Introduction
Cancer cells consume large amounts of nutrients to
promote survival and drive tumor progression (1). Therefore, metabolic reprogramming with
substrate dependence represents an attractive target for cancer
treatment (2). The most distinct
metabolic cancer hallmark is a heightened dependence on glycolytic
flux (2). This feature is widely
exploited by positron-emission tomography imaging of tumors using
2-[18F]fluoro-2-deoxy-D-glucose (18F-FDG)
(3). Furthermore, enhanced
glycolysis of cancer cells can be therapeutically targeted by
2-deoxyglucose (2-DG). This glucose analogue is phosphorylated to
2-DG-6-phosphate, which competitively inhibits glucose-6-phosphate
from entering the glycolytic pathway (4,5). Thus,
cancer cells exposed to high 2-DG doses undergo growth arrest and
apoptotic death. However, targeting of glycolysis with 2-DG has
exhibited limited clinical efficacy (6), which likely reflects the metabolic
plasticity of cancer cells.
When glycolytic flux is blocked, cancer cells can
reciprocally increase oxidative metabolism to accrue sufficient
energy and biomass for growth and survival (7). In addition to 2-DG, thiazolidinediones
comprise a group of energy restriction mimetic agents that can
target cancer metabolism (4,8).
Recent studies support the repurposing of troglitazone (TGZ) as a
promising thiazolidinedione for cancer therapy (9). Our group previously demonstrated that
TGZ acutely impairs mitochondrial oxidative respiration in a
peroxisome proliferator-activated receptor-γ-independent manner
(10). The two pathways for glucose
metabolism (i.e., mitochondrial oxidative phosphorylation vs.
cytoplasmic lactate production) branch out from pyruvate. In a
recent study, pioglitazone was observed to block cytosolic pyruvate
from entering the mitochondria for oxidative phosphorylation in
hepatocytes (11). Import of
pyruvate across the inner mitochondrial membrane requires a
transport system known as the mitochondrial pyruvate carrier (MPC).
Cells stripped of MPC function have been reported to exhibit a
profound suppression of glucose oxidation and cell growth (12). Thus, TGZ may be an alternative
candidate to 2-DG in therapeutic targeting of cancer cells that are
more dependent on mitochondrial rather than glycolytic glucose
metabolism.
Another issue to consider is that depriving multiple
rather than a single bioenergetic pathway may be required to
achieve a sufficient cytotoxic effect in cancer cells with lower
dependency on mitochondrial glucose metabolism. The principal
substrate used by cancer cells deprived of glucose to fuel ATP
production and biomass formation is glutamine. Indeed, some cancer
cells obtain more than half of their ATP through oxidation of
α-ketoglutarate derived from glutamine (13). Other cancer cells may become
addicted to glutamine for survival (14). This suggests the potential
usefulness of combining glutamine deprivation with inhibition of
mitochondrial glucose metabolism for tumor treatment (15).
Successful targeting of tumor metabolism for imaging
and treatment may emerge from a better understanding of how
antimetabolic agents affect cancer cell glucose uptake and
survival. The present study investigated the effects of TGZ on
cancer cells with divergent sensitivity to 2-DG. T47D cells were
investigated, as TGZ was previously observed to exert strong
suppressive effects on the mitochondrial function and oxidative
phosphorylation of these cells (10). CT26 cells were selected as a cancer
cell type exhibiting low TGZ sensitivity based on pilot
experiments. Furthermore, the role of mitochondrial pyruvate
availability in these effects and the potential synergistic action
of glutamine deprivation were further explored.
Materials and methods
Cell culture and materials
The T47D human breast, HT29 human colon and CT26
mouse colon cancer cell lines (American Type Cell Culture), the
human colon cancer cell lines HCT15 and HCT116 and the human breast
cancer cell line MDA-MB-231 (Korean Cell Line Bank) were maintained
in RPMI-1640 medium (Gibco BRL; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Serana Europe GmbH), 2 mM
L-glutamine, and 100 U/ml penicillin-streptomycin in 5%
CO2 at 37°C. The HT29 cell line was authenticated at our
institution by short tandem repeat profiling. For T47D cells,
experiments were performed in phenol red-free media supplemented
with 5% charcoal-stripped serum to avoid the effects of trace
amounts of estrogen.
Cells were treated for the indicated durations with
2-DG (Sigma-Aldrich; Merck KGaA), TGZ (Biorbyt), UK5099 (Santa Cruz
Biotechnology, Inc.), and L-glutamic acid Υ-(p-nitroanilide)
hydrochloride (GPNA; Sigma-Aldrich; Merck KGaA) by addition to the
culture media. mPyr (Santa Cruz Biotechnology, Inc.) was added to
the culture media 4 h prior to treatment with TGZ or UK5099.
18F-FDG uptake
measurement
Cells in 12-well plates treated for the indicated
durations were incubated with 370 kBq 18F-FDG added to
the media. Following incubation for 30 min in 5% CO2 at
37°C, the cells were rapidly washed twice with cold
phosphate-buffered saline (PBS) and lysed with 0.1 N sodium
hydroxide. Cell-associated radioactivity was measured on a
γ-counter (PerkinElmer, Inc.) and expressed as % uptake relative to
the controls.
