with emodin, AZT or a combination
of the two quickly increased to a peak at 0.5 h, and then gradually
declined. With prolongation of the processing time, the expression
of EGR1 in K562 cells decreased to a minimum at 8 h, which
was similar to the levels in untreated levels. When the cells were
treated with emodin or AZT alone for 0.5 h, the EGR1 gene
expression in K562 cells increased by 48.33 or 14.1%, respectively,
while treatment of K562 cells with the combination of emodin and
AZT for 0.5 h increased the expression of EGR1 gene by
128.64%, which was significantly higher compared with the effect of
emodin or AZT alone (Fig. 3).
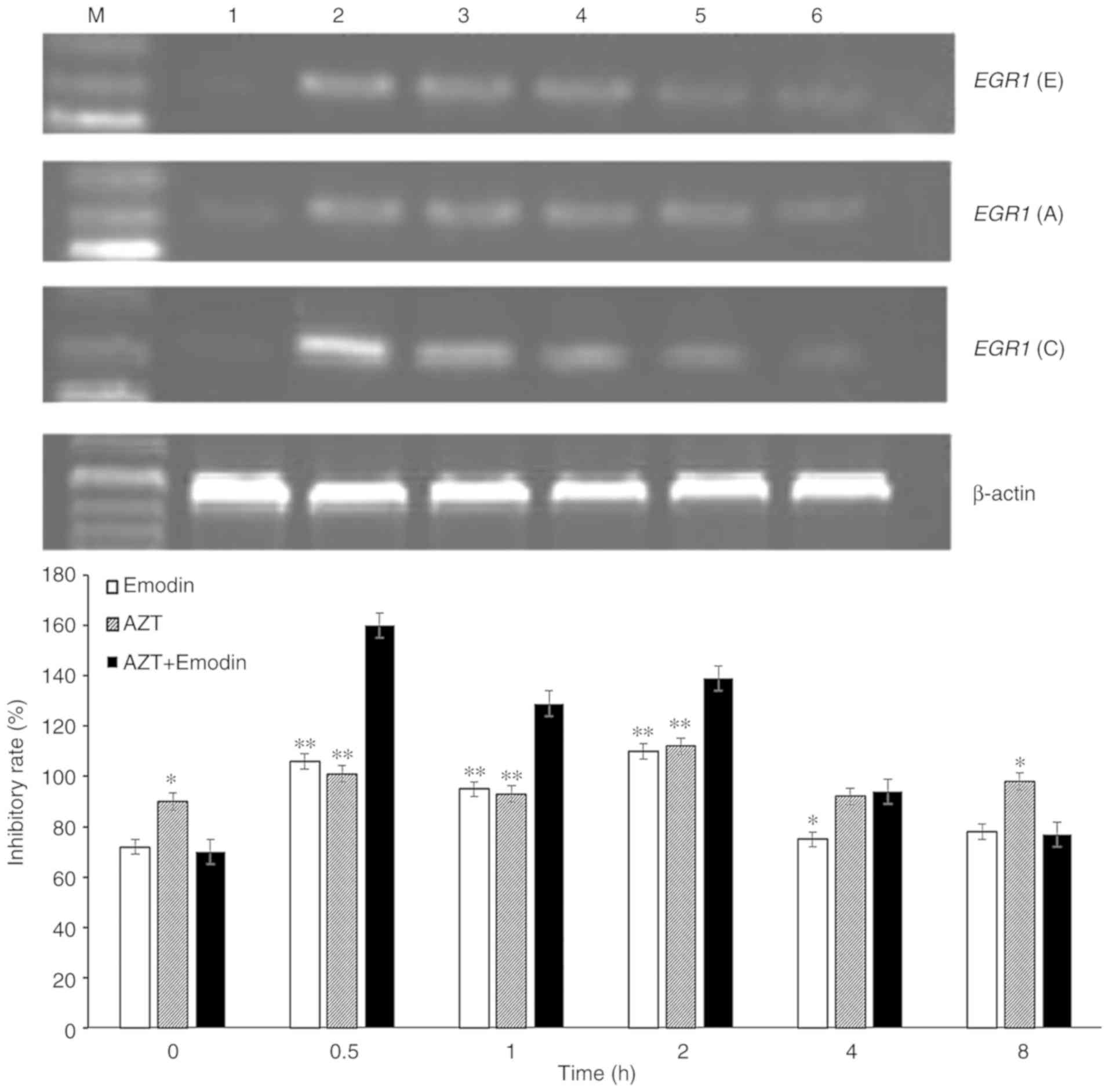 | Figure 3.Expression of EGR1 mRNA in
K562 cells treated with emodin alone, AZT alone and a combination
