Introduction
Esophageal cancer is ranked as the ninth most
commonly diagnosed cancer and the sixth leading cause of
cancer-associated mortality worldwide, with an estimated 572,034
new cases (3.2% of the total) and 508,585 deaths (5.3% of the
total) in 2018 (1). It has two main
subtypes: Esophageal squamous cell carcinoma (ESCC) and esophageal
adenocarcinoma (EAC), which are epidemiologically and biologically
different. Although EAC is more common in the USA and several
countries across Europe, ESCC is the predominant histology subtype
of esophageal cancer globally, and occupies >80% of cases of
esophageal cancer (2). Currently,
surgery is the definitive treatment method for early stage
esophageal cancer. For those patients with advanced/unresectable
tumors, neoadjuvant chemoradiotherapy has become the standard
treatment option (3,4). Even for those early stage patients,
pre/post-operative chemoradiotherapy is also widely practiced in
the clinical setting. Although some promising outcomes have been
observed in clinic trials with neoadjuvant or adjuvant therapy
(5–7), the overall 5-year survival of this
disease still ranges from 15 to 25% (4), and resistance to chemoradiotherapy is
one of the primary reasons for this. Thus, an improved
understanding of the molecular mechanisms that affect tumor cell
sensitivity to radiotherapy and chemotherapy would be beneficial
for future personalized treatment plans to improve patient
survival.
MicroRNAs (miRNAs/miRs) are a class of small
noncoding RNAs that regulate gene expression at the
post-transcriptional level, and play important roles in various
biological processes, including the development of diseases
(8). One of the most important
tumor-promoting miRNAs is miR-155, which is processed from the B
cell integration cluster (9). It
has been revealed that aberrant expression of miR-155-5p presents
as an oncogenic feature in several types of hematological
malignancies and solid tumors (10–14),
including ESCC (15,16). It could promote cell proliferation,
inhibit apoptosis, and induce EMT, invasion and migration, tumor
metastasis and recurrence (17–19).
Meta-analyses have demonstrated that miR-155 could be a potential
biomarker for lung cancer detection (20), and the combined detection of
multiple miRNA levels in ESCC tissues have significant prognostic
values (21). However, conflicting
studies have been published regarding the role of its dysregulation
in radio- and chemo-resistance. For example, interference of
miR-155-5p caused resistance to chemotherapy drugs and ionizing
radiation in parental human epidermoid carcinoma cells (22) and breast cancer (23), respectively. However, the opposite
behaviors have also been observed, indicating a greatly increased
sensitivity to chemotherapy drugs (11,24)
and radiation (25).
Given that there has been little data on the
potential role of miR-155-5p in the radio- and chemo-resistance of
ESCC cells, the present study determined the endogenous miR-155-5p
expression levels and chemoradio-resistance profiles in ESCC cell
lines. The results revealed that miR-155-5p was positively
correlated with radio- and chemo-resistance. A systematic analysis
was subsequently performed in order to reveal its role in response
to radiation and drugs, and the underlying mechanism. The data
generated in the present study are helpful for finding effective
targets for ESCC chemo-radio sensitization.
Materials and methods
Cell culture and transfection
The human esophageal squamous cancer cell lines
(KYSE-30, KYSE-140, KYSE-410, KYSE-450, KYSE-510 and TE-1), which
were kindly provided by Professor Zhan (National Laboratory of
Molecular Oncology), were cultured in RPMI-1640 medium plus 10%
fetal bovine serum (cat. no. 10099-141; Thermo Fisher Scientific,
Inc.) at 37°C in 5% CO2. All cell lines were genetically
authenticated using STR profiling by Genesky Biotechnologies,
Inc.
Mimic/antagomiR/siRNA/plasmid DNA
transfection
All mimics, antagomiR, siRNA and the scramble
sequence control (NC), as well as riboFECT CP transfection kit
(cat. no. C10511-05) were obtained from Guangzhou Ribobio Co., Ltd.
Briefly, 4×105 cells were seeded into each well of
6-well plates and cultured overnight, and then they were
transfected with 50 nM mimic/siRNA, or 100 nM antagomir using a
riboFECT CP transfection kit. A total of 1.2 µg of the GFP-tagged
overexpression MAP3K10 construct (cat no. HBLV-MAP3K10-GFP, Hanbio
Biotechnology Co., Ltd.) was transfected into KYSE-410 cells using
Attractene transfection reagent (cat no. 301005; Qiagen) according
to the manufacturer's instructions. All transfections were carried
out at room temperature, after which cells were cultured at 37°C
for 24 h. Then they were seeded into 96-well or 6-well plates and
underwent subsequent analysis 24 h later.
The siRNA sequences used for MAP3K10 interference in
the present study were as follows (5′→3′): CCUGGAAACUGGUCUCCUUdTdT
and dTdTGGACCUUUGACCAGAGGAA.
Clonogenic survival assay for
radiation
ESCC cells in the exponential growth phase were
seeded at a density of 250 (0 Gy), 500 (1 Gy), 1,000 (2 Gy), 2,000
(4 Gy), 4,000 (6 Gy) cells/well on six-well plates in triplicate.
After 24 h of incubation, adhesive cells were exposed to a 6 MV
X-ray in CX-SN5340 (VARIAN) at 0 Gy, 1 Gy, 2 Gy, 4 Gy and 6 Gy with
an average dose rate of 300 cGy/min. After incubation for an
additional 14 days, the cultures were fixed in methanol and stained
with crystal violet. The number of colonies containing >50 cells
were counted under a light microscope. The surviving fraction was
calculated as previously described (26).
Chemoresistance profiling
(IC50 determination)
Vinorelbine (Changchun Guoao Pharmaceutical Co.,
Ltd.), paclitaxel (Sichuan Taiji Pharmaceutical Co., Ltd.),
docetaxel (Jiangsu Aokangsai Pharmaceutical Co., Ltd.),
5-flurorocil (Tianjin Jinyao Pharmaceutical Co., Ltd.), mitomycin
(Zhejiang Haizheng Pharmaceutical Co., Ltd.), nedaplatin (Jiangsu
Aokangsai Pharmaceutical Co., Ltd.) and cisplatin (Jiangsu Haosen
Pharmaceutical Co., Ltd.) at the clinical-grade (NCI Dictionary of
Cancer Terms, http://www.cancer.gov/dictionary) were used in the
present study. Chemoresistance profiling (IC50
measurements): Cells in the logarithmic growth phase were seeded in
triplicate in 96-well plates at a density of 5×103/well
and treated with 4-fold serially diluted drugs for 72 h. Cell
survival was then measured using a Cell Counting Kit-8-based
(CCK-8; cat. no. B34302; Bimake) cell proliferation assay. The
IC50 (the concentration of drug required for 50% of the
cells to be killed) was calculated with the no-drug control as the
reference.
Cell proliferation assay
Cells in the logarithmic growth phase were seeded in
96-well plates at a cell density of 2×103/well (in
triplicate) to allow adhesion. At 0, 24, 48, 72 and 96 h, cells
were incubated with 10 µl CCK-8 at 37°C for an extra 2 h. The
optical density was then measured with a microplate reader (Tecan
Group Ltd.) at 450 nm. The cells were then cultured with fresh
medium until the next round of measurements. The mean and standard
deviation of the triplet measurements were calculated and
plotted.
Wound-healing assay
Confluent cells were serum-starved in RPMI-1640
medium for 10–12 h and scratched using the tip of a 10-µl pipette.
After being washed twice with PBS to remove non-adherent cells, the
plates were changed to 500 µl RPMI-1640 medium plus 10% FBS. The
wound area was photographed at 0 and 22 h under an Olympus IX73
inverted microscope. A cell-free region was drawn and measured by
CellSens Standard software (Olympus). The average and standard
deviation were calculated from no less than three different wounds
from one of three attempts.
Invasion assay
A BioCoat™ Matrigel invasion chamber (cat. no.
40480; BD Biosciences) was used according to the manufacturer's
protocol. Briefly, 4×104 cells were trypsinized, washed,
suspended in 200 µl serum-free RPMI-1640 medium, and seeded in the
upper portion of the invasion chamber. The lower portion of the
chamber contained 500 µl of RPMI-1640 medium plus 10% FBS, which
served as a chemo-attractant. After 36 h, the non-invasive cells
were removed from the upper surface of the membrane with a cotton
swab. The invasive cells on the lower surface of the membrane were
stained with 0.1% crystal violet for 30 min at room temperature,
and counted in four separate areas with an inverted microscope.
miRNA target prediction
Two miRNA target prediction and functional study
databases, TargetScan (http://www.targetscan.org/) and miRDB (http://mirdb.org/), were employed to search for the
potential targets of miR-155-5p. The overlapped targets were
selected for further validation.
Luciferase reporter assay
A full length of the human MAP3K10 3′-UTR region
(284 bp) with the miR-155-5p targeting sequence was cloned at the
downstream region of the firefly luciferase gene in the pGL3-basic
vector (cat. no. E1751; Promega Corporation) to construct
pGL3-luc-MAP3K10.
Cells were seeded into 96-well plates at
~1×104 cells/well and transfected with a mixture of 100
ng pGL3-luc-MAP3K10, 10 ng Renilla plus 5 pmol mimic or
scrambled control (NC) nucleotides, with the riboFECT CP
transfection reagents according to the manufacturer's protocol.