Lactate production assay
Culture media collected from cells was assayed for
L-lactate concentration with a Cobas kit (Roche/Hitachi) in
accordance with the manufacturer's instructions. This assay uses an
enzymatic reaction that converts lactate to pyruvate and hydrogen
peroxide. The hydrogen peroxide then undergoes an enzymatic
reaction to generate a colored dye that is measured by a
Roche/Hitachi analyzer.
Sulforhodamine-B (SRB) assay
Cell survival and proliferation were measured using
SRB assays. Briefly, cells were fixed with 10% (w/v)
trichloroacetic acid and stained with SRB for 30 min. After
appropriate dilution, unstained viable cells were counted using a
counting chamber and hemocytometer. After excess dye was removed by
repeated washing with 1% (v/v) acetic acid, protein-bound dye was
dissolved in 10 mM of Tris base solution and measured for optical
density at 510 nm on a microplate reader (Versa Max; Molecular
Devices) (16). In a pilot
experiment, SRB assays were performed to test the survival of
several cancer cell lines in response to a 48-h exposure to TGZ.
Other SRB experiments were performed after 48 or 72 h of
treatment.
Immunoblotting of activated
p70S6K
Cells were lysed with cold PRO-PREP lysis buffer
(Intron Biotechnology, Inc.) with a proteinase inhibitor cocktail
(Sigma-Aldrich; Merck KGaA) and a phosphatase inhibitor cocktail
(Thermo Fisher Scientific, Inc.). A total of 10 µg protein was
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and subsequently transferred to a polyvinylidene
difluoride membrane. The membrane was then blocked with 5% non-fat
milk in Tris-buffered saline with 0.5% Tween-20 for 1 h at room
temperature and incubated with a rabbit polyclonal antibody against
phospho-p70S6K (9205S; 1:1,000 dilution; Cell Signaling Technology,
Inc.) overnight. This was followed by incubation with
HRP-conjugated anti-rabbit IgG (cat. no. 7074S; 1:2,000 dilution;
Cell Signaling Technology, Inc.) at room temperature for 1 h.
Immune reactive proteins were detected with an enhanced
chemiluminescence system and measured by a GS-800TM calibrated
densitometer and Quantity One software 4.6.3 (Bio-Rad Laboratories,