of emodin and AZT for 0, 0.5, 1, 2, 4 and 8 h. Lanes; M, DNA marker
lane; lane 1, blank control; lane 2, 0.5 h; lane 3, 1 h; lane 4, 2
h; lane 5, 4 h; and lane 6, 8 h. EGR1 (E): 32 µmol/l emodin;
EGR1 (A): 320 µmol/l AZT; EGR1 (C): 32 µmol/l emodin
+ 320 µmol/l AZT. *P<0.05, **P<0.01 compared with the
combination of emodin and AZT. EGR1, early growth response
−1; AZT, azidothymidine. |
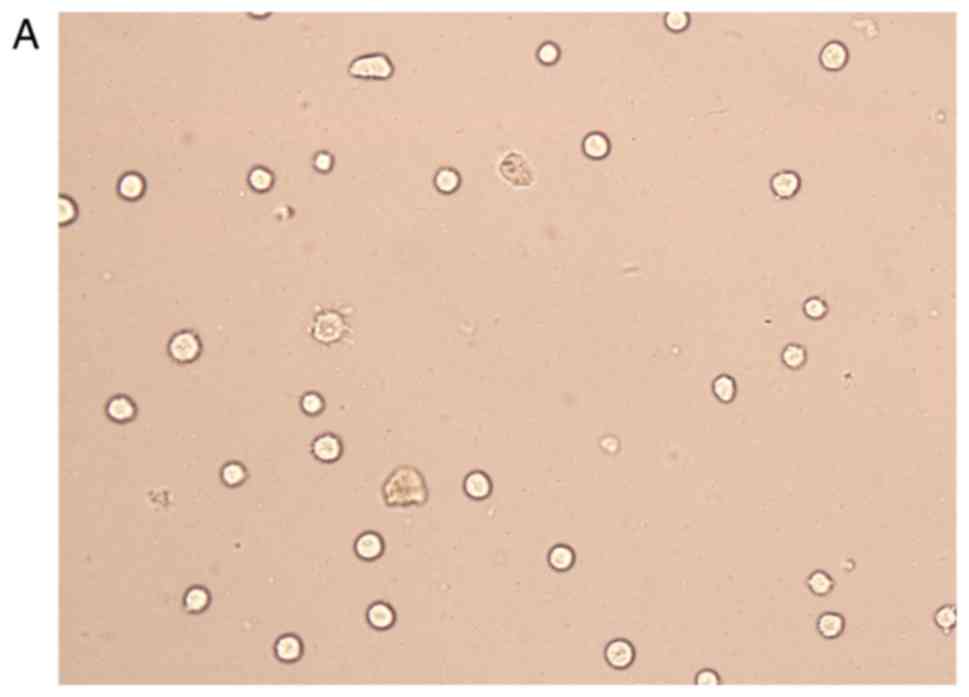
Cell transfection
The transfection efficiency was observed with FAM
fluorescently labeled siRNA, and it was found that the transfected
cells emitted green fluorescence under a fluorescence microscope.
The transfection efficiency was >75%, and the cells were used in
subsequent experiments (Fig. 4).
