Introduction
Lung cancer is the most common cause of
cancer-related deaths, and every year 1.6 million cases result to
fatalities due to this disease (1).
Lung cancer is histologically classified into non-small cell lung
cancer (NSCLC) which is observed in up to 85% of all lung cancer
cases diagnosed, and small-cell lung cancer (SCLC) which accounts
for 15% of the remaining cases (2).
Only 15% of lung cancer cases are diagnosed at an early stage for
which a complete surgical resection is a curative treatment
(3). Lung tumors that are present
in the advanced stage of the disease are additionally treated with
chemotherapy and radiotherapy that may aid the reduction of the
tumor size and the symptoms of the disease. However, this type of
therapy may cause serious side-effects. During the past 10 years,
targeted therapy has been used with EGFR tyrosine kinase inhibitors
(TKIs) for patients with activating EGFR mutations, and with ALK
inhibitors for patients with abnormal fusion of the anaplastic
lymphoma kinase (ALK). These compounds have demonstrated increased
therapeutic efficacy and reduced side effects compared with those
of chemotherapy in NSCLC patients. However, the patients treated
with TKIs develop drug resistance usually within 6–12 months
(4). Therefore, the development of
more effective therapeutic agents is urgently required for the
management of NSCLC.
The tumor suppressor protein p53 is a transcription
factor that downregulates the expression levels of the genes
associated with tumor development and progression. TP53 exerts
functions in the regulation of the cell cycle, apoptosis,
senescence and cell metabolism, and is activated in response to
cellular stress stimuli, such as DNA damage, oncogenic signaling
and mitotic stress (5). Mutations
in the p53 gene occur frequently in various types of human
cancer including lung cancer (6).
The most common type of p53 mutations are missense mutations
which localize to the DNA binding domain of the gene sequence (exon
5–8). Mutations in the p53 gene lead to loss of tumor
suppressive function and/or gain of oncogenic function that
contribute to tumor cell growth, survival, metastasis and drug
resistance (7). In lung cancer
patients, it has been demonstrated that p53 mutations are
associated with poor prognosis (8).
Mutant p53 protein accumulates in tumors and the restoration of its
activity can cause cell growth inhibition (9).
In the present study, the tumor inhibitory effects
of SCH 529074, one of the p53 restoring compounds initially
discovered using drug screening based on p53 DNA binding assays
(10), was investigated. SCH 529074
was capable of restoring DNA binding activity of mutant p53 and
inhibiting Mdm2 (murine double minute 2) ubiquitination of
wild-type (WT) p53. Moreover, this compound affected p53 WT cells
(10,11). Due to the high prevalence of p53
mutations in NSCLC and the lack of effective treatment for this
cancer type, the reactivation of mutant p53 may provide a novel
therapeutic approach. The aim of the present study was to assess
the role of SCH 529074 in NSCLC cells with different p53 mutations
and explore the molecular alterations caused by this type of drug
treatment.
Materials and methods
Cell lines and cell culture
The NSCLC cell lines (A549, H157, H322 and H1975)
were obtained from the American Type Culture Collection (ATCC)
whereas colon cancer cell lines (HCT116 and HCT116
p53−/−) were a gift from Professor Berit Jungnickel
(Institute of Cell Biology, Faculty of Biology and Pharmacy,
Friedrich Schiller University, Jena). The NSCLC cell lines were
grown in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine
serum (Biochrom). The colon cancer cell lines were grown in
Leibovitz's L-15 medium supplemented with 10% (v/v) fetal bovine
serum as previously described (12). The cells were maintained in a
humidified atmosphere with 5% CO2 at 37°C.
Mutation analysis
Genomic DNA was extracted from the cell lines using
the Maxwell® 16 FFPE Tissue LEV DNA Purification Kit
(Promega Corp.) according to the manufacturer's instructions. The
Maxwell® 16 LEV Instrument was used for genomic
analysis. The concentration and purity (260/280 nm ratio) of the
genomic DNA were measured using Nano Drop (VWR).
The exons 5–9 of the p53 gene were amplified
using gene specific primers (Table
SI) and Hotstart Taq polymerase (VWR). The PCR products were
purified using DNA Clean & Concentrator™-5 kit (Zymo Research)
according to the manufacturer's recommendations. Purified PCR
products (70–90 ng) were subsequently sequenced bidirectionally
based on capillary electrophoresis (LGC Genomics GmbH). The
sequencing profiles were analyzed with the Finch TV 1.4.0 software
program (Geospiza; PerkinElmer).