Inc.). After visualizing phosphorylated proteins, the membranes
were subjected to a stripping procedure and were re-incubated with
rabbit polyclonal antibody against total protein (cat. no. 9202S;
1:1,000 dilution; Cell Signaling Technology, Inc.).
Statistical analysis
All data are presented as mean ± standard deviation.
The level of significance of the differences between groups was
analyzed using a Student's t-test for two groups and analysis of
variance with Tukey's post-hoc test for ≥3 groups. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
2-DG suppresses survival of CT26 cells
more efficiently compared with T47D cells
In a pilot experiment, 48 h treatment with 40 µM TGZ
reduced the survival of HCT15 and HCT116 human colon cancer cells
and MDA-MB-231 human breast cancer cells to 60.5±5.0, 37.9±4.8 and
76.8±4.8% of controls, respectively (Fig. S1). HT29 and CT26 mouse colon cancer
cells exhibited survival rates of 77.5±7.5 and 94.2±2.5% of
controls, respectively, by the same treatment (Fig. S1). Based on these results, CT26
cells were selected as a cell type with low TGZ sensitivity.
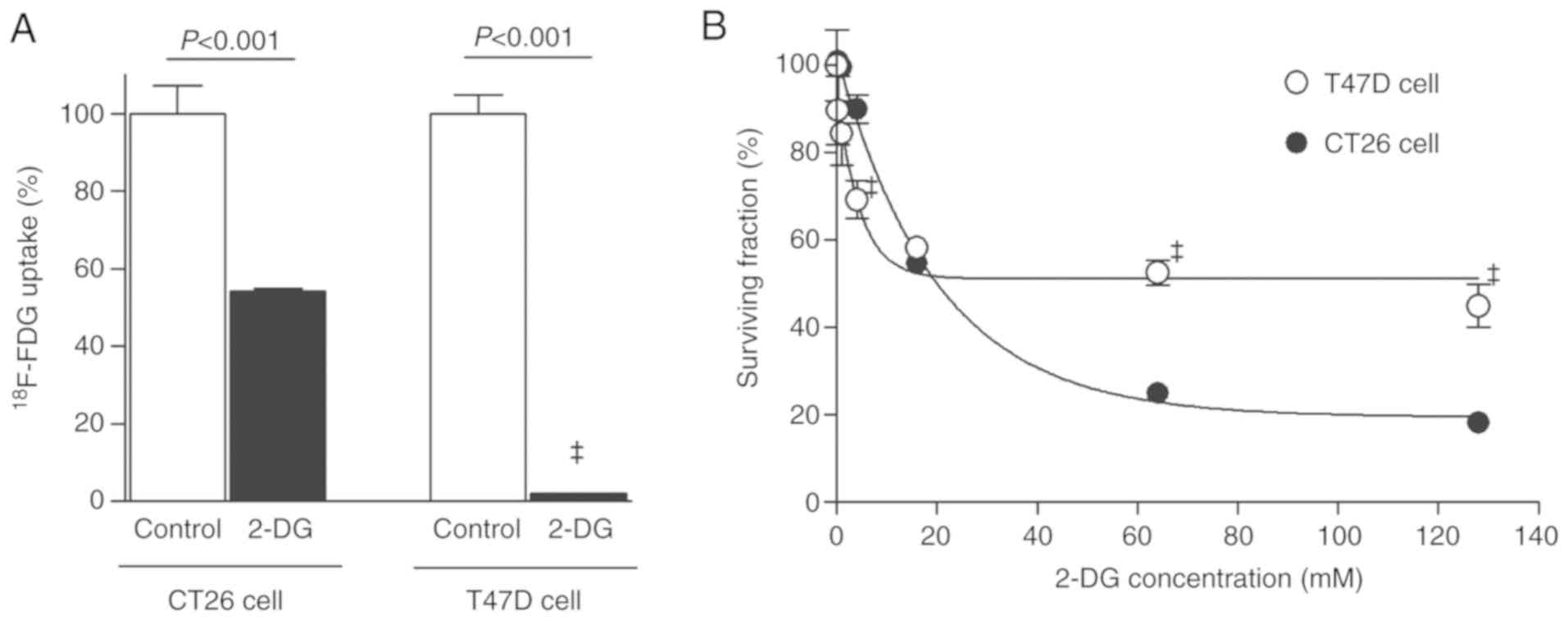
Treatment with 50 mM 2-DG for 1 h only modestly reduced
18F-FDG uptake by CT26 cancer cells to 54.2±0.7% of
controls, whereas it completely suppressed 18F-FDG
uptake of T47D cancer cells to 2.0±0.1% of controls (Fig. 1A). However, 2-DG suppressed the
survival of CT26 cells more potently compared with T47D cells.
Hence, 72 h of treatment with 64 mM 2-DG decreased the survival of
the two cell types to 25.0±0.8 and 52.6±2.6% of controls,
respectively (both P<0.0001; Fig.
1B).
These results revealed a dissociation between
2-DG-mediated glycolysis inhibition, which was more efficient in
T47D cells, and suppression of survival, which was more efficient
in CT26 cells.
TGZ stimulates glucose uptake and
suppresses survival more efficiently in T47D sells
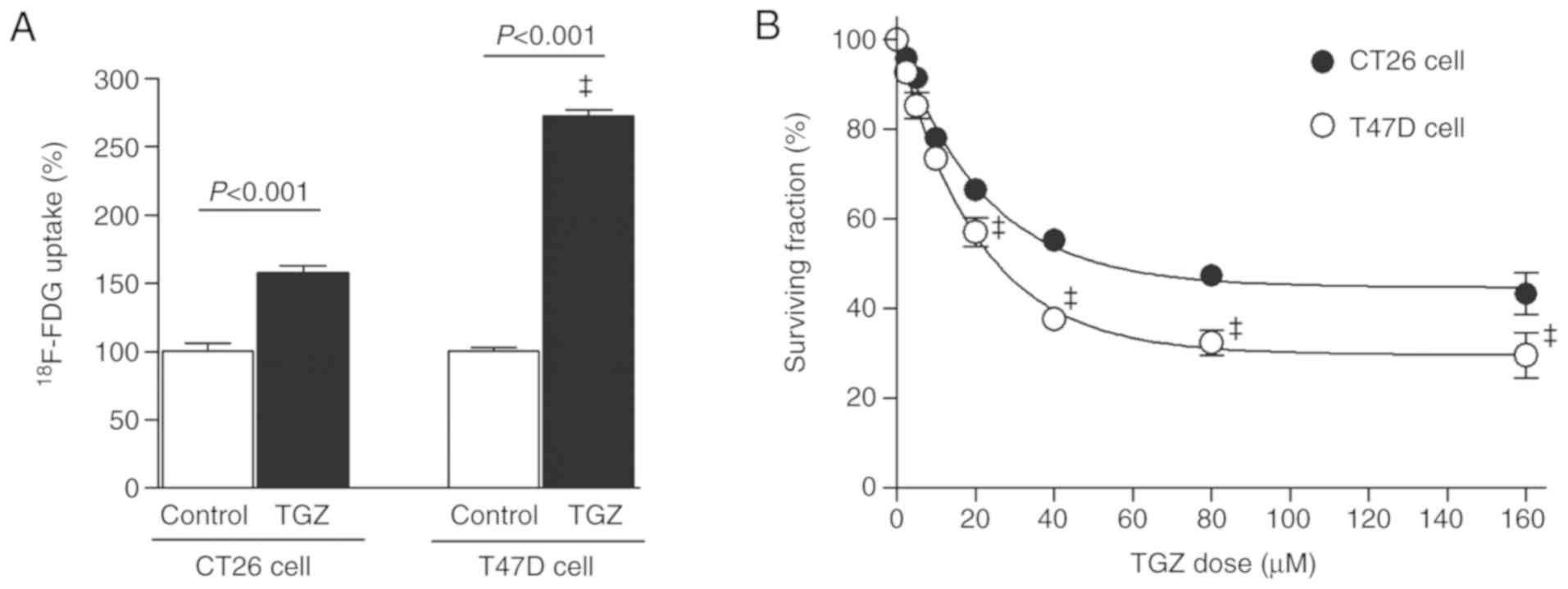
Treatment with 10 µM TGZ for 1 h rapidly increased
18F-FDG uptake of T47D cells to a significantly greater
extent (272.5±4.1% of controls) compared with that of CT26 cells
(157.8±4.9%; P<0.001; Fig.