After trypan blue staining, with a count of live cells up to 75%,
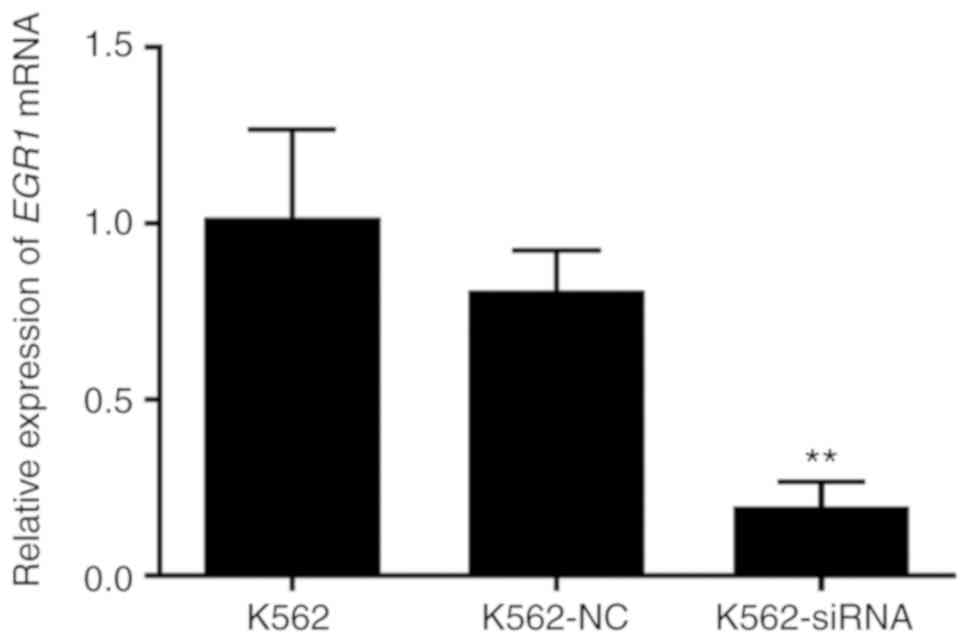
the cells were used for follow-up studies. The results of the PCR
analysis demonstrated that, compared with the non-specific control
and the blank control groups, the expression level of EGR1
in the siRNA group was decreased at 24 h after transfection, and
the difference was statistically significant (P<0.01); there was
no significant difference with the specific control group. This
indicates that siRNA blocks the expression of EGR1. After 24
h of transfection (Fig. 5), the
expression of EGR1 was significantly lower compared with
that prior to transfection, and the difference was statistically
significant (P<0.01).
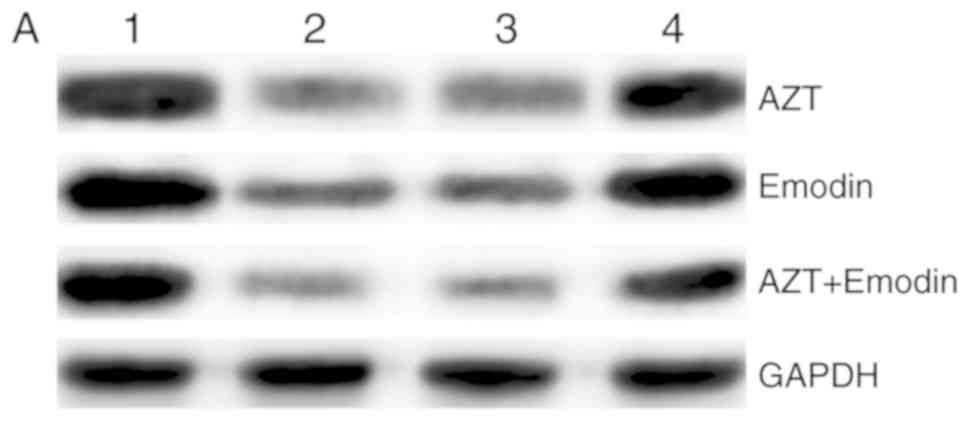
Detection of β-catenin protein
expression in the Wnt/β-catenin pathway by western blotting
The results of western blotting are shown in
Fig. 6A and B. Compared with the
blank control group, the protein expression of β-catenin in K562
cells treated with emodin (32 µM) and AZT (320 µM) was decreased,
and the difference was statistically significant (P<0.05).
Compared with the single-drug and the control groups, the
expression level of the β-catenin protein in K562 cells treated by
the combination of emodin (32 µM) and AZT (320 µM) was
significantly decreased (P<0.05). Compared with untransfected
K562 cells, the expression of the β-catenin protein was
significantly higher in the three groups with positively
transfected siRNA, and the differences were statistically
significant (P<0.05). There was no significant difference in the
expression level of the β-catenin protein between the three groups
of negatively transfected siRNA and untransfected K562 cells
(P>0.05).