Both firefly and Renilla luciferase activities were assessed
18 h after transfection by the Dual-Luciferase Reporter Assay
system (cat. no. E1910; Promega Corporation) using a Promega GloMax
20/20 luminometer. The relative firefly luciferase activities of
the UTR constructs and C-Jun N-terminal kinase (JNK) pathway
reporter constructs (cat. no. CCA-901L; Qiagen) were analyzed as
previously described (27).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cells at the
logarithmic growth phase using TRNzol-A+ reagent (cat. no. DP421;
Tiangen Biotech Co., Ltd.), and 1 µg RNA was converted to cDNA with
a 100-nM mixture of miR-155 specific stem-loop primer and U6
specific reverse primers (synthetized by ShingGene) using the
HiScript II 1st Strand cDNA Synthesis kit (cat. no. R211-01;
Vazyme) for a 20-µl reaction. The steady state of the miR-155-5p
and U6 was simultaneously quantified using a dual-RT-qPCR assay
with the differentially fluorescent-labeled TaqMan probes for
miR-155-5p (FAM) and U6 (HEX) (ShingGene) and a FTC-3000P PCR
instrument (Funglyn Biotech, Inc.). Briefly, 3.125 mM
Mg2+, 0.2 mM dNTP, 6.25 U Hotstart Taq DNA polymerase, 1
µM Forward Primer, 1 µM Reverse Primer and 0.2 µM TaqMan Probe were
used for each round of RT- PCR. The conditions used were as
follows: 95°C for 5 min, followed by 40 cycles at 95°C for 15 sec,
and 60°C for 1 min (28). The
relative expression level of miR-155-5p was normalized to U6 using
the ΔΔCq method (29). The
sequences of the primers and probes were listed as follows (5′→3′):
miR-155-5p RT, GCGCGTGAGCAGGCTGGAGAAATTAACCACGCGCACCCC, miR-155-5p
forward, TCGTTAATGCTAATCGTG, reverse, GAGCAGGCTGGAGAA and probe,
FAM-ACCACGCGCACCC; U6 forward, CTCGCTTCGGCAGCACATA, RT and reverse,
CGCTTCACGAATTTGCGTG and probe, HEX-CCTTGCGCAGGGGCCATGC.
Western blot analysis
Cells were lysed in a solution of 60 mM Tris-HCl, pH
6.8, 2.00% sodium dodecyl sulfate, 20.00% glycerol, 0.25%
bromophenol blue, 1.25% 2-mercaptoethanol and heated at 100°C for
10 min. Protein concentrations were determined using a BCA protein
assay kit. After being separated by 10 or 12% SDS-PAGE, the protein
(40–50 µg) was transferred to a PVDF membrane (cat. no. IPVH00010;
EMD Millipore). The membranes were blocked in 1X PBS buffer
containing 5% BSA (cat. no. A1933; Sigma-Aldrich; Merck KGaA) and
0.05% Tween-20 (cat. no. A100777; Sangon Biotech Co., Ltd.) for 1 h
at room temperature, and then incubated with the following primary
antibodies: γ-H2AX (rabbit anti-human monoclonal; 1:1,000; cat. no.
9718; Cell Signaling Technology, Inc.); H2AX (rabbit anti-human
polyclonal; 1:1,000; cat. no. 10856-1-AP); Lamin B1 (rabbit
anti-human polyclonal; 1:1,000; cat. no. 12987-1-AP); RAD51 (rabbit
anti-human polyclonal; 1:1,000; cat. no. 14961-1-AP); and Ku80
(rabbit anti-human polyclonal; 1:1,000; cat. no. −1-AP; all from
ProteinTech Group, Inc.); MAP3K10 (sheep anti-human polyclonal;
1:1,000; cat. no. AF5066; R&D Systems); GAPDH (mouse anti-human
monoclonal; 1:2,000; cat. no. 60004-1-Ig); and α-tubulin (mouse
anti-human monoclonal; 1:2,000; cat. no. 11224-1-AP; both from
ProteinTech Group, Inc.) overnight at 4°C. The blots were washed
with PBST three times for 10 min each and incubated with secondary
antibodies anti-rabbit IgG (1:3,000; cat. no. SA00001-2); and
anti-mouse IgG (1:3,000, cat. no. SA00001-1; both from ProteinTech
Group, Inc.; and anti-sheep IgG, 1:1,000, cat. no. HAF016, R&D
Systems) for 1 h at room temperature. The target bands were
revealed by SuperSignal West Pico PLUS chemiluminescence substrate
(cat. no. 34580; Thermo Fisher Scientific, Inc.), and the relative
density of each protein over Lamin B1, GAPDH or α-tubulin was
quantified using a Gel-Pro Analyzer 3.1 (Media Cybernetics).
5-Aza-2′-deoxycytidine treatment
KYSE-140 and KYSE-30 cells were treated with 50 mM
5-aza-2′--deoxycytidine (5-aza-dC; cat. no. A3656; Sigma-Aldrich;
Merck KGaA) for 72 h with a change of culture medium every 24 h as
previously described (30).
BSP analysis
Genomic DNA was isolated using a PureGenome™ kit
(cat. no. P-9040-M; Aline Bioscience) and quantitated via
electrophoresis on an agarose gel. The bisulfate conversion was
achieved using an EZ DNA Methylation-Gold kit (cat. no. D5006; ZYMO
Research). The CpG island upstream of miR-155 gene was amplified by
two pairs of primers. The sequences of the primers were listed as
follows (5′→3′): 1st forward, GTTTGGTYGGTTATGAGTTATAAGTGAG and
reverse, CAAAAACRTCTCCTTAATTCCCC; 2nd forward
AAGGAGAYGTTTTTGGTATTGTAGG and reverse, GACACCACTAAATCCCCAAAAAAC.
Briefly, 500 nM of each primer, 4 mM dNTP, 2 mM MgCl2,
6.25 U Hotstart Taq DNA polymerase and 2% DMSO were used for each
round of PCR. The conditions used were as follows: 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec, and 60°C for 1 min. The
PCR fragments from the converted DNA were cloned and verified by
sequencing as previously described (31).
Online data for gene expression in
esophageal cancer
A ESCC cohort from The Cancer Genome Atlas (TCGA)
was included for survival rate analysis using Kaplan-Meier survival
analysis. Oncomine database (https://www.oncomine.org/resource/login.html) was used
to mine the data of MAP3K10 gene expression in ESCC and EAC.
Statistical analysis
Data are presented as the means, and error bars
indicate the standard deviation (SD). All statistical analyses were
performed with Excel (Microsoft, Inc.) or GraphPad Prism 6
(GraphPad Software Inc.). One-way ANOVA followed by Dunnett's post
hoc test and two-tailed Student's t-test were used to
calculate statistical significance. A P-value of <0.05 was
considered to indicate a statistically significant difference.
Results
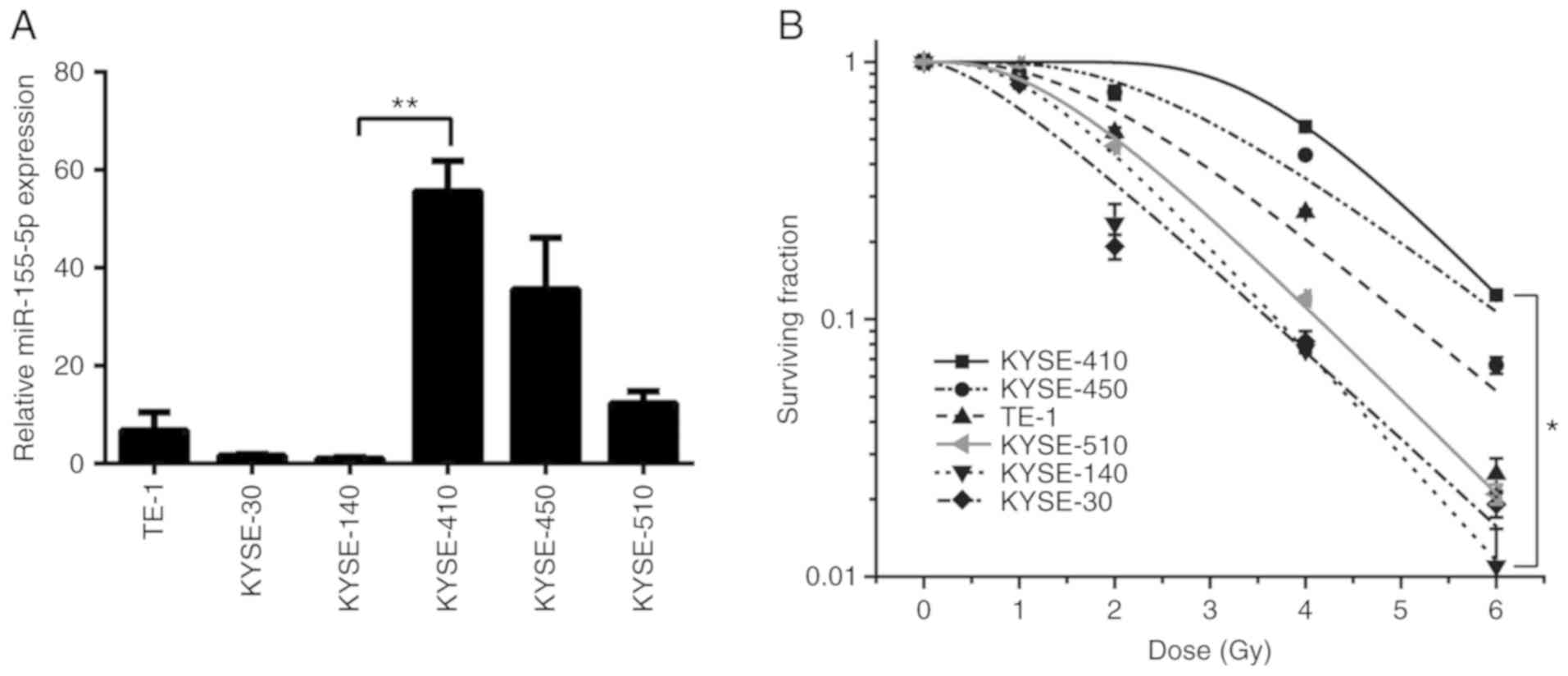
miR-155-5p is differentially expressed
in ESCC cells and positively associated with radioresistance
The expression level of miR-155-5p was examined in
six ESCC cell lines via RT-qPCR (Fig.