2A).
The antitumor effect of TGZ on T47D cells was also
more prominent compared with that on CT26 cells. Thus, the
surviving fraction after 72 h of treatment with 80 µM TGZ was
37.7±1.9 and 55.2±1.1% of controls for the respective cell types
(both P<0.001; Fig. 2B).
The metabolic and cytotoxic effects of
TGZ are completely reversed by methyl pyruvate (mPyr)
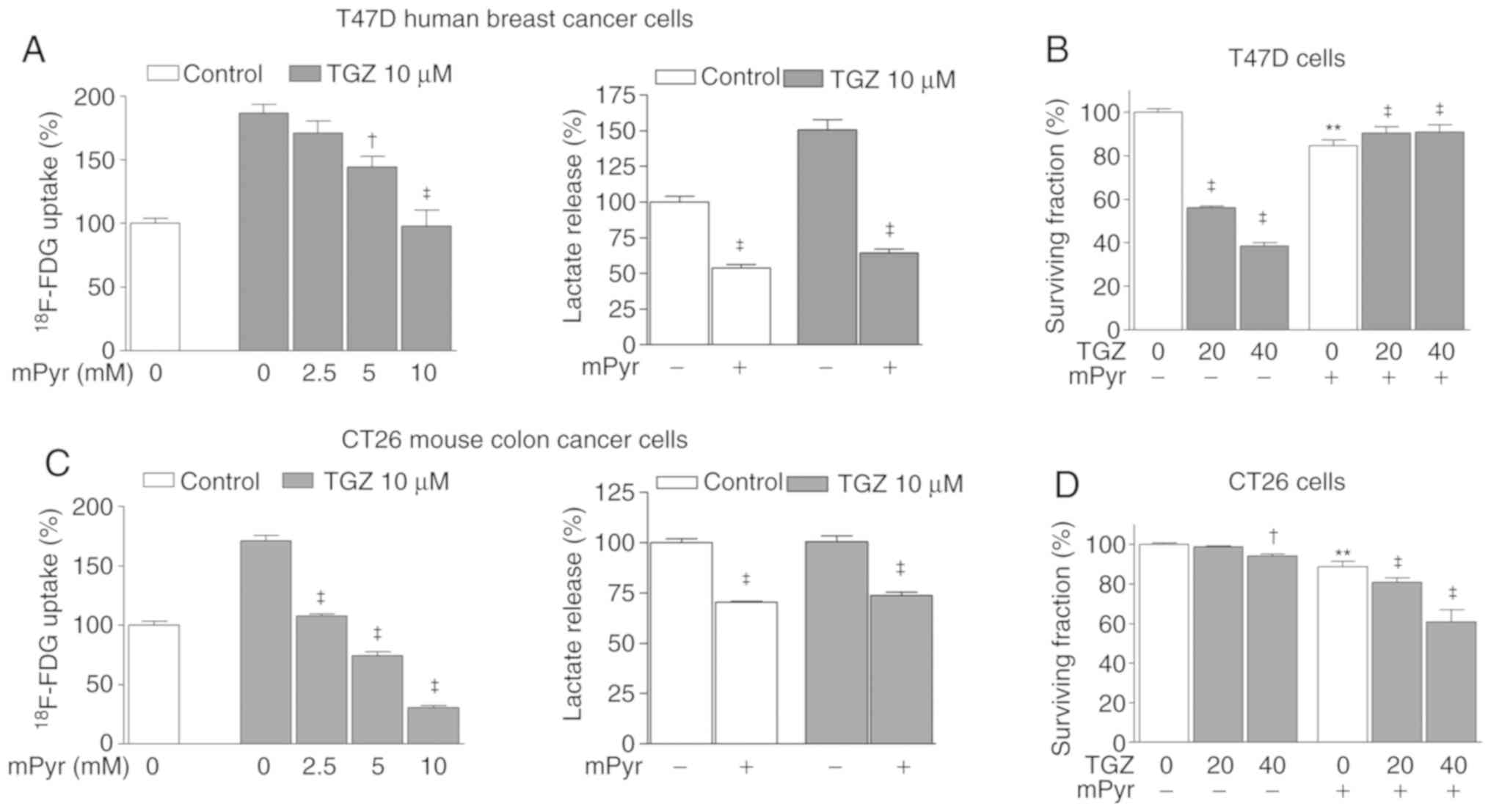
The role of mitochondrial pyruvate on the metabolic
and antitumor effects of TGZ on T47D and CT26 cells was next
investigated. Cells were treated with mPyr, a pyruvate derivative
that diffuses freely into the mitochondria independently of
carrier-mediated transport. The results revealed that graded doses
of mPyr completely abrogated the ability of TGZ to increase
18F-FDG uptake of both T47D and CT26 cells (Fig. 3A and C). Increased lactate
production of T47D cells to 150.5±7.3% of controls by TGZ was also
completely reversed by mPyr (Fig.