Discussion
The results of the present study indicated that the
combination of emodin and AZT exerted obvious synergistic
inhibitory effects (CI <1) on leukemia K562 cells in
vitro. The synergy between the two drugs exhibited a type of
concentration dependent trend. The combination of emodin and AZT at
higher concentrations exhibited more significant synergy. Compared
with emodin or AZT alone, the combination of emodin and AZT at
lower doses produced proliferation inhibition and apoptosis
induction similar to those achieved by higher doses of emodin or
AZT alone. This indicated that the effective combination of the two
drugs may be associated with lower toxicity and can significantly
improve therapeutic efficacy. In addition, the proliferation
inhibition is more extensive than the apoptosis rate, which may be
caused by AZT or emodin inducing autophagy, and autophagy is a
protective mechanism for the apoptosis of leukemia K562 cells
(28–31). The evaluation of the synergy between
emodin and AZT may provide evidence for clinical application and
future treatment studies.
The combination of telomerase inhibitors with
small-molecule substances, such as anthraquinone, quinoline,
berberine and their analogues, has become an important focus of
antitumor drug research in recent years (32).
As a natural monomer supplementary chemotherapy
drug, emodin may inhibit tumor cell growth in pancreatic, prostate
and colorectal cancer, as well as other malignant tumors (33). Emodin may induce apoptosis in leukemia
cells through Bcl-2/Bax, the caspase-3 protein family, the human
telomerase reverse transcriptase Htert, c-Myc, and other
apoptosis-related molecules (34).
Emodin may also reverse multi-drug resistance of tumor cells and
increase the body's sensitivity to antitumor drugs. It was
previously demonstrated that emodin can reduce the expression of
the multi-drug resistance gene MRP1 through the PI3K/AKT pathway
(35) and reverse the multi-drug
resistance of CML cells (36). In
addition, emodin was shown to markedly enhance the sensitivity of
tumor cells to arsenic trioxide (37), 5-fluorouracil (38), platinum (39), paclitaxel (40), gemcitabine (41) and curcumin (42), as well as other drugs, in a variety of
cancers.
The present results confirmed the synergistic
interaction between emodin and AZT, thus supporting the use of this
combination at low doses with low toxicity. Administration of
emodin in combination with a telomerase inhibitor may be an
effective approach to the treatment of refractory and recurrent
leukemia.
In recent years, EGR1, a tumor suppressor
gene, has been increasingly attracting research attention due to
its role in the occurrence and development of tumors. EGR1
is one of the most important members of the immediate early gene
family. The expression of EGR1 decreases or even disappears
in a variety of human malignancies, and its expression level is
associated with tumor sensitivity to chemotherapy (43). Apoptosis induction by EGR1 may
be relevant to its broad spectrum multi-directional regulation and
cell signaling pathway network. It is generally believed that
EGR1 acts as a tumor suppressor by regulating the expression
of downstream target genes, such as transforming growth factor-β,
cyclin D1, c-Jun, phosphatase and tensin homolog, p53 and p21
(44,45). In the present study, changes in the
transcriptional level of the EGR1 gene were observed using
RT-PCR analysis. It was observed that the gene expression of
EGR1 was increased in K562 cells that were treated with
emodin or AZT alone, or the combination of emodin and AZT, and that
the increase in the expression of the EGR1 gene induced by
the combination of the two drugs was more significant compared with
that by either drug alone; furthermore, this effect was
time-dependent. Western blotting demonstrated that the protein
expression of β-catenin in the Wnt/β-catenin signaling pathway that
was promoted by the combination of emodin and AZT was significantly
higher compared with that of either drug alone. After transfection
of siRNA with the EGR1 gene, the expression of the β-catenin
protein in the Wnt/β-catenin signaling pathway was increased,
indicating that the EGR1 gene may regulate the Wnt/β-catenin
signaling pathway. The difference between the expected and the
actual result may be due to the higher expression of EGR1
induced by the two-drug combination, which may exert antitumor
effects directly or through regulating other downstream target
genes not investigated in the present study. We will assess the
expression of other related proteins in the Wnt/β-catenin signaling
pathway in our subsequent research.