1A). The results revealed that the expression of miR-155-5p
varied among the different cells, with KYSE-410 cell expression
~55.3-fold higher than KYSE-140 cells. As radiotherapy is widely
applied to patients with ESCC and has a central role in the
therapeutic strategy against ESCC, the sensitivity of six ESCC cell
lines to radiation was then evaluated using a clonogenic survival
assay (Fig. 1B). The surviving
fraction of KYSE-410 cells was greater than that of the other five
cell lines, indicating that KYSE-410 was the most radioresistant,
whereas, KYSE-30 and KYSE-140 presented with the highest
radiosensitivity. These indicated a positive association between
radioresistant capacity and the expression of miR-155-5p in ESCC
cells. Thus, KYSE-140 (the most radiosensitive cells whose miR-155
was the lowest) and KYSE-410 (the most radioresistant cells whose
miR-155 is the highest) were selected cell lines for the following
study.
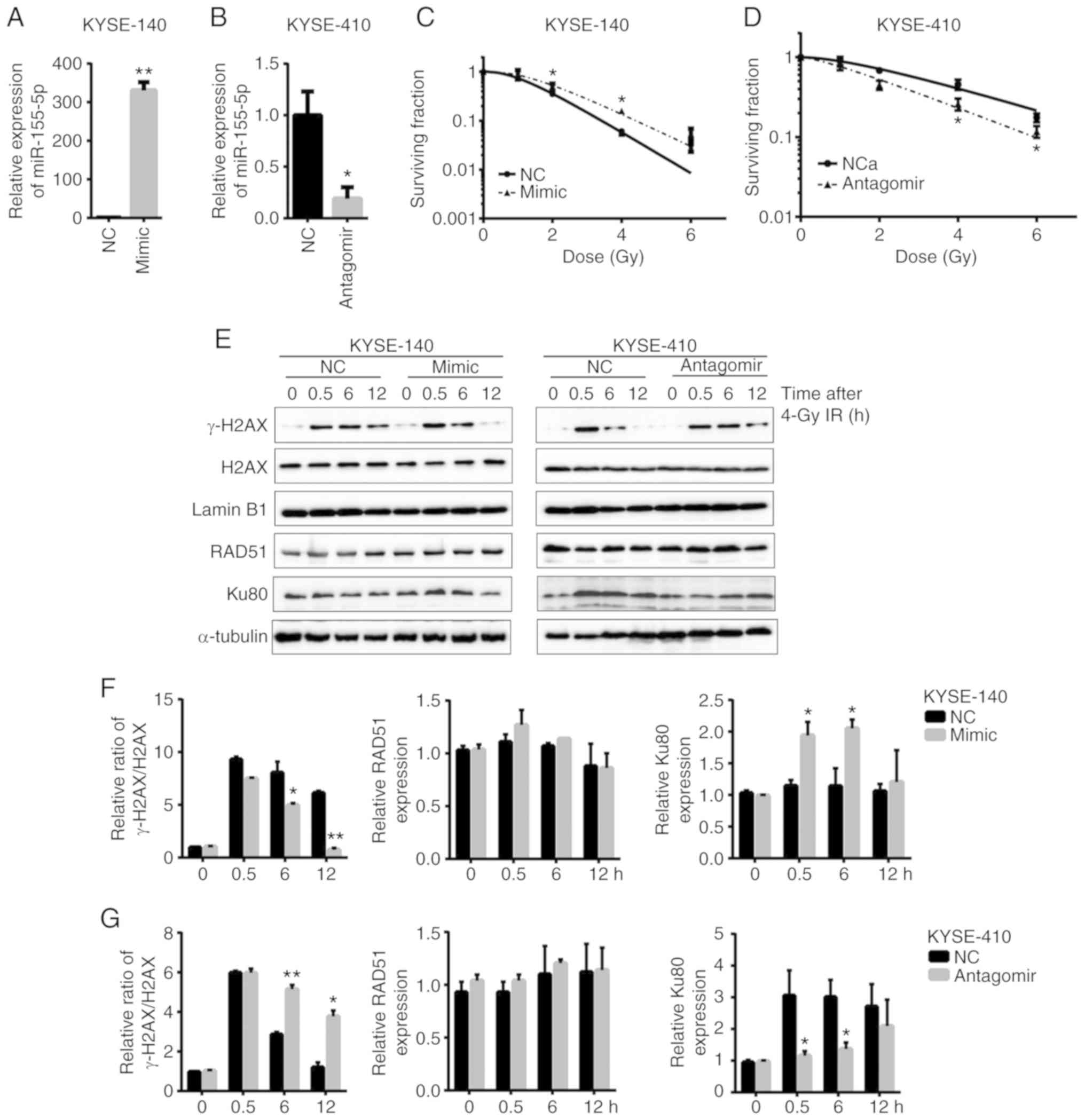
miR-155-5p renders ESCC cells
resistant to radiation by repairing the DNA damage more
efficiently
In order to investigate the role of miR-155-5p in
the radiotherapy response of ESCC cells, the present study
ectopically expressed miR-155-5p and the corresponding control
scrambled RNA in radiosensitive KYSE-140 cells, which were then
irradiated after seeding on cell culture plates for clonogenic
survival assays. Following overexpression of miR-155-5p to
>300-fold (Fig. 2A), the
survival rate of the mimic-transfected KYSE-140 cells at each dose
was higher than that of NC (Fig.
2C), indicating that miR-155-5p enhanced radioresistance.
Conversely, silencing of miR-155-5p to 20% in KYSE-410 cells
(Fig. 2B) decreased the cell
survival rate against radiation (Fig.
2D), indicating that inhibition of miR-155 sensitized ESCC
cells to radiation.
It has previously been proposed that ionizing
radiation damages tumor cells through several mechanisms, mainly by
DNA damage, particularly double-strand breaks (DSBs) (32,33).
Cell survival following DNA damage relies on DNA repair, the
abrogation of which causes genomic instability and cell death. In
order to confirm that a defect in the repair of DSBs is involved in
the radioresistance mediated by miR-155-5p in ESCC cells, cells
were exposed to X-rays at 4 Gy in the present study. The expression
level of phosphorylated histone family member X (γ-H2AX), which is
a powerful biomarker to monitor DSBs in cells (34), was detected at various time-points
after radiation. Significant induction of γ-H2AX was observed at
0.5 h after radiation compared to the cells without radiation. It
decreased more rapidly from 6 h post-radiation in miR-155-5p
overexpressing-KYSE-140 cells, compared to the NC group.
Conversely, the level of γ-H2AX decreased more slowly in the
anti-miR-155-5p KYSE-410 cells from 6 h after radiation, compared
to the NC group (Fig. 2E-G). This
revealed that there was an early onset and high capacity of DNA
repair by upregulation of miR-155-5p.
DSB induced in mammalian cells is repaired by two
repair pathways. One is non-homologous end joining (NHEJ), the
other is homologous recombination (HR). Deficiency in proteins
involved in the DNA damage repair is considered a major determinant
of response to radiotherapy and chemotherapy (35). Thus, the present study investigated
whether increased expression and/or activity of DNA repair proteins
confer resistance to radiation. The protein levels of RAD51 and
Ku80, the key components of HR and NHEJ, respectively, were
examined. The results revealed the expression of Ku80 was increased
from 0.5 h after radiation in the miR-155-5p mimic-transfected
KYSE-140 cells, but it remained almost unchanged in the NC group.
Consistently, accumulation of Ku80 occurred at 0.5 h after
radiation in KYSE-410 cells, but its level was significantly lower
in anti-miR-155 KYSE-140 cells compared to the NC group (Fig. 2E-G). Thus, NHEJ rather than HR
played a major role in repairing the DSB induced by radiation in
ESCC cells. Collectively, miR-155-5p promoted DNA damage repair and
induced resistance against radiation via upregulation of Ku80 and
activation of NHEJ repair.