3A). Although increased lactate production by TGZ was less
evident in CT26 cells, mPyr reduced lactate production in both
TGZ-treated and control cells (Fig.
3C). Finally, while 40 µM of TGZ significantly reduced the
survival of T47D cells to 38.7±0.7% of controls, the same dose of
TGZ caused only a small reduction in CT26 cell survival.
Furthermore, while mPyr completely rescued T47D cells from the
anticancer effect of TGZ (Fig. 3B),
it further reduced the survival of TGZ-treated CT26 cells (Fig. 3D). This signifies a lack of
protective effect of mPyr in these cells.
The metabolic and antitumor effects of
TGZ on T47D cells are closely simulated by UK5099
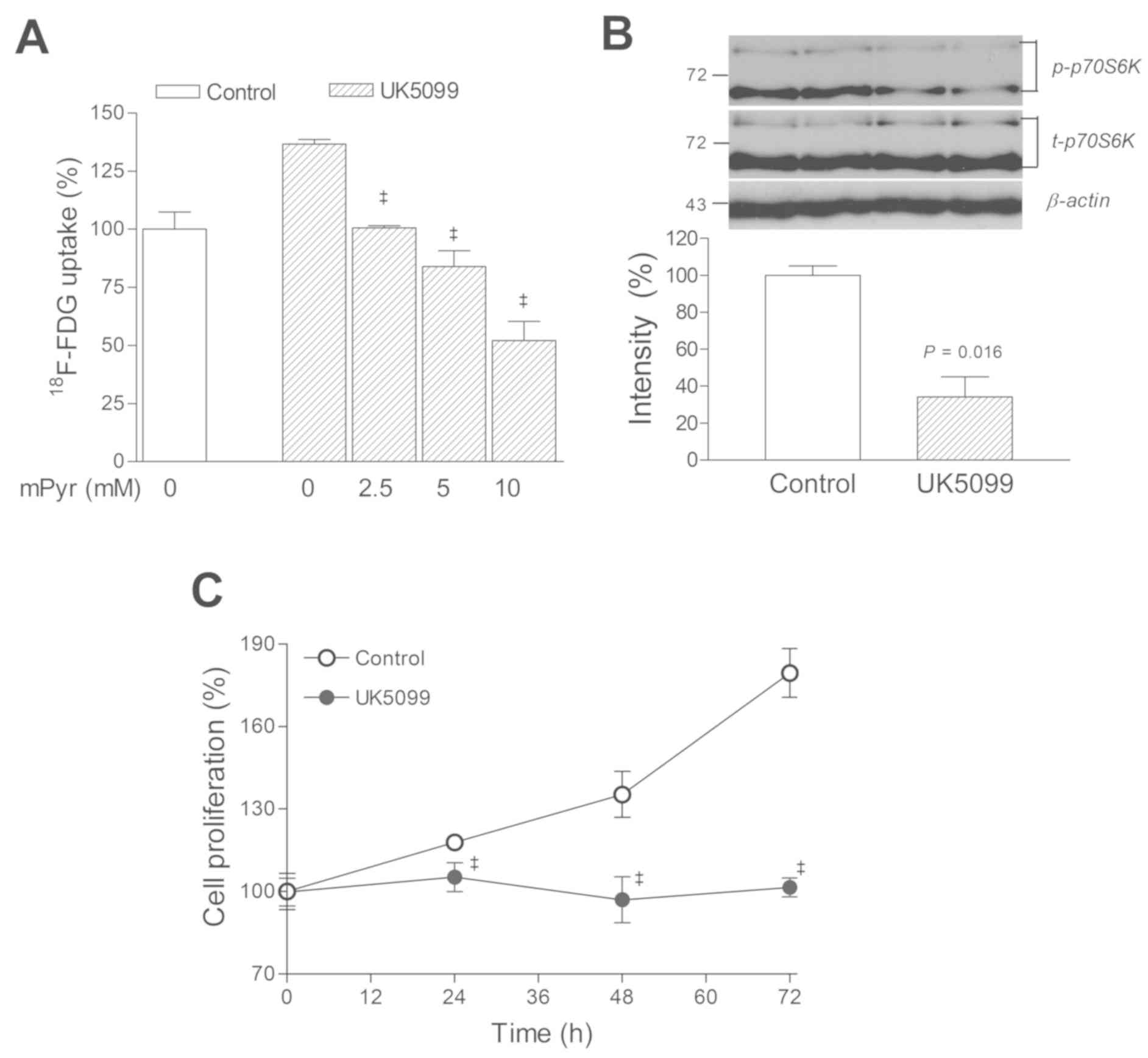
To verify whether blocking MPC function alone was
sufficient to increase glucose uptake and decrease the survival of