In summary, the present study demonstrated that the
combination of emodin and AZT can enhance tumor cell proliferation
inhibition, apoptosis induction and decrease the expression of the
Wnt/β-catenin signaling pathway more efficiently compared with
emodin or AZT alone, which may be mediated by increased expression
of EGR1. However, it is unclear whether the therapeutic dose
in the combination therapy of the two drugs is a safe dose, and the
exact underlying mechanism has not been fully elucidated. We will
determine therapeutic dosage and explore the underlying mechanism
in CML clinical samples in our future research.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81460456) and the
Natural Science Foundation of Gansu Province (grant no.
1308RJZA169).
Availability of data and materials
All the datasets generated and analyzed in the
present study are included in this published article.
Authors' contributions
CC contributed to the experimental design. WM and LY
contributed significantly to conducting the experiments. FL
performed the data analyses and wrote the manuscript. CZ helped
perform the analysis with constructive discussion. All the authors
have read and approved the final version of this manuscript for
publication and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schiffer CA: BCR-ABL tyrosine kinase
inhibitors for chronic myelogenous leukemia. N Engl J Med.
357:258–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El Jurdi N, Bankoff M, Klein A and Saif
MW: Perforation of the Colon during imatinib mesylate (Gleevec)
treatment in a patient with chronic myeloid leukemia (CML). Cureus.
8:e6602016.PubMed/NCBI
|
|
4
|
Kantarjian H, O'Brien S, Cortes J, Wierda
W, Faderl S, Garcia-Manero G, Issa JP, Estey E, Keating M and
Freireich EJ: Therapeutic advances in leukemia and myelodysplastic
syndrome over the past 40 years. Cancer. 113:1933–1952. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen R, Zhang J, Hu Y, Wang S, Chen M and
Wang Y: Potential antineoplastic effects of aloe-emodin: A
comprehensive review. Am J Chin Med. 42:275–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ismail S, Haris K, Abdul Ghani AR,
Abdullah JM, Johan MF and Mohamed Yusoff AA: Enhanced induction of
cell cycle arrest and apoptosis via the mitochondrial membrane
potential disruption in human U87 malignant glioma cells by aloe
emodin. J Asian Nat Prod Res. 15:1003–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chun-Guang W, Jun-Qing Y, Bei-Zhong L,
Dan-Ting J, Chong W, Liang Z, Dan Z and Yan W: Anti-tumor activity
of emodin against human chronic myelocytic leukemia K562 cell lines
in vitro and in vivo. Eur J Pharmacol. 627:33–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin CH, Lin YM, Lai YL and Lee SY:
Mechanical properties, accuracy, and cytotoxicity of UV-polymerized
3D printing resins composed of BisEMA, UDMA, and TEGDMA. J Prosthet
Dent. 2019.(Epub ahead of print). View Article : Google Scholar
|
|
9
|
Jin RR, Chao R, Xi YM, Chen C, Chu HY, Li
M and Zhang H: Effects of AZT on leukemia cell line KG-1a
proliferation and telomerase activity. Zhongguo Shi Yan Xue Ye Xue
Za Zhi. 20:277–281. 2012.(In Chinese). PubMed/NCBI
|
|
10
|
Gu S, Chen C, Jiang X and Zhang Z:
Resveratrol synergistically triggers apoptotic cell death with
arsenic trioxide via oxidative stress in human lung adenocarcinoma
A549 cells. Biol Trace Elem Res. 163:112–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Gao P and Zheng J: Arsenic
trioxide inhibits cell proliferation and human papillomavirus
oncogene expression in cervical cancer cells. Biochem Biophy Res
Commun. 451:556–561. 2014. View Article : Google Scholar
|
|
12
|
Sun R, Eriksson S and Wang L:
Identification and characterization of mitochondrial factors
modulating thymidine kinase 2 activity. Nucleosides Nucleotides
Nucleic Acids. 29:382–385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mattson DM, Ahmad IM, Dayal D, Parsons AD,