Drugs
SCH 529074
[N3-[2-[[4-[Bis(4-chlorophenyl)methyl]-1-piperazinyl]methyl]-4-quinazolinyl]-N1,
N1-dimethyl-1,3-propanediamine] was purchased from Tocris
Bioscience. Doxorubicin was purchased from LC Laboratories.
Chloroquine (CQ) was purchased from Sigma Aldrich; Merck KGaA.
Functionality assay of p53
The functionality of p53 in the investigated cell
lines was determined by doxorubicin treatment. The cells were
seeded in 6-well plates and treated with increasing concentrations
of doxorubicin (0.5, 1 and 2 µM) for 24 h. The cells were harvested
and the cell lysate was prepared for western blot analysis.
Trypan blue exclusion test for cell
viability
The cells (1×105/well) were seeded into
12-well plates. The following day, the cells were treated with
different concentrations of SCH 529074 (2 and 4 µM) or DMSO (0.1%)
for 24 h and stained with 0.4% trypan blue solution at room
temperature for 5 min (Invitrogen; Thermo Fisher Scientific, Inc.).
The number of viable cells was determined by an automated cell
counter Countess (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions.
Colony formation assay
The cells were seeded into 6-well plates at a
density of 2,000 cells/well. Following 24 h of culture, the cells
were treated with 1 µM of SCH 529074 or DMSO and were incubated for
8–12 days until colonies were formed. The colonies (at least 50
cells per colony were fixed with 100% of methanol and stained with
crystal violet (0.5%) at room temperature for 30 min. The stained
colonies were visualized by light microscopy (Zeiss AG).
Cell cycle and apoptosis analyses by
flow cytometry
To determine the effect of SCH 529074 on the cell
cycle, the cells (4×105/well) were seeded into 6-well
plates and treated with SCH 529074 (2 and 4 µM) or DMSO (0.1%). The
following day, the cells (1–2×106) were fixed in 70%
ethanol following trypsinization and stored at −20°C overnight. The
cells were then stained with propidium iodide (PI) solution (1
µg/µl; BD Biosciences) and analyzed by flow cytometry as previously
described (13).
Caspase-3/7 activity assay
The caspase-3/7 activity assay was applied to
determine the extent of apoptosis. The cells
(2×104/well) were seeded into 96-well plates and treated
with SCH 529074 (4 µM) or DMSO (0.1%) for 24 h. A total of 100 µl
of caspase-3/7 reagents was added to each well and the samples were
shaken for 30 sec. The reagents were obtained from the
Caspase-Glo® 3/7 Assay kit (Promega Corp.). Following
incubation of the cells at room temperature (RT) for 2 h,
luminescence was recorded with the LUMIstar Galaxy (BMG
Labtech).
Quantitative real-time RT-PCR
(RT-qPCR)
Total RNA was isolated from the cells using peqGOLD
TriFastTM (Peqlab) following the manufacturer's instructions. A
total of 500 ng of RNA was used for first-strand cDNA synthesis by
the QuantiTect Reverse Transcription Kit (Qiagen, Inc.). With
regard to the expression analysis, qPCR was conducted on the
Rotor-Gene Q (Qiagen, Inc.) using FastStart Universal SYBR Green
Master (Roche Diagnostics) with the following conditions: 95°C 5
sec, 40 cycles of 95°C 5 sec and 60°C 1 min. The housekeeping gene
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was
used for normalization of the gene expression data. The
2−ΔΔCq method was used for quantification (14). The data were derived from three
independent experiments. The sequences of the primers are listed in
Table SI.
Western blotting
The cells (4×105/well) were seeded into
6-well plates and incubated overnight. The following day, the cells
were treated with SCH 529074 (2, 4 and 6 µM) or DMSO for 24 h,
harvested and lysed using RIPA buffer (Sigma Aldrich; Merck KGaA).