cancer cells, the specific MPC inhibitor UK5099 was used. In T47D
cells, the results of UK5099 treatment closely matched those
obtained by using TGZ. Hence, 18F-FDG uptake was
increased to 136.7±1.9% of controls following 1 h of exposure to
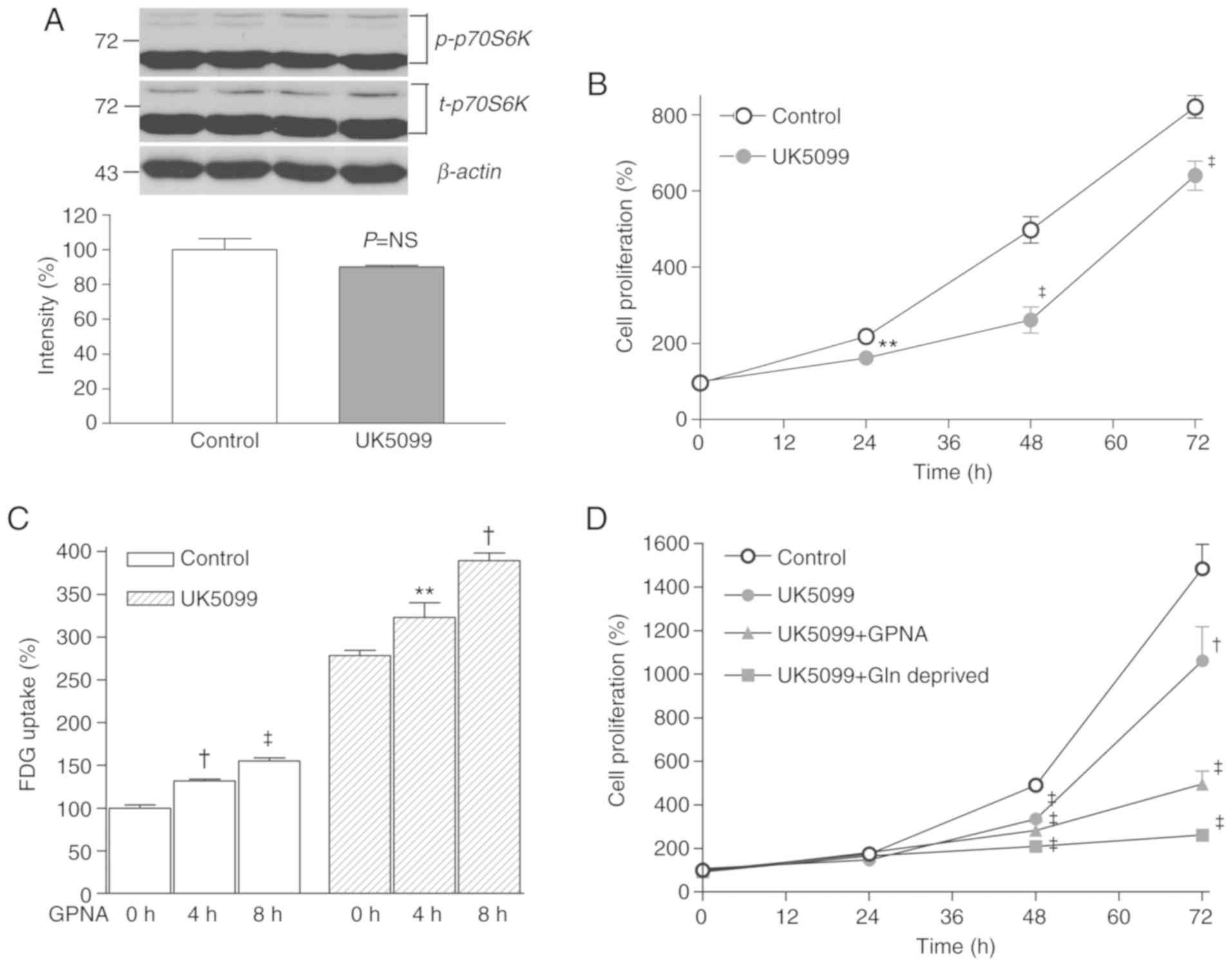
UK5099, and this effect was completely reversed by mPyr (Fig. 4A). Furthermore, treatment with 50 µM
UK5099 markedly reduced p70S6K activation to 27.4±8.7% of controls
(Fig. 4B) and completely blocked
cell proliferation (Fig. 4C).
Suppression of CT26 cell proliferation
requires glutamine restriction combined with UK5099 treatment
Subsequently, the effects of UK5099 on CT26 cells
that exhibited lower TGZ sensitivity were examined. The results
demonstrated that 50 µM of UK5099 did not decrease p70S6K
activation and only modestly suppressed the proliferation of CT26
cells (Fig. 5A).