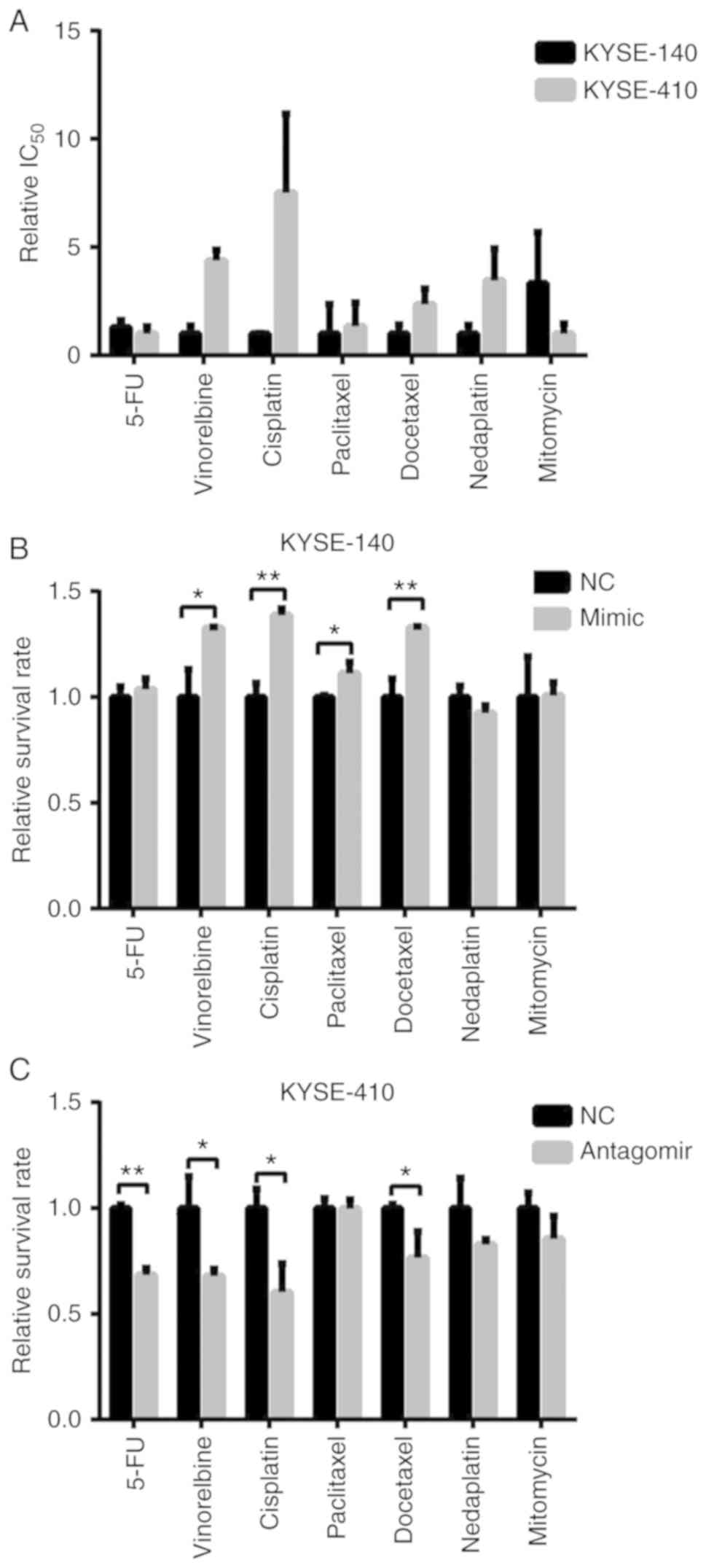
Increased expression of miR-155-5p
promotes multi-drug resistance in ESCC cells
Generally, resistance occurs not only to radiation
but also to traditional chemotherapeutic drugs in cancer cells.
Thus, the present study performed drug-resistance profiling in two
cell lines against the following drugs: Paclitaxel, docetaxel,
vinorelbine, cisplatin, nedaplatin, mitomycin and 5-fluorouracil.
The dose required for the IC50 after a treatment of 72 h
was determined. In agreement with the radio-resistance profiles,
KYSE-410 cells were more resistant to multi-drugs than KYSE-140
cells (Fig. 3A), which indicated
that miR-155-5p was also involved in the chemo-resistance of ESCC
cells.
In order to demonstrate its role in the ESCC
chemoresistance, the present study examined the drug-induced cell
death in mimic/antagomiR-transfected cells. The results revealed
that introducing miR-155-5p increased the cell viability of
KYSE-140 cells after treatment with vinorelbine, cisplatin,
paclitaxel and docetaxel (Fig. 3B).
Conversely, knockdown of miR-155-5p sensitized KYSE-410 cells to
5-flourouracil, vinorelbine, cisplatin and docetaxel (Fig. 3C). Therefore, miR-155-5p enhanced
ESCC cell resistance to vinorelbine, cisplatin and docetaxel in a
drug-specific manner.
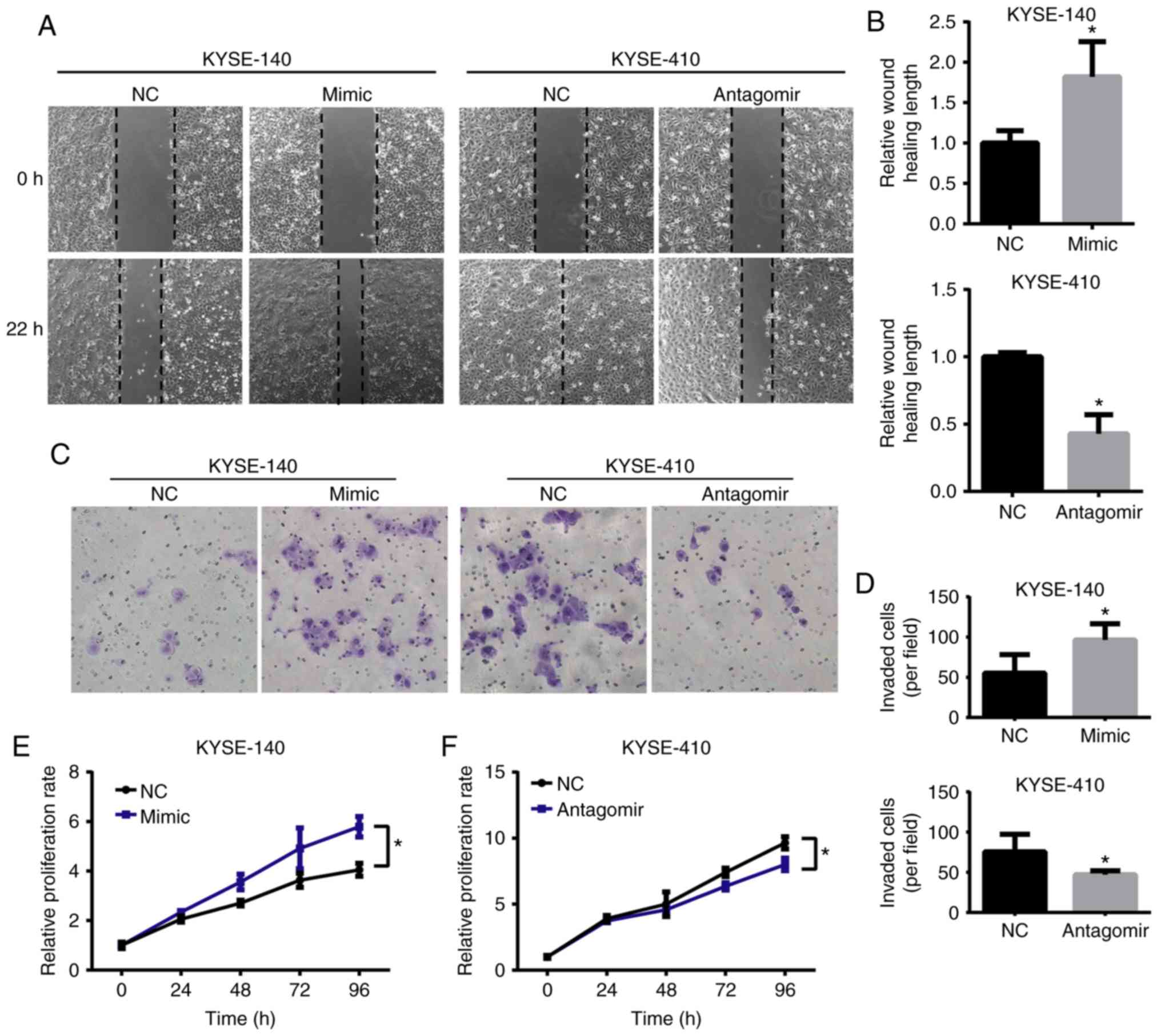
miR-155 enhances migration, invasion
and proliferation of ESCC cells
The present study further investigated whether
miR-155-5p interfered with the potential motility and proliferation
of ESCC cells. A marked positive correlation between the expression
of miR-155-5p and the motility of ESCC cells was observed.