The concentration of protein was determined by using a BCA protein
assay kit. Total protein (30–50 µg) was separated by 8% of sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to a AmershamTM ProtranTM 0.2 µM nitrocellulose
membrane using a TransBlot® Transfer System (Bio-Rad
Laboratories, Inc.). The membranes were blocked for 1 h with 5% BSA
or 5% non-fat dry milk in TBST and probed overnight with primary
antibodies for p53, p-p53, p21, PUMA, LC3 A/B and actin (Table SII) at 4°C. The membranes were
washed with TBST and incubated with secondary antibodies for 1 h at
RT. Finally, the immunoreactivity was visualized using an enhanced
chemiluminescence detection system (GE Healthcare). The signals
were quantified using Image Studio Lite Version 5.2.5 (LI-COR
Biosciences).
Statistical analysis
Statistical analysis was performed using the
software package SPSS 19.0 (IBM Corp). The differences between the
single drug-treated and control samples were assessed using the
Student's t-test. For combination drug therapy, two-way ANOVA (with
Tukey's multiple comparisons test) was performed using GraphPad
Prism 7.04 (GraphPad Software, Inc.). P<0.05 was considered to
indicate statistically significant differences.
Results
TP53 mutation status and p53 protein
expression in NSCLC cell lines
TP53 mutation was confirmed in three (H1975, H322
and H157) cell lines. Direct sequencing of the p53 gene
confirmed the presence of the ‘CGT>CAT’ (R273H), ‘CGG>CTG’
(R248L) and ‘CAG>TAG’ (298stop) mutations in H1975, H322 and
H157 cells, respectively (Fig.
S1A). The WT TP53 status was observed in both A549 and HCT116
cells (Fig. S1B). High levels of
the mutant p53 protein were detected in H1975, H322, A549 and
HCT116 cells. As anticipated the p53 protein was present in neither
HCT116 p53−/− nor H157 cells due to deletion or
non-sense mutation of p53 in the two cell lines. The expression of
p53 mRNA in these two cell lines (data not shown) was also
not detected. To assess the functionality of p53 in these cell
lines, the levels of the phosphorylated p53 protein (p-p53) and the
expression levels of the p53 target gene p21 were analyzed