Therefore, upon combining glutamine restriction with
UK5099 treatment in these cells, 18F-FDG uptake, which
was modestly stimulated by 50 µM UK5099, was further increased by
adding GPNA, a specific inhibitor of glutamine uptake through the
alanine-serine-cysteine-preferring transporter 2 (ASCT2) (Fig. 5B). Furthermore, the ability of 50 µM
UK5099 to suppress CT26 cell proliferation was significantly
potentiated by either adding GPNA or depleting glutamine from the
culture media (Fig. 5B).
Discussion
Tumor cells have characteristic metabolic
requirements that may represent attractive targets for cancer
therapy (1,2). In the present study, it was observed
that 2-DG inhibited glycolysis in T47D cells more prominently
compared with CT26 cells, but its cytotoxic effect was potent in
CT26 cells whereas it was only modest in T47D cells. This indicates
that T47D cell survival is less dependent on glycolytic metabolism
and suggests the possibility of a greater dependence on oxidative
phosphorylation.
The selection of T47D cells for our experiments was
based on our previous observation that oxidative phosphorylation in
T47D cells is strongly suppressed by TGZ treatment (10). This was accompanied by reduced
mitochondrial membrane potential and increased reactive oxygen
species generation, indicating a perturbation of mitochondrial
function. The present study was performed as a follow-up to
investigate the therapeutic potential of TGZ on these cells. CT26
cells were selected based on a pilot experiment, where various
cancer cell lines were tested, and CT26 cells exhibited the
smallest reduction in survival by TGZ treatment. Therefore, the
CT26 cell line selected to investigate the reason why its response
to TGZ differed from that of T47D cells. Although identification of
a human cancer cell line with a similar response to TGZ as CT26
cells would have been of interest, this was not attempted in the
present study.
The present study demonstrated that TGZ treatment
resulted in greater glucose uptake and cytotoxicity in T47D cells
compared with CT26 cells. These findings support the hypothesis
that T47D cells rely more on mitochondrial glucose metabolism for
survival.
When T47D cells were treated with mPyr, the
metabolic and antitumor effects of TGZ were completely reversed.
mPyr is a form of pyruvate that freely enters the mitochondria
without the need for carrier-mediated transport. A recent study
using hepatocytes demonstrated that pioglitazone inhibits pyruvate
import into the mitochondria (11).
Pyruvate is a key metabolite at the major junction of carbohydrate
metabolism between cytosolic glycolysis and the mitochondrial Krebs
cycle (17). Transport of pyruvate
through the inner mitochondrial membrane occurs through the
recently identified MPC (12,17).
Given the importance of oxidative pyruvate
metabolism for adequate ATP production and biomass conversion,
suppressing mitochondrial pyruvate availability may underlie the
antitumor effects exerted by TGZ. To compare the effects of
blocking mitochondrial pyruvate import, T47D cells were treated
with the specific MPC inhibitor UK5099. Similar to TGZ, UK5099
alone sufficiently augmented glucose uptake and glycolysis, and
this metabolic effect was completely abrogated by mPyr. Treatment
with UK5099 alone also completely blocked T47D cell proliferation.
These findings are reminiscent of a recent study wherein a specific
MPC inhibitor reduced tumor cell oxidative glucose metabolism and
extracellular lactate uptake, thereby leading to cytotoxicity
(18). Taken together, the findings
of the present study indicate that suppression of mitochondrial
pyruvate availability is the major mechanism through which TGZ
exerts metabolic and cytotoxic effects on T47D cells that exhibit a
greater dependency on mitochondrial metabolism.
In comparison, CT26 cells, which exhibited a greater
dependency on glycolysis and lower sensitivity to TGZ, displayed
only a modest reduction of proliferation in response to UK5099. It
was also observed that UK5099 suppressed the activation of p70S6K
in T47D cells, but not in CT26 cells. This serine/threonine kinase
is a downstream target of mechanistic target of rapamycin (mTOR)
signaling that is used as a marker of activation of the mTOR
pathway. Activation of p70S6K is important in mRNA translation that
is required for cell cycle progression, proliferation, and survival
pathways in tumors (6,19). A recent study demonstrated that the
level of activated p70S6K can predict the response to glycolysis
inhibition by identifying cancer cells that use mTOR signaling to
reprogram energy metabolism in order to survive (20). Therefore, maintenance of p70S6K
activation may have contributed to the ability of CT26 cells to
survive UK5099 treatment.
Depriving multiple key substrates of cancer
bioenergetics, rather than a single substrate, may be more
effective for tumor treatment (5).
It is well-known that cancer cells become more dependent on
glutamine metabolism when pyruvate is restricted or MPC function is
impaired (21). Glutamine
metabolism generates acetyl CoA and oxaloacetate, thereby
supporting tricarboxylic acid cycle activity. In CT26 cells, UK5099
alone stimulated glucose uptake and suppressed proliferation to
only a moderate extent, but these effects were significantly
enhanced by additional restriction of glutamine utilization. This
indicates that, in cells that manage to survive and grow under
conditions of low mitochondrial pyruvate availability, additional
glutamine restriction may achieve a more efficient antitumor
effect.