Following overexpression of miR-155, KYSE-140 cells migrated
1.8-fold faster than the control groups (Fig. 4A and B). In contrast, miR-155-5p
downregulation decreased the migratory speeds of KYSE-410 cells by
~50% (Fig. 4A and B). Additionally,
the number of invaded cells were increased in the
miR-155-5p-overexpressing KYSE-140 cells and decreased in
anti-miR-155-5p KYSE-410 cells (Fig. 4C
and D), confirming the promoting role of miR-155-5p in
motility. Furthermore, the present study demonstrated that the
proliferation, quantified by a CCK-8 assay over a period of 4 days,
was increased in mimic-transfected KYSE-140 cells (Fig. 4E) and decreased in
antagomir-transfected KYSE-410 cells compared with the control
groups (Fig. 4F). All these data
indicated that in addition to radio- and chemo-resistance,
miR-155-5p also contributed to high migration and invasion
capacities and an increased proliferation rate in ESCC cells,
confirming the oncomiR role of miR-155-5p. Consistently, by
analyzing the survival rate of a ESCC cohort form TCGA, we found
that high expression of miR-155-5p was associated with poor overall
survival in patients with ESCC (Fig.
S1).
DNA methylation around the
transcription start site of miR-155HG exhibits differences in ESCC
cell lines and is negatively correlated with the expression of
miR-155
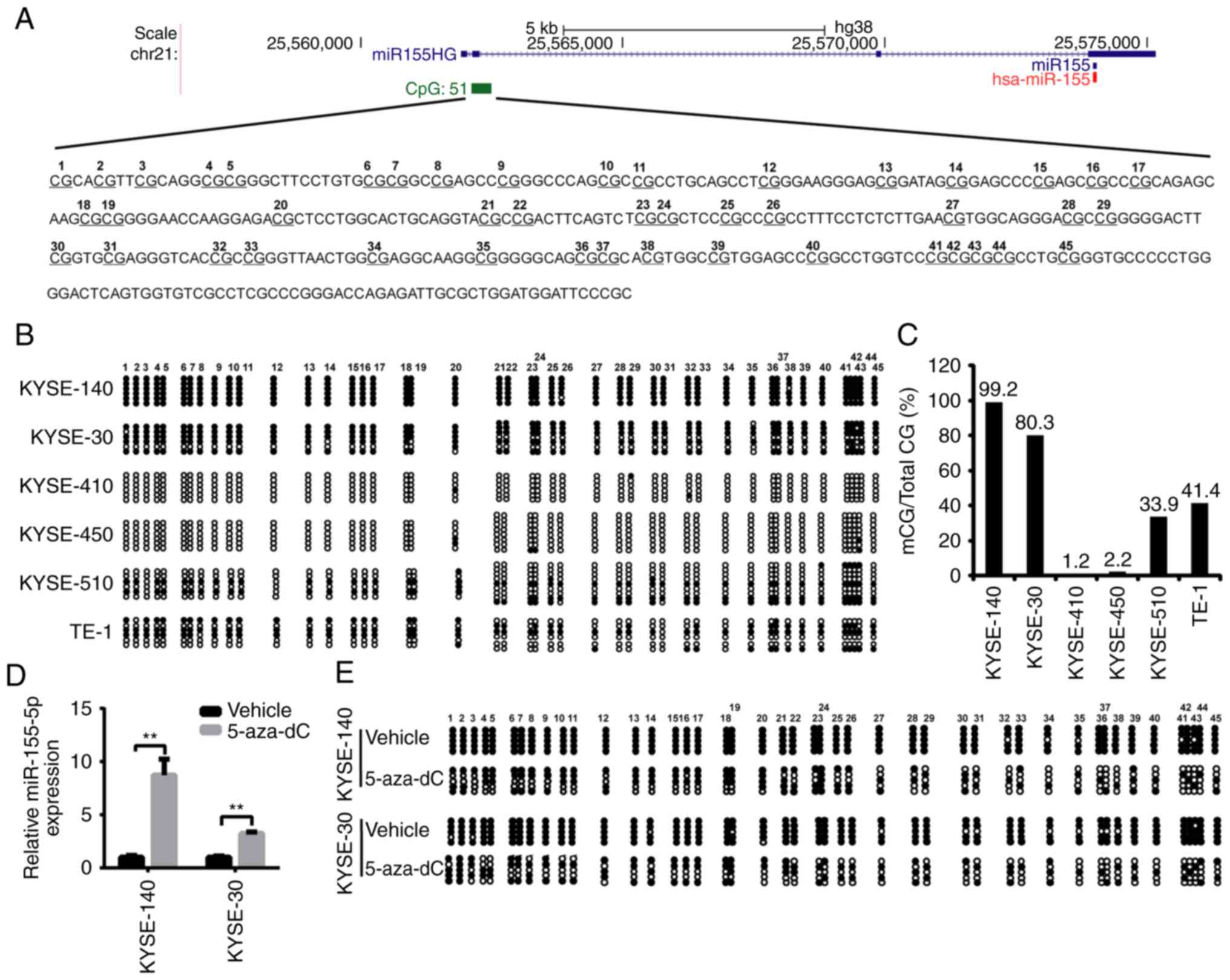
miR-155 is encoded by the non-protein-coding BIC
gene (now designated, MIR155 host gene or MIR155HG). MIR155HG
promoter sequence and the first exon harbor a CpG island (CGI)
containing 51 CpGs (Fig. 5A). In
order to elucidate the mechanisms implicated in the regulation of
miR-155 expression, the present study hypothesized that the DNA
methylation status of CpGs, which is the best-characterized
epigenetic mechanism (36), may be
involved in the regulation of miR-155 expression levels. A
bisulfite conversion sequencing (BSP) analysis of this region was
performed. Two pairs of primers were used to amplify the 45 CpG
sites in the CGI. The results revealed that DNA methylation
differed among the six cell lines. CGI was hypermethylated in
KYSE-140 and KYSE-30 cells, moderately methylated in KYSE-510 and
TE-1 cells, but barely methylated in KYSE-410 and KYSE-450 cells
(Fig. 5B and C). To further confirm
the transcriptional repression of miR-155 by DNA methylation, the
methylase inhibitor 5-zaz-dC was used in the present study. After
treatment with 5-aza-dC, the expression of miR-155-5p was increased
by >3-fold in the KYSE-140 and KYSE-30 cells (Fig. 5D). In addition, evaluation of DNA
methylation status revealed that the two cell lines exhibited a
partially demethylated pattern in the CpG island upstream of the
miR-155 gene (Fig. 5E). In
conclusion, the transcription of miR-155 gene was repressed by DNA
methylation of the MIR155HG promoter.
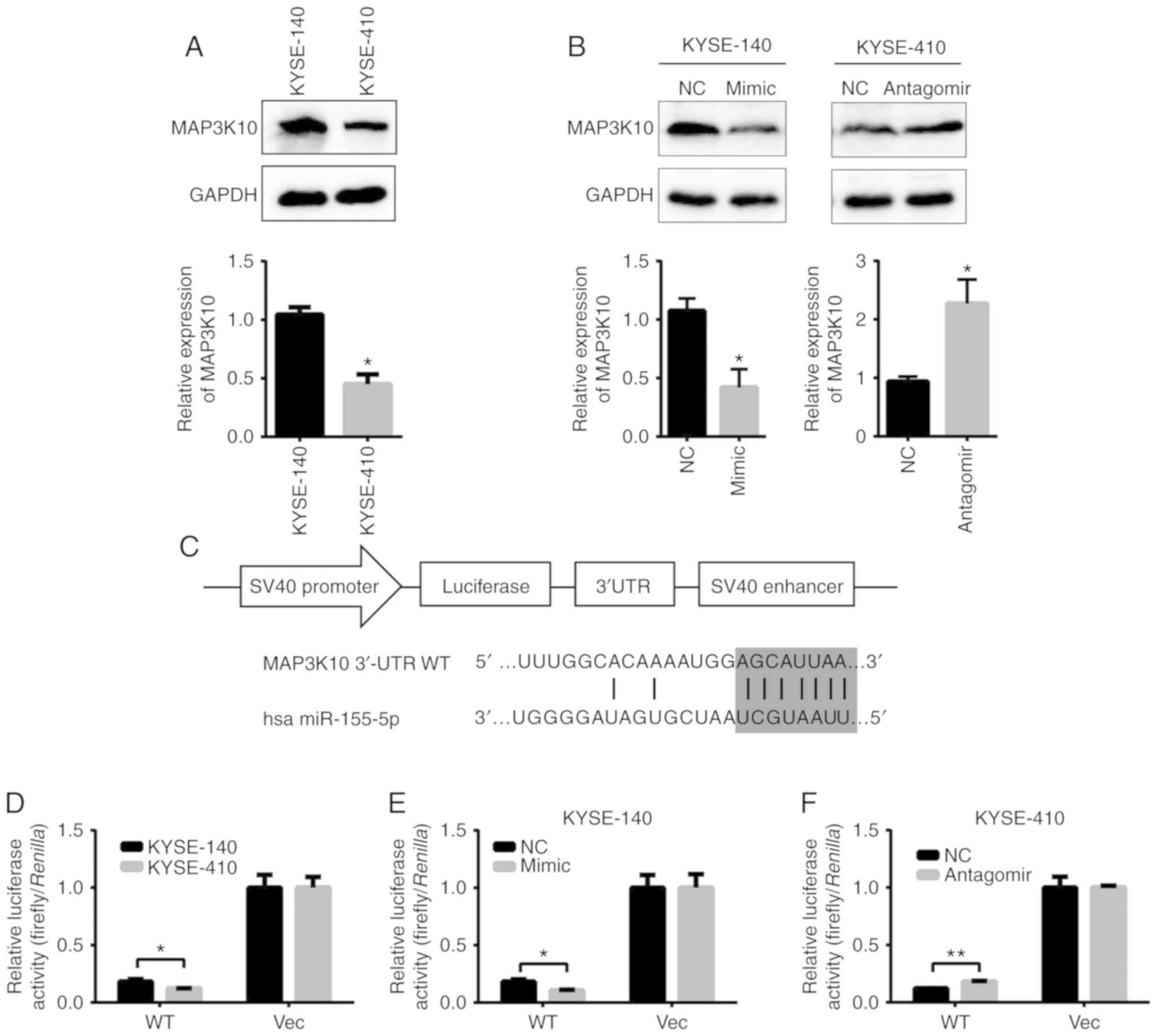
MAP3K10 is the target gene of miR-155
in ESCC cells
The present study assessed the level of overlapped
predicted target genes of miR-155-5p in the Arraystar datasets
(data not shown) of KYSE-140 and KYSE-410 cells. MAP3K10 was
observed to be expressed in an opposite manner to miR-155-5p.