following doxorubicin treatment. The induction of the p-p53 levels
and the expression levels of p21 were present in the WT cell lines
(A549 and HCT116), and only weak signals were detected in the
mutant cell lines (H1975 and H322). The p53-deficient cell line
(HCT116 p53−/−) or the cell line with the 298stop (H157)
codon did not express p53 or p21 proteins (Fig. S2).
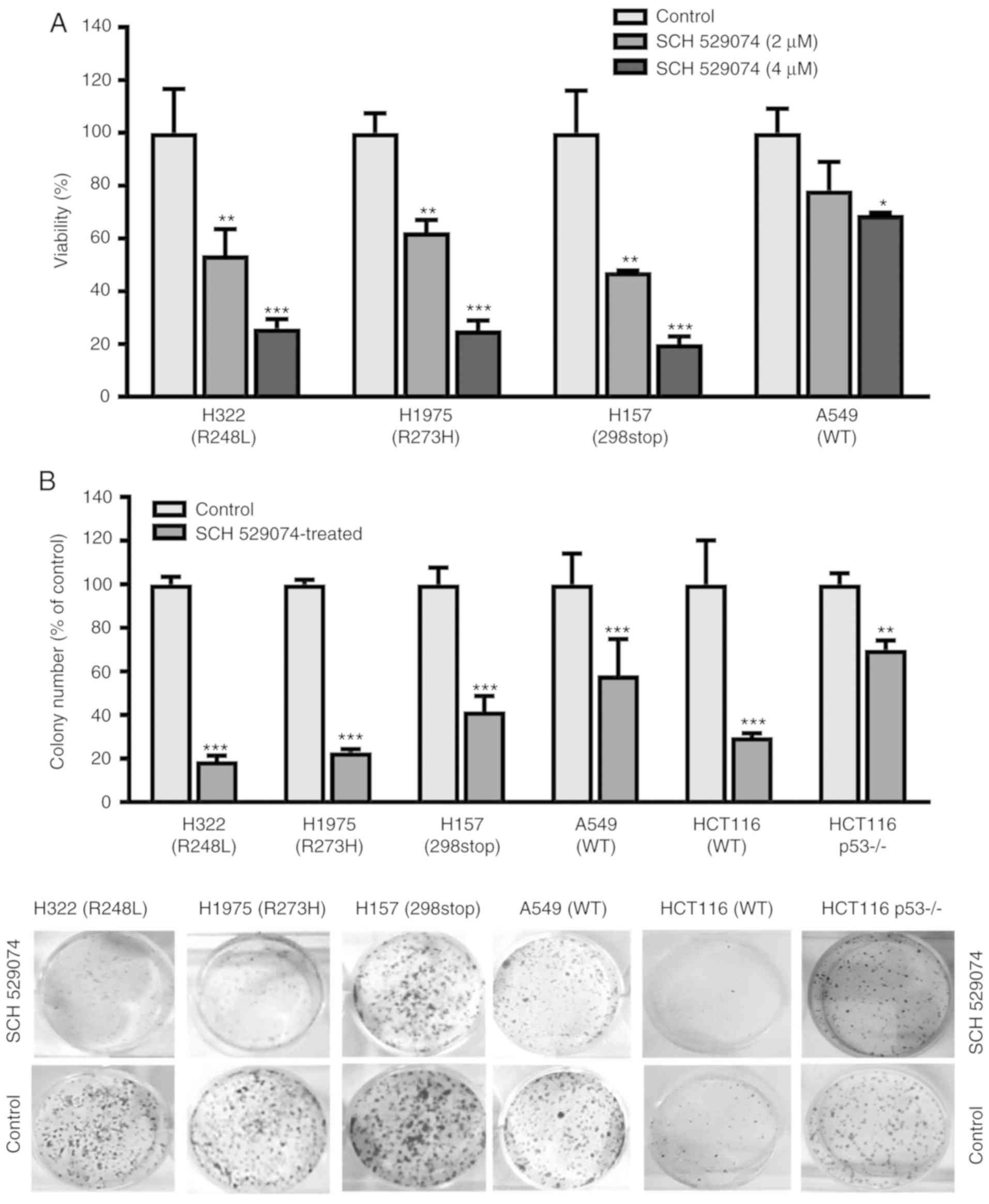
SCH 529074 inhibits cancer cell
viability and colony formation
To evaluate the effects of the drugs on tumor cell
growth, the cell survival assay was performed. Reduced cell
viability was observed in all tested NSCLC cell lines treated with
2 and 4 µM of SCH 529074 (Fig. 1A).
Upon treatment with the higher concentration (4 µM) of this
compound, the viability of the cells was significantly decreased to
20–25% in p53 mutant cells (H157, H1975 and H322, P<0.001) and
to 68% in the p53 WT cell line A549 (P=0.032) compared with that
observed in the control cells.
Subsequently, the toxicity of SCH 529074 to NSCLCs
was investigated by colony formation assay, which is commonly used
for determining the efficacy of cytotoxic drugs and for testing the
ability of the cells to undergo unlimited divisions (15). The data indicated that SCH 529074
treatment significantly reduced the ability of cancer cells to form
colonies (Fig. 1B). The colony
formation activity of SCH 529074-treated cells was reduced to 19
and 23% (P<0.001) in H322 and H1975 cells, respectively,
compared with that of the control cells. In H157 and A549 cells,
this parameter was reduced to 41% (P<0.001) and 58% (P=0.0003),
respectively, compared with that of the control cells. Similarly,
colony formation activity was reduced to 30% (P<0.001) in HCT116
cells and to 70% (P=0.0034) in HCT116 p53−/- cells
compared with that of the control cells. These data indicated that
p53 mutant cells were more sensitive to SCH 529074 treatment
compared with the p53 WT NSCLC cells. Notably, upon SCH529074
treatment, the p53 null mutant cell line (H157) was sensitive to a
short-term effect as demonstrated by the cell viability assay
(Fig. 1A) and more resistant to
long-term effects as demonstrated by the colony formation assay
(Fig. 1B).