Aykin-Burns N, Li L, Orcutt KP, Spitz DR, Dornfeld KJ and Simons
AL: Cisplatin combined with zidovudine enhances cytotoxicity and
oxidative stress in human head and neck cancer cells via a
thiol-dependent mechanism. Free Radic Biol Med. 46:232–237. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chau D, Ng K, Chan TS, Cheng YY, Fong B,
Tam S, Kwong YL and Tse E: Azacytidine sensitizes acute myeloid
leukemia cells to arsenic trioxide by up-regulating the arsenic
transporter aquaglyceroporin 9. J Hematol Oncol. 8:462015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bar-Shavit R, Turm H, Salah Z, Maoz M,
Cohen I, Weiss E, Uziely B and Grisaru-Granovsky S: PAR1 plays a
role in epithelial malignancies: Transcriptional regulation and
novel signaling pathway. IUBMB Life. 63:397–402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim JH, Liu X, Wang J, Chen X, Zhang H,
Kim SH, Cui J, Li R, Zhang W, Kong Y, et al: Wnt signaling in bone
formation and its therapeutic potential for bone diseases. Ther Adv
Musculoskelet Dis. 5:13–31. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wagner ER, Zhu G, Zhang BQ, Luo Q, Shi Q,
Huang E, Gao Y, Gao JL, Kim SH, Rastegar F, et al: The therapeutic
potential of the wnt signaling pathway in bone disorders. Curr Mol
Pharmacol. 4:14–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clevers H, Loh KM and Nusse R: Stem cell
signaling. An integral program for tissue renewal and regeneration:
Wnt signaling and stem cell control. Science. 346:12480122014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holland JD, Klaus A, Garratt AN and
Birchmeier W: Wnt signaling in stem and cancer stem cells. Curr
Opin Cell Biol. 25:254–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Undi RB, Gutti U, Sahu I, Sarvothaman S,
Pasupuleti SR, Kandi R and Gutti RK: Wnt signaling: Role in
regulation of haematopoiesis. Indian J Hematol Blood Transfus.
32:123–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu S, Li F, Xing S, Zhao T, Peng W and Xue
HH: Hematopoietic and leukemic stem cells have distinct dependence
on tcf1 and lef1 transcription factors. J Biol Chem.
291:11148–11160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jamieson CH, Ailles LE, Dylla SJ,
Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating
A, et al: Granulocyte-macrophage progenitors as candidate leukemic
stem cells in blast-crisis CML. New Engl J Med. 351:657–667. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fu J, Si L, Zhuang Y, Zhang A, Sun N, Li
D, Hao B and Ju X: Wnt/betacatenin inhibition reverses multidrug
resistance in pediatric acute lymphoblastic leukemia. Oncol Rep.
41:1387–1394. 2019.PubMed/NCBI
|
|
27
|
Yang W, Nam K, Ju JH, Lee KM, Oh S and
Shin I: S100A4 negatively regulates β-catenin by inducing the
Egr-1-PTEN-Akt-GSK3β degradation pathway. Cell Signal.
26:2096–2106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang PH, Huang CY, Chen MC, Lee YT, Yue
CH, Wang HY and Lin H: Emodin and aloe-emodin suppress breast
cancer cell proliferation through ER α inhibition. Evid Based
Complement Alternat Med. 2013:3761232013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han H, Li J, Feng X, Zhou H, Guo S and
Zhou W: Autophagy-related genes are induced by histone deacetylase
inhibitor suberoylanilide hydroxamic acid via the activation of
cathepsin B in human breast cancer cells. Oncotarget.
8:53352–53365. 2017.PubMed/NCBI
|
|
30
|
Liu H, Gu LB, Tu Y, Hu H, Huang YR and Sun
W: Emodin ameliorates cisplatin-induced apoptosis of rat renal
tubular cells in vitro by activating autophagy. Acta Pharmacol Sin.
37:235–245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z,
Wang S, Zhang G, Yang P, Cao Z and Ju D: Asparaginase induces
apoptosis and cytoprotective autophagy in chronic myeloid leukemia
cells. Oncotarget. 6:3861–3873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y and Zhang Y: Functional and
mechanistic analysis of telomerase: An antitumor drug target.