Further RT-qPCR and western blot analyses demonstrated that the
protein level of MAP3K10 was significantly higher in the KYSE-140
than that in the KYSE-410 cells (western blot analysis, 1.00:0.51;
Fig. 6A). Furthermore, miR-155-5p
mimic transfection decreased the levels of MAP3K10 by ~60% in
KYSE-140 cells, and its level was increased by ~2.3-fold in the
antagomiR-transfected KYSE-410 cells (Fig. 6B).
For confirmation that MAP3K10 is the direct target
of miR-155-5p, its 3′-UTR regions were placed downstream of the
firefly luciferase gene in pGL3 (Promega Corporation) to create the
pGL3-MAP3K10 UTR construct (Fig.
6C). Both pGL3-MAP3K10 UTR and pGL3 were transfected into
KYSE-140 and KYSE-410 cells, in order to observe the functional
state of miR-155-5p in these cells. pGL3-MAP3K10-UTR, but not pGL3,
produced a 1.5-fold higher luciferase activity in KYSE-140 than in
KYSE-410 cells, in an opposite pattern of miR-155-5p expression
(Fig. 6D). Furthermore, the
luciferase activity of pGL3-MAP3K10-UTR WT was decreased by 45% in
the mimic-transfected KYSE-140 cells (Fig. 6E) and increased by 50% in the
antagomiR-transfected KYSE-410 cells, but not in the
pGL3-transfected control (Fig. 6F).
Collectively, MAP3K10 is in fact a direct target of miR-155-5p and
may execute its effect on ESCC radio- and chemo-resistance.
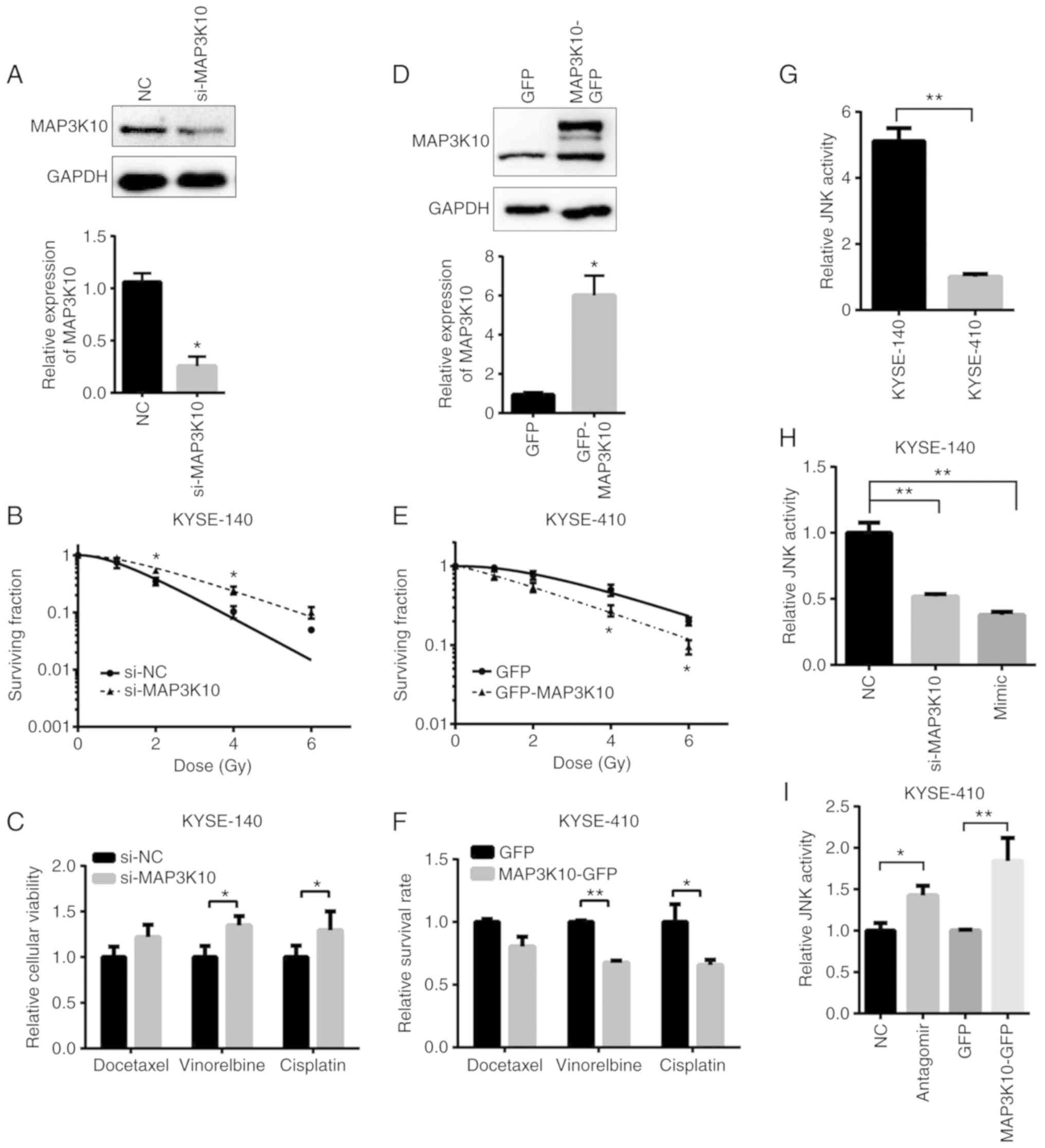
MAP3K10 suppresses radio- and
chemo-resistance of ESCC
In order to investigate the role of MAP3K10, siRNA
transfection-mediated knockdown of MAP3K10 was performed in
KYSE-140 cells in the present study. The expression of MAP3K10 was
decreased to 28% by siRNA at the protein level (Fig. 7A). Suppression of MAP3K10 not only
increased the cell viability when compared with the control groups
after exposure to radiation (Fig.
7B), but also significantly desensitized KYSE-140 cells to the
cell death triggered by vinorelbine and cisplatin (Fig. 7C). Conversely, overexpression of
MAP3K10 in KYSE-410 cells (Fig. 7D)
decreased resistance against radiation (Fig. 7E) and drugs (Fig. 7F). In contrast to the effect imposed
by the miR-155a-5p mimic, both MAP3K10 knockdown and overexpression
failed to cause a significant change of sensitivity to docetaxel,
which indicated that other target genes of miR-155-5p may
participate in this process.
Furthermore, the present study also observed that
the proliferation of ESCC cells was suppressed by MAP3K10 (Fig. S2E). However, migration speed and
invasion capacity were not influenced by forced reversal of MAP3K10
(Fig. S2A-D), indicating that the
miR-155-5p regulated the motility of ESCC cells via other target
genes. Additionally, MAP3K10 was revealed to be slightly
downregulated in ESCC tumor samples but significantly upregulated
in esophageal adenocarcinoma (EAC) compared with normal tissues
based on Oncomine database (Fig.
S3), which indicated that it may not contribute to
tumorigenesis in ESCC as that in EAC, but induced
chemoradio-resistance in ESCC.
MAP3K10 is a member of the serine/threonine kinase
family, which preferentially activates the C-Jun N-terminal kinase
(JNK) signaling pathway (37). The
present study examined the JNK pathway activity by Qiagen™ pathway
reporter assay and revealed that it was ~5-fold higher in KYSE-140
cells than in KYSE-410 cells (Fig.
7G). Furthermore, suppression of MAP3K10 by siRNA or mimic
transfection decreased the JNK signaling activity by ~50% in
KYSE-140 cells (Fig. 7H).
Conversely, the activity was upregulated by >1.4-fold in
antagomiR- and MAP3K10-GFP-transfected KYSE-410 cells (Fig. 7I). Therefore, MAP3K10 mediated the
promoting effect of miR-155 on resistance against both radiation
and drug treatment in ESCC cells, via its effect on the JNK
signaling pathway.