In conclusion, TGZ was effective against T47D cancer
cells with lower 2-DG sensitivity by suppressing mitochondrial
pyruvate availability. Although glycolysis-dependent CT26 cancer
cells were less responsive to TGZ or UK5099, antitumor efficacy was
enhanced by the combined suppression of glutamine utilization.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Science, ICT and Future Planning
(NRF-2018R1D1A1B07043260).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
Conceptualization: KHJ and KHL. Formal analysis:
KHJ, SHM, YSC and KHL. Funding acquisition: KHL. Methodology: JHL,
JWP and KHJ. Writing of the original draft: KHJ. Manuscript review
and editing: KHL. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All the authors declare that they have no competing
interests.
References
|
1
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vander Heiden MG: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Farwell MD, Pryma DA and Mankoff DA:
PET/CT imaging in cancer: Current applications and future
directions. Cancer. 120:3433–3445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuntz S, Mazerbourg S, Boisbrun M, Cerella
C, Diederich M, Grillier-Vuissoz I and Flament S: Energy
restriction mimetic agents to target cancer cells: Comparison
between 2-deoxyglucose and thiazolidinediones. Biochem Pharmacol.
92:102–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim EH, Lee JH, Oh Y, Koh I, Shim JK, Park
J, Choi J, Yun M, Jeon JY, Huh YM, et al: Inhibition of
glioblastoma tumorspheres by combined treatment with 2-deoxyglucose
and metformin. Neuro Oncol. 19:197–207. 2017.PubMed/NCBI
|
|
6
|
Pusapati RV, Daemen A, Wilson C, Sandoval
W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M and
Settleman J: mTORC1-dependent metabolic reprogramming underlies
escape from glycolysis addiction in cancer cells. Cancer Cell.
29:548–562. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swerdlow RH, E L, Aires D and Lu J:
Glycolysis-respiration relationships in a neuroblastoma cell line.
Biochim Biophys Acta. 1830:2891–2898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei S, Kulp SK and Chen CS: Energy
restriction as an antitumor target of thiazolidinediones. J Biol
Chem. 285:9780–9791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mazerbourg S, Kuntz S, Grillier-Vuissoz I,
Berthe A, Geoffroy M, Flament S, Bordessa A and Boisbrun M:
Reprofiling of troglitazone towards more active and less toxic
derivatives: A new hope for cancer treatment? Curr Top Med Chem.
16:2115–2124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moon SH, Lee SJ, Jung KH, Quach CH, Park
JW, Lee JH, Cho YS and Lee KH: Troglitazone stimulates cancer cell
uptake of 18F-FDG by suppressing mitochondrial respiration and
augments sensitivity to glucose restriction. J Nucl Med.
57:129–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shannon CE, Daniele G, Galindo C,
Abdul-Ghani MA, DeFronzo RA and Norton L: Pioglitazone inhibits
mitochondrial pyruvate metabolism and glucose production in
hepatocytes. FEBS J. 284:451–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vacanti NM, Divakaruni AS, Green CR,
Parker SJ, Henry RR, Ciaraldi TP, Murphy AN and Metallo CM:
Regulation of substrate utilization by the mitochondrial pyruvate
carrier. Mol Cell. 56:425–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reitzer LJ, Wice BM and Kennell D:
Evidence that glutamine, not sugar, is the major energy source for
cultured HeLa cells. J Biol Chem. 254:2669–2676. 1979.PubMed/NCBI
|
|
14
|
Yuneva M, Zamboni N, Oefner P,
Sachidanandam R and Lazebnik Y: Deficiency in glutamine but not
glucose induces MYC-dependent apoptosis in human cells. J Cell
Biol. 178:93–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takeuchi Y, Nakayama Y, Fukusaki E and
Irino Y: Glutamate production from ammonia via glutamate
dehydrogenase 2 activity supports cancer cell proliferation under
glutamine depletion. Biochem Biophys Res Commun. 495:761–767. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rampelt H and van der Laan M: Metabolic
remodeling: A pyruvate transport affair. EMBO J. 34:835–837. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Corbet C, Bastien E, Draoui N, Doix B,
Mignion L, Jordan BF, Marchand A, Vanherck JC, Chaltin P, Schakman
O, et al: Interruption of lactate uptake by inhibiting
mitochondrial pyruvate transport unravels direct antitumor and
radiosensitizing effects. Nat Commun. 9:12082018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang C, Ko B, Hensley CT, Jiang L, Wasti
AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al:
Glutamine oxidation maintains the TCA cycle and cell survival
during impaired mitochondrial pyruvate transport. Mol Cell.
56:414–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gibbons JJ, Abraham RT and Yu K: Mammalian
target of rapamycin: Discovery of rapamycin reveals a signaling
pathway important for normal and cancer cell growth. Semin Oncol.
36 (Suppl 3):S3–S17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hidalgo M and Rowinsky EK: The
rapamycin-sensitive signal transduction pathway as a target for
cancer therapy. Oncogene. 19:6680–6686. 2000. View Article : Google Scholar : PubMed/NCBI
|