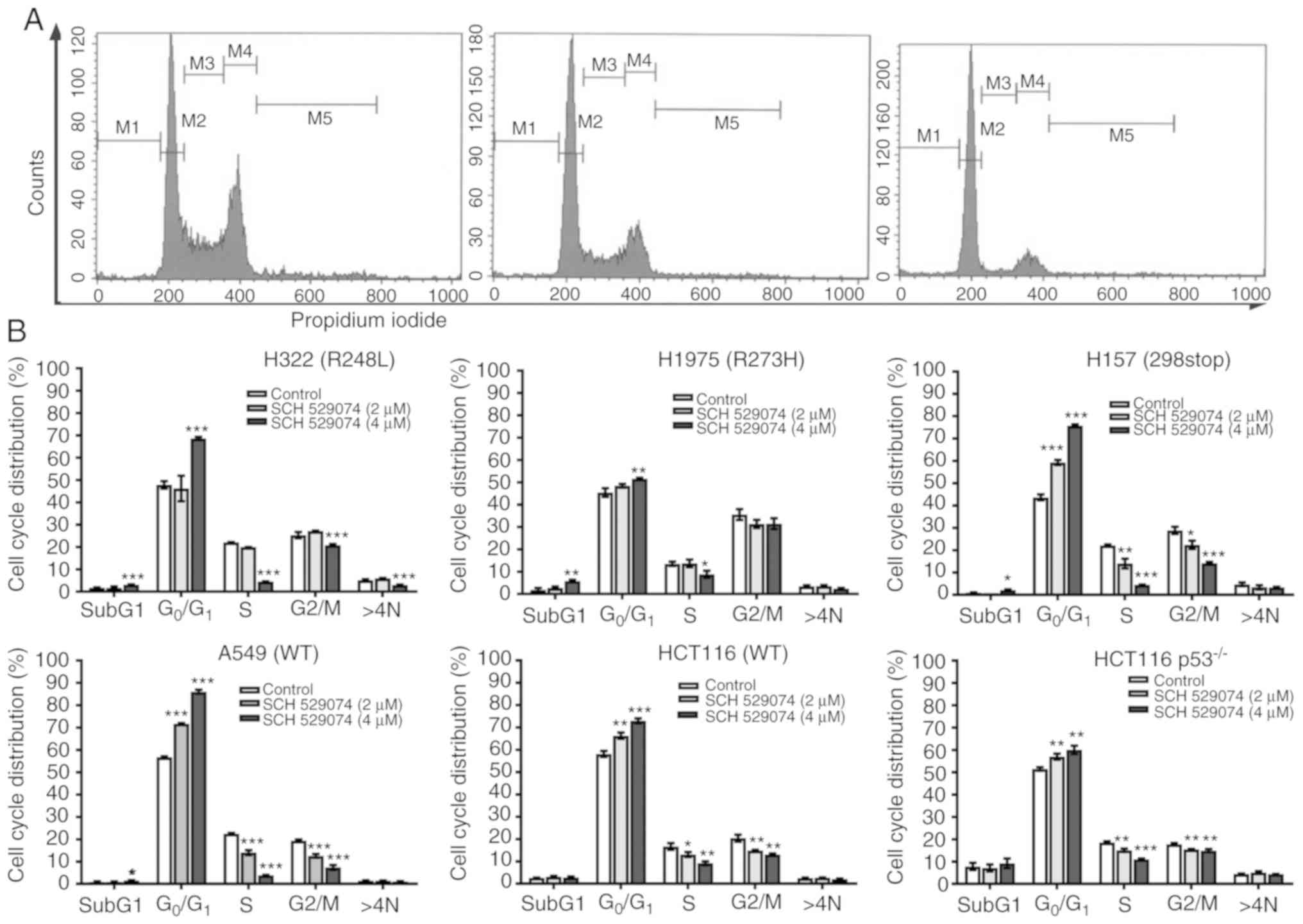
SCH 529074 induces G0/G1 cell cycle
arrest
Due to the important role of p53 in cell cycle
regulation and the observed growth inhibitory effects of SCH
529074, its effects were further investigated on cell cycle
progression. Fig. 2A revealed the
cell cycle profiles of H157 cells following different treatment
concentrations. NSCLC cells (H157, A549, HCT116 and HCT116
p53−/−) were arrested at the G0/G1 phase (59%,
P<0.001; 72%, P<0.001; 66%, P<0.001; and 57%, P<0.001)
compared with the control cells following low concentration (2 µM)
of drug treatment (Fig. 2B). This
G0/G1 phase arrest was not observed in the two p53 mutant NSCLC
cell lines (H1975 and H322). Treatment of the cells with high SCH
529074 concentration (4 µM) resulted in a significantly increased
G0/G1 phase and a decreased S phase. These effects were observed in
all the different cell lines assessed regardless of the p53 status.
In addition, an increased sub-G1 subpopulation was further observed
in all NSCLC cell lines following treatment with 4 µM of SCH 529074
(Fig. 2B) indicating the induction
of apoptosis in these cells.