Pharmacol Ther. 163:24–47. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu A, Sha L, Shen Y, Huang L, Tang X and
Lin S: Experimental study on anti-metastasis effect of emodin on
human pancreatic cancer. Zhongguo Zhong Yao Za Zhi. 36:3167–3171.
2011.(In Chinese). PubMed/NCBI
|
|
34
|
Wei TN, Hu JD, Chen YY, Chen XJ, Liu TB
and Lu LH: Effect of emodin on induction of apoptosis in jurkat
cells and its possible mechanisms. Zhongguo Shi Yan Xue Ye Xue Za
Zhi. 17:1203–1206. 2009.(In Chinese). PubMed/NCBI
|
|
35
|
Tazzari PL, Cappellini A, Ricci F,
Evangelisti C, Papa V, Grafone T, Martinelli G, Conte R, Cocco L,
McCubrey JA and Martelli AM: Multidrug resistance-associated
protein 1 expression is under the control of the phosphoinositide 3
kinase/Akt signal transduction network in human acute myelogenous
leukemia blasts. Leukemia. 21:427–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Min H, Niu M, Zhang W, Yan J, Li J, Tan X,
Li B, Su M, Di B and Yan F: Emodin reverses leukemia multidrug
resistance by competitive inhibition and downregulation of
P-glycoprotein. PLoS One. 12:e01879712017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang J, Tang XM, Li H, Shi GY, Zhu P, Jin
HF and Yi J: Emodin sensitizes HeLa cell to arsenic trioxide
induced apoptosis via the reactive oxygen species-mediated
signaling pathways. Shi Yan Sheng Wu Xue Bao. 36:465–475. 2003.(In
Chinese). PubMed/NCBI
|
|
38
|
Zhao LM, Zhang LM, Liu JJ, Wan LJ, Chen
YQ, Zhang SQ, Yan ZW and Jiang JH: Synthesis and antitumor activity
of conjugates of 5-fluorouracil and emodin. Eur J Med Chem.
47:255–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang W, Sun YP, Huang XZ, He M, Chen YY,
Shi GY, Li H, Yi J and Wang J: Emodin enhances sensitivity of
gallbladder cancer cells to platinum drugs via glutathion depletion
and MRP1 downregulation. Biochem Pharmacol. 79:1134–1140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li J, Liu P, Mao H, Wanga A and Zhang X:
Emodin sensitizes paclitaxel-resistant human ovarian cancer cells
to paclitaxel-induced apoptosis in vitro. Oncol Rep.
21:1605–1610. 2009.PubMed/NCBI
|
|
41
|
Zeng Y, Liu A and Tong HF: Effect of
emodin combined gemcitabine on the growth and apoptosis of
pancreatic cancer cell line BxPC-3 in vitro. Zhongguo Zhong Xi Yi
Jie He Za Zhi. 31:552–554. 2011.(In Chinese). PubMed/NCBI
|
|
42
|
Sun Y, Wang X, Zhou Q, Lu Y, Zhang H, Chen
Q, Zhao M and Su S: Inhibitory effect of emodin on migration,
invasion and metastasis of human breast cancer MDA-MB-231 cells
in vitro and in vivo. Oncol Rep. 33:338–346. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Calogero A, Porcellini A, Lombari V,
Fabbiano C, Arcella A, Miscusi M, Ponti D and Ragona G: Sensitivity
to cisplatin in primary cell lines derived from human glioma
correlates with levels of EGR-1 expression. Cancer Cell Int.
11:52011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ferraro B, Bepler G, Sharma S, Cantor A
and Haura EB: EGR1 predicts PTEN and survival in patients with
non-small-cell lung cancer. J Clin Oncol. 23:1921–1926. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ragione FD, Cucciolla V, Criniti V, Indaco
S, Borriello A and Zappia V: P21Cip1 gene expression is modulated
by Egr1: A novel regulatory mechanism involved in the resveratrol
antiproliferative effect. J Biol Chem. 278:23360–23368. 2003.
View Article : Google Scholar : PubMed/NCBI
|