Discussion
The overall prognosis for ESCC is poor, due to
diagnosis at advanced stages of disease, high incidences of tumor
recurrence and metastasis, and the insensitivity to radiotherapy
and chemotherapy. Several clinical trials have demonstrated that
the response to chemoradiation is crucial for the prognosis of
patients (5–7); thus, finding molecular markers that
can predict the benefits of chemoradiotherapy for patients with
ESCC can prevent discomfort and toxicity. The present study
demonstrated that miR-155-5p expression under the negative control
of DNA methylation conferred ESCC cell resistance against both
radiation and chemotherapy drugs in vitro, in agreement with
the in vivo data that high expression of miR-155 in patients
with ESCC revealed a worse prognosis than those with low expression
(38). Consistent with its status
as an oncomiR in other tumors (39–41),
it was also revealed that miR-155-5p could enhance proliferation,
migration and invasion of ESCC cells. In addition, it has been
previously reported that miR-155 promotes cancer progression via
inhibition of apoptosis, inducement of EMT and metastasis, and
increased risk for recurrence (17–19).
It has been proposed that repair of DNA damage is
essential for the maintenance of genomic stability and tumor cell
survival following radiation (33).
In the present study, it was revealed that miR-155-5p accelerated
DNA damage repair, and NHEJ was the major repair pathway for DSBs
in ESCC that was responsible for the high efficient DNA repair. As
anticipated, enhanced DNA damage repair capacity led to resistance
to radiation. These results are consistent with the viewpoint that
NHEJ acts during any phase of the cell cycle and is the primary
mechanism for the repair of DSBs induced by radiation (42); however, this is contrary to a study
on breast cancer in which it was revealed that miR-155-5p decreased
the efficiency of homologous recombination repair and enhanced
sensitivity to radiation by targeting RAD51 directly (23). The present study demonstrated that
the expression of RAD51 was not down-/up-regulated in the
miR-155-5p mimic/antagomir-transfected cells. This may be due to
the different interactions of miRNA-mRNAs in different types of
cancer (43). Thus, miR-155-5p
binds to other target genes in ESCC and promotes
chemoradio-resistance. Given that the level of Ku80 changed only
when radiation occurred in the present study, it was supposed that
Ku80 is indirectly regulated by miR-155-5p through other mediators
after radiation. Further investigations are required in order to
confirm the association between Ku80 and miR-155-5p.
Radioresistance may occur simultaneously with
chemoresistance in patients with cancer (44). In fact, in the present study,
miR-155-5p also enhanced chemoresistance, and this impact was drug
type-specific. The intrinsic response of cancer cells to
chemotherapy drugs may be different due to the different drug
properties. First, the anticancer effect of chemotherapeutic drugs
was achieved through various mechanisms. For example, cisplatin
induces covalent crosslinks between DNA bases, interferes with DNA
repair mechanisms and causes DNA damage and subsequently apoptosis
in cancer cells (45). Docetaxel
not only inhibits depolymerization of microtubules, but also
induces apoptosis by binding to Bcl-2 or Bcl-xL and thus arresting
the function of each (46). Second,
unlike the target therapeutics, the pathways challenged by the
conventional chemotherapeutics remain unclear. It has previously
been reported that different drugs affect specific signaling
pathways in tumor cells, and thus the response of these pathways to
drugs was revealed to be both cell type- and drug type-specific
(47). Furthermore, it was revealed
that certain drugs such as 5-fluorouracil had no effect on
miR-155-5p mimic, but had an effect on antagomir. It is proposed
that the signaling pathways involved in 5-fluorouracil transport
and metabolism may be mutant or defective in KYSE-140 cells. In
this case, forced reversion of miR-155-5p may not help to change
the cell survival rate under 5-fluorouracil treatment. It is a
question worth further investigation. Therefore, miR-155-5p induced
resistance to docetaxel, cisplatin and vinorelbine in ESCC cells,
potentially by involving certain signaling pathways.
A miRNA executes its biological function via
repression in a sequence-specific manner of up to ~2,000
protein-coding genes at both stability and translation levels of
mRNAs. The present study defined the role of MAP3K10, the direct
target of miR-155-5p and relayed the impact of miR-155-5p on the
ESCC radio- and chemo-resistance through regulation of JNK pathway
activity. A previous study revealed that knockdown of MAP3K10
sensitized pancreatic cancer cells to gemcitabine (48). However, the opposite was observed in
the present study. siRNA-mediated suppression of MAP3K10 enhanced
rather than suppressed the multi-chemoresistance and
radio-resistance of ESCC cells. The functional disparity in cancer
biology of MAP3K10 is likely attributed to the system difference of
studies concerning the type of cancer with different expression
patterns of MAP3K10. Compared with tumor-adjacent normal tissue,
increased MAP3K10 expression was observed in pancreatic ductal
adenocarcinoma (PDAC) tissues and cells (48). However, it was observed to be
slightly downregulated in ESCC tumor samples following a
hierarchical clustering analysis of gene expression through the
Oncomine database and gene microarray data analysis (49). In another study, genome-wide gene
expression profiling revealed that MAP3K10 was significantly
upregulated in esophageal adenocarcinoma (EAC) compared with normal
tissues when assessing with DNA microarray technology (50). As mutations in MAP3K10 are rare in
esophageal cancer, according to TCGA analysis, it is proposed that
the epigenetic modification of MAP3K10 at the post-transcriptional
level may be a crucial factor leading to its misregulation in
esophageal cancer. Therefore, these results indicated that MAP3K10
may not participate in tumorigenesis in ESCC such as in PDAC and
EAC, but confer sensitivity to radiation and drugs in ESCC.
In summary, the present study revealed that
miR-155-5p, whose expression under the control of DNA methylation
confers resistance to radiation and chemotheraputic drugs, enhanced
proliferation, migration and invasion, and promoted DNA damage
repair by repairing the DSBs more efficiently. MAP3K10
significantly contributed to the positive control of ESCC
chemoradio-resistance and proliferation via the JNK pathway. The
present study provides a new set of diagnostic targets for the
guided personalized chemotherapy of ESCC.
Supplementary Material
Supporting Data
Acknowledgements
We thank Dr Zhang for his help in revising the final
manuscript. Special thanks to Professor Zhan from the National
Laboratory of Molecular Oncology, for kindly providing the ESCC
cell lines.
Funding
The present study was supported by the National
Natural Science Foundation of China (81602230, 81502191 and
81402327), and the Anhui Provincial Natural Science Foundation
(1508085SMH233, 1508085QH178 and 1508085QH183). The funders had no
role in the design of the study; in the collection, analyses, or
interpretation of data, in the writing of the manuscript, and in
the decision to publish the results.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LL and LQ conceived and designed the study. WL, HZ,
XL, RX, HD and QY acquired the data. LL and QY analyzed and
interpreted the data (e.g., statistical analysis, biostatistics,
computational analysis). WL, LL and LQ wrote, reviewed, and/or
revised the study. All authors read and approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smyth EC, Lagergren J, Fitzgerald RC,
Lordick F, Shah MA, Lagergren P and Cunningham D: Oesophageal
cancer. Nat Rev Dis Primers. 3:170482017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Napier KJ, Scheerer M and Misra S:
Esophageal cancer: A Review of epidemiology, pathogenesis, staging
workup and treatment modalities. World J Gastroint Oncol.
6:112–120. 2014. View Article : Google Scholar
|
|
4
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgan MA, Lewis WG, Crosby TD, Escofet X,
Roberts SA, Brewster AE, Harvard TJ and Clark GW: Prospective
cohort comparison of neoadjuvant chemoradiotherapy versus
chemotherapy in patients with oesophageal cancer. Br J Surg.
94:1509–1514. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tepper J, Krasna MJ, Niedzwiecki D, Hollis
D, Reed CE, Goldberg R, Kiel K, Willett C, Sugarbaker D and Mayer
R: Phase III trial of trimodality therapy with cisplatin,
fluorouracil, radiotherapy, and surgery compared with surgery alone
for esophageal cancer: CALGB 9781. J Clin Oncol. 26:1086–1092.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Hagen P, Hulshof MC, van Lanschot JJ,
Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ,
Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, et al: Preoperative
chemoradiotherapy for esophageal or junctional cancer. N Engl J
Med. 366:2074–2084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Developmental Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tam W: Identification and characterization
of human BIC, a gene on chromosome 21 that encodes a noncoding RNA.
Gene. 274:157–167. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eis PS, Tam W, Sun L, Chadburn A, Li Z,
Gomez MF, Lund E and Dahlberg JE: Accumulation of miR-155 and BIC
RNA in human B cell lymphomas. Proc Natl Acad Sci USA.
102:3627–3632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zang YS, Zhong YF, Fang Z, Li B and An J:
miR-155 inhibits the sensitivity of lung cancer cells to cisplatin
via negative regulation of Apaf-1 expression. Cancer Gene Ther.
19:773–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johansson J, Berg T, Kurzejamska E, Pang
MF, Tabor V, Jansson M, Roswall P, Pietras K, Sund M, Religa P and
Fuxe J: miR-155-mediated loss of C/EBPbeta shifts the TGF-β
response from growth inhibition to epithelial-mesenchymal
transition, invasion and metastasis in breast cancer. Oncogene.