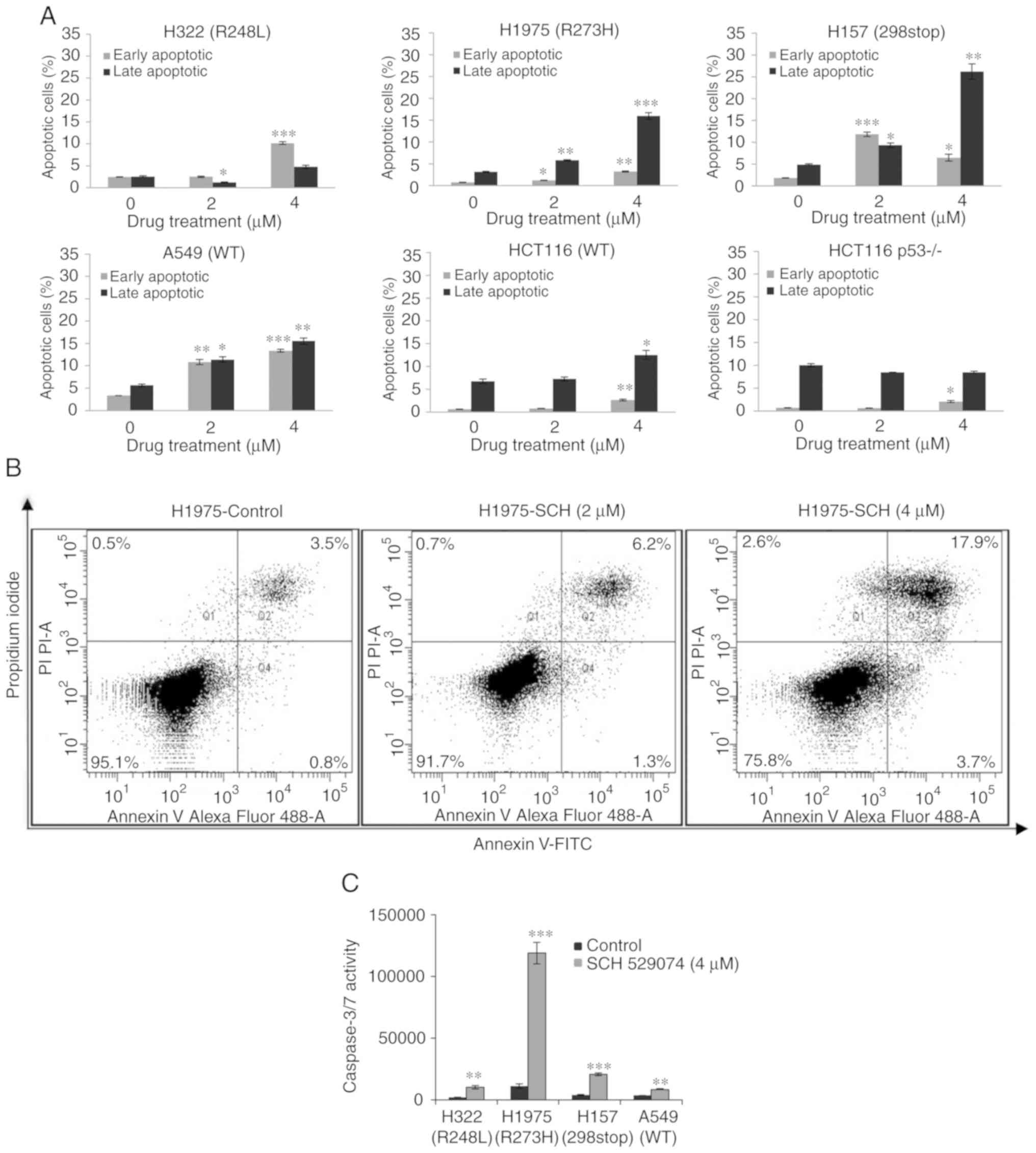
SCH 529074 induced apoptosis in cancer cells. To
confirm the effects of SCH 529074 on cancer cell apoptosis, flow
cytometric analysis was carried out following Annexin V/PI double
staining. Upon SCH 529074 treatment (4 µM), the early apoptotic
fraction was increased to ~10% (P<0.001) in H322 cells compared
with those of the control cells (Fig.
3A and B). In H1975 cells treated with SCH 529074 (2 µM), the
early and late apoptotic rates were significantly increased
compared with that of the control cells (P<0.05 and P<0.01),
and following treatment with high concentration of the compound (4
µM), both early and late apoptotic rates were significantly
enhanced (P<0.01 and P<0.001). In H157 cells, 2 µM of SCH
529074 treatment induced early and late apoptosis (P<0.001 and
P<0.05), and 4 µM of this compound also induced early and late
apoptosis (P<0.05 and P<0.01). Similarly, in A549 cells, 2
and 4 µM of SCH 529074 significantly increased early and late
apoptosis (P<0.01 and P<0.05 for 2 µM; P<0.001 and
P<0.01 for 4 µM). These data revealed that SCH 529074 could
induce apoptosis in all assessed NSCLC cell lines irrespective of
their p53 mutational status. In line with that, in colon cancer
cells, a significant induction of early and late apoptosis was
observed in HCT116 cells at a concentration of 4 µM (P<0.01 and
P<0.05), and in HCT116 p53−/− cells, treatment with 4
µM of SCH 529074 significantly induced early apoptosis
(P<0.05).
To examine whether the induction of apoptosis was
caspase-dependent, a caspase Glo® 3/7 activity assay was
used. All NSCLC cell lines demonstrated a significantly higher
caspase-3/7 activity following SCH 529074 treatment compared with
that of the control cells (P<0.01 and P<0.001; Fig. 3C).
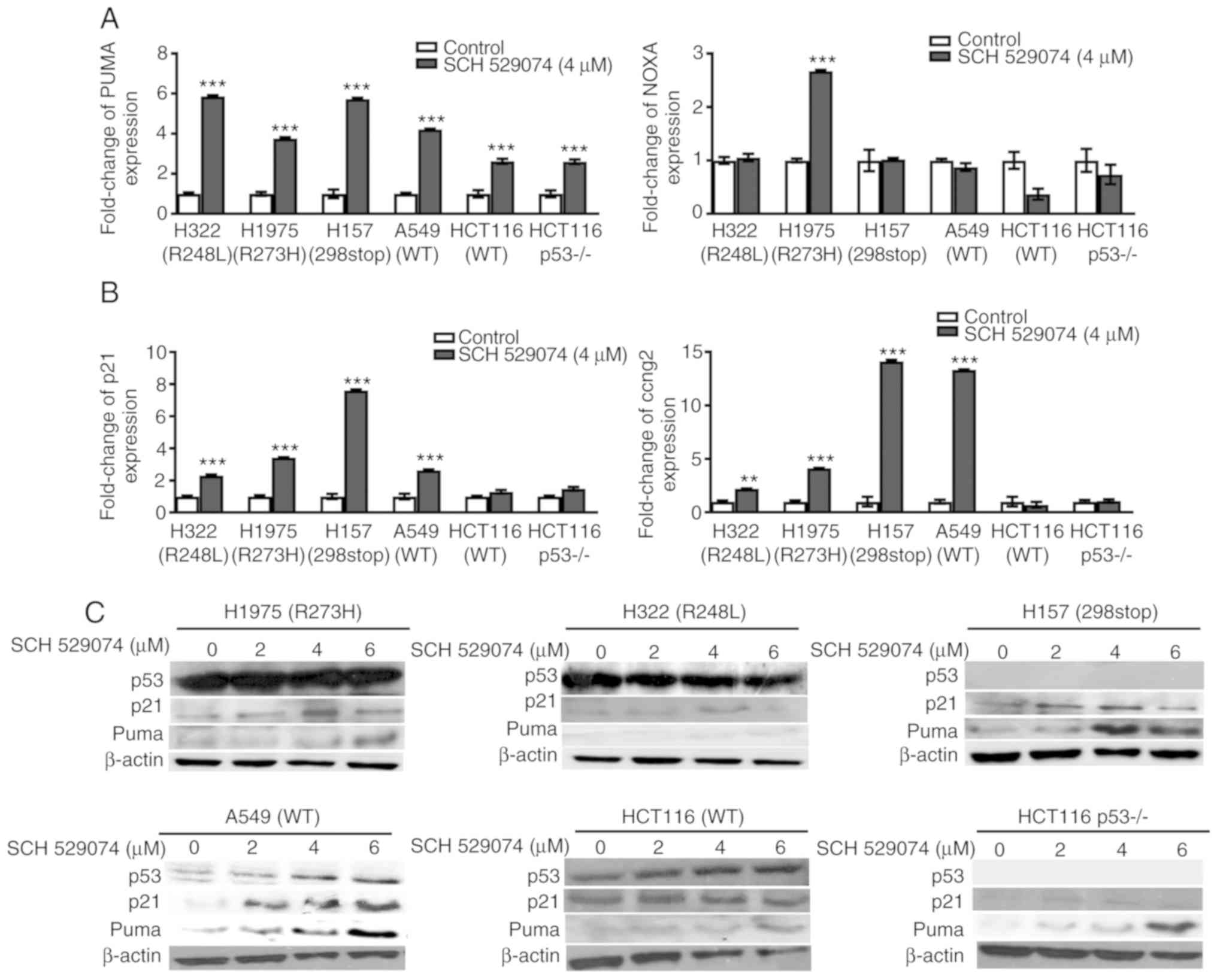
SCH 529074 alters the expression of
pro-apoptotic and cell cycle regulatory genes
TP53 has been revealed to induce apoptosis and
regulate the cell cycle according to external stimuli, which