32:5614–5624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gironella M, Seux M, Xie MJ, Cano C,
Tomasini R, Gommeaux J, Garcia S, Nowak J, Yeung ML, Jeang KT, et
al: Tumor protein 53-induced nuclear protein 1 expression is
repressed by miR-155, and its restoration inhibits pancreatic tumor
development. Proc Natl Acad Sci USA. 104:16170–16175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao H, Huang S, Liu A and Chen Z:
Up-regulated expression of miR-155 in human colonic cancer. J
Cancer Res Ther. 14:604–607. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu R, Liao J, Yang M, Shi Y, Peng Y, Wang
Y, Pan E, Guo W, Pu Y and Yin L: Circulating miR-155 expression in
plasma: A potential biomarker for early diagnosis of esophageal
cancer in humans. J Toxicol Environ Health A. 75:1154–1162. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Cheng C, Yuan X, He JT, Pan QH
and Sun FY: microRNA-155 acts as an oncogene by targeting the tumor
protein 53-induced nuclear protein 1 in esophageal squamous cell
carcinoma. Int J Clin Exp Pathol. 7:602–610. 2014.PubMed/NCBI
|
|
17
|
Zhang W, Ji W and Zhao X: miR-155 promotes
anaplastic thyroid cancer progression by directly targeting SOCS1.
BMC Cancer. 19:10932019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li DP, Fan J, Wu YJ, Xie YF, Zha JM and
Zhou XM: miR-155 up-regulated by TGF-β promotes
epithelial-mesenchymal transition, invasion and metastasis of human
hepatocellular carcinoma cells in vitro. Am J Transl Res.
9:2956–2965. 2017.PubMed/NCBI
|
|
19
|
Zhang J, Ye Y, Chang DW, Lin SH, Huang M,
Tannir NM, Matin S, Karam JA, Wood CG, Chen ZN and Wu X: Global and
Targeted miRNA expression profiling in clear cell renal cell
carcinoma tissues potentially links miR-155-5p and miR-210-3p to
both tumorigenesis and recurrence. Am J Pathol. 188:2487–2496.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shao C, Yang F, Qin Z, Jing X, Shu Y and
Shen H: The value of miR-155 as a biomarker for the diagnosis and
prognosis of lung cancer: A systematic review with meta-analysis.
BMC Cancer. 19:11032019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao S, Zhao ZY, Zhang ZY, Zhang Y and Wu
R: Prognostic value of MicroRNAs in esophageal carcinoma: A
meta-analysis. Clin Transl Gastroenterol. 9:2032018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pouliot LM, Chen YC, Bai J, Guha R, Martin
SE, Gottesman MM and Hall MD: Cisplatin sensitivity mediated by
WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer
Res. 72:5945–5955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gasparini P, Lovat F, Fassan M, Casadei L,
Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro
CL, et al: Protective role of miR-155 in breast cancer through
RAD51 targeting impairs homologous recombination after irradiation.
Proc Natl Acad Sci USA. 111:4536–4541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao K, X, Chen X, Zhu Q, Yin F, Ruan Q,
Xia J and Niu Z: Inhibition of miR-140-3p or miR-155-5p by
antagomir treatment sensitize chordoma cells to chemotherapy drug
treatment by increasing PTEN expression. Eur J Pharmacol.
854:298–306. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Babar IA, Czochor J, Steinmetz A, Weidhaas
JB, Glazer PM and Slack FJ: Inhibition of hypoxia-induced miR-155
radiosensitizes hypoxic lung cancer cells. Cancer Biol Ther.
12:908–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng F, Qian L, Lv L, Ding B, Zhou G,
Cheng X, Niu S and Liang Y: miR-193a-3p regulation of
chemoradiation resistance in oesophageal cancer cells via the PSEN1
gene. Gene. 579:139–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lv L, Deng H, Li Y, Zhang C, Liu X, Liu Q,
Zhang D, Wang L, Pu Y, Zhang H, et al: The DNA
methylation-regulated miR-193a-3p dictates the
multi-chemoresistance of bladder cancer via repression of
SRSF2/PLAU/HIC2 expression. Cell Death Dis. 5:e14022014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang F, Hajkova P, Barton SC, O'Carroll D,
Lee C, Lao K and Surani MA: 220-plex microRNA expression profile of
a single cell. Nat Protoc. 1:1154–1159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang JX, Chen ZH, Xu Y, Chen JW, Weng HW,
Yun M, Zheng ZS, Chen C, Wu BL, Li EM, et al: Downregulation of
MicroRNA-644a promotes esophageal squamous cell carcinoma
aggressiveness and stem cell-like phenotype via dysregulation of
PITX2. Clin Cancer Res. 23:298–310. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma K, He Y, Zhang H, Fei Q, Niu D, Wang D,
Ding X, Xu H, Chen X and Zhu J: DNA methylation-regulated
miR-193a-3p dictates resistance of hepatocellular carcinoma to
5-fluorouracil via repression of SRSF2 expression. J Biol Chem.
287:5639–5649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Powell SN and Bindra RS: Targeting the DNA
damage response for cancer therapy. DNA Repair (Amst). 8:1153–1165.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ivashkevich A, Redon CE, Nakamura AJ,
Martin RF and Martin OA: Use of the gamma-H2AX assay to monitor DNA
damage and repair in translational cancer research. Cancer Lett.
327:123–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bouwman P and Jonkers J: The effects of
deregulated DNA damage signalling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lujambio A, Ropero S, Ballestar E, Fraga
MF, Cerrato C, Setién F, Casado S, Suarez-Gauthier A,
Sanchez-Cespedes M, Git A, et al: Genetic unmasking of an
epigenetically silenced microRNA in human cancer cells. Cancer Res.
67:1424–1429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nagata K, Puls A, Futter C, Aspenstrom P,
Schaefer E, Nakata T, Hirokawa N and Hall A: The MAP kinase kinase
kinase MLK2 co-localizes with activated JNK along microtubules and
associates with kinesin superfamily motor KIF3. EMBO J. 17:149–158.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nagy A, Lanczky A, Menyhart O and Gyorffy
B: Author Correction: Validation of miRNA prognostic power in
hepatocellular carcinoma using expression data of independent
datasets. Sci Rep. 8:115152018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li N, Cui T, Guo W, Wang D and Mao L:
miR-155-5p accelerates the metastasis of cervical cancer cell via
targeting TP53INP1. Onco Targets Ther. 12:3181–3196. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qu Y, Zhang H, Sun W, Han Y, Li S, Qu Y,
Ying G and Ba Y: MicroRNA-155 promotes gastric cancer growth and
invasion by negatively regulating transforming growth factor-β
receptor 2. Cancer Sci. 109:618–628. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ji H, Tian D, Zhang B, Zhang Y, Yan D and
Wu S: Overexpression of miR-155 in clear-cell renal cell carcinoma
and its oncogenic effect through targeting FOXO3a. Exp Ther Med.
13:2286–2292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang C and Lees-Miller SP: Detection and
repair of ionizing radiation-induced DNA double strand breaks: New
developments in nonhomologous end joining. Int J Radiat Oncol Biol
Phys. 86:440–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chiu YC, Tsai MH, Chou WC, Liu YC, Kuo YY,
Hou HA, Lu TP, Lai LC, Chen Y, Tien HF and Chuang EY: Prognostic
significance of NPM1 mutation-modulated microRNA-mRNA regulation in
acute myeloid leukemia. Leukemia. 30:274–284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang Y, Park MA, Heo SW, Park SY, Kang KW,
Park PH and Kim JA: The radio-sensitizing effect of xanthohumol is
mediated by STAT3 and EGFR suppression in doxorubicin-resistant
MCF-7 human breast cancer cells. Biochim Biophys Acta.
1830:2638–2648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pienta KJ: Preclinical mechanisms of
action of docetaxel and docetaxel combinations in prostate cancer.
Semin Oncol. 28:3–7. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu Q, Zhang C, Ding X, Deng H, Zhang D,
Cui W, Xu H, Wang Y, Xu W, Lv L, et al: Preclinical optimization of
a broad-spectrum anti-bladder cancer tri-drug regimen via the
Feedback System Control (FSC) platform. Sci Rep. 5:114642015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
An Y, Cai B, Chen J, Lv N, Yao J, Xue X,
Tu M, Tang D, Wei J, Jiang K, et al: MAP3K10 promotes the
proliferation and decreases the sensitivity of pancreatic cancer
cells to gemcitabine by upregulating Gli-1 and Gli-2. Cancer Lett.
329:228–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Su H, Hu N, Yang HH, Wang C, Takikita M,
Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ, et al: Global
gene expression profiling and validation in esophageal squamous
cell carcinoma and its association with clinical phenotypes. Clin
Cancer Res. 17:2955–2966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim SM, Park YY, Park ES, Cho JY, Izzo JG,
Zhang D, Kim SB, Lee JH, Bhutani MS, Swisher SG, et al: Prognostic
biomarkers for esophageal adenocarcinoma identified by analysis of
tumor transcriptome. PLoS One. 5:e150742010. View Article : Google Scholar : PubMed/NCBI
|