further cause activation of the p53 responsive genes (16). As a p53 activator, SCH 529074 may
induce transcription of the p53 target genes. To examine this, the
expression of the p53 downstream apoptosis-associated genes
PUMA, NOXA and BAX as well as of the cell
cycle-associated genes p21 and CCNG2, were analyzed.
As revealed in Fig. 4A, PUMA
was significantly increased in all the cancer cell lines assessed
irrespective of the p53 mutation status. The expression levels of
NOXA were only upregulated in H1975 cells (R273H).
BAX expression was not significantly altered following drug
treatment (data not shown). The mRNA levels of p21 and
CCNG2 were increased in all NSCLC cell lines but not in
colon cancer cell lines (Fig. 4B).
To confirm these results, the protein levels of PUMA and p21 were
further analyzed by western blotting. Consistent with the mRNA
data, the protein levels of PUMA and p21 were revealed to be
increased upon 4 or 6 µM of SCH 529074 treatment in the cancer cell
lines regardless of their p53 status (Figs. 4C and S3).
SCH 529074 treatment increases the
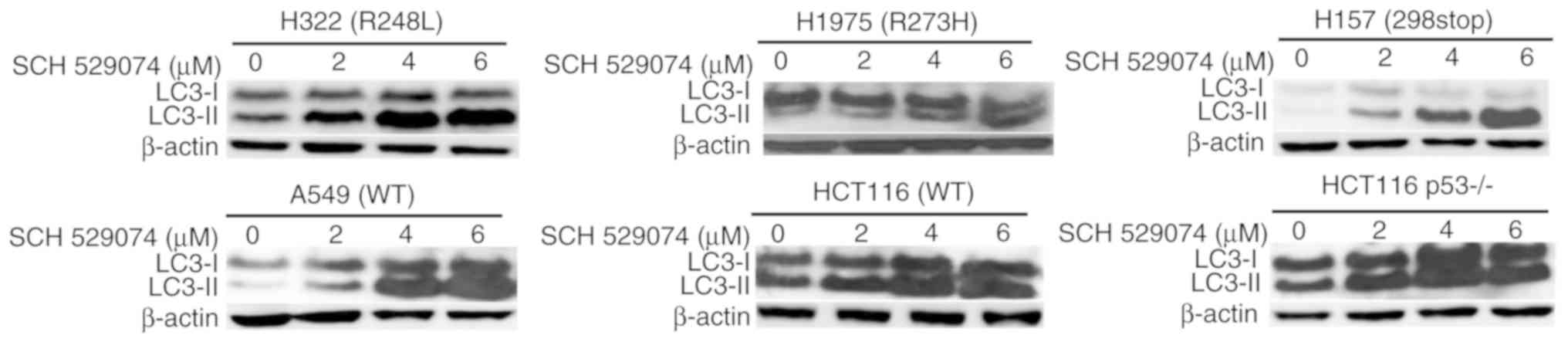
levels of LC3II
Since p53 can regulate autophagy (17), further experiments assessed the
ability of SCH 529074 to affect autophagy. During autophagy, the
cytoplasmic form of LC3-I is converted to the
autophagosome-associated form of LC3II, which is considered a
marker for autophagy (18). As
revealed in Fig. 5, treatment with
SCH 529074 led to increased LC3-II levels in all tumor cell lines
assessed regardless of their p53 mutation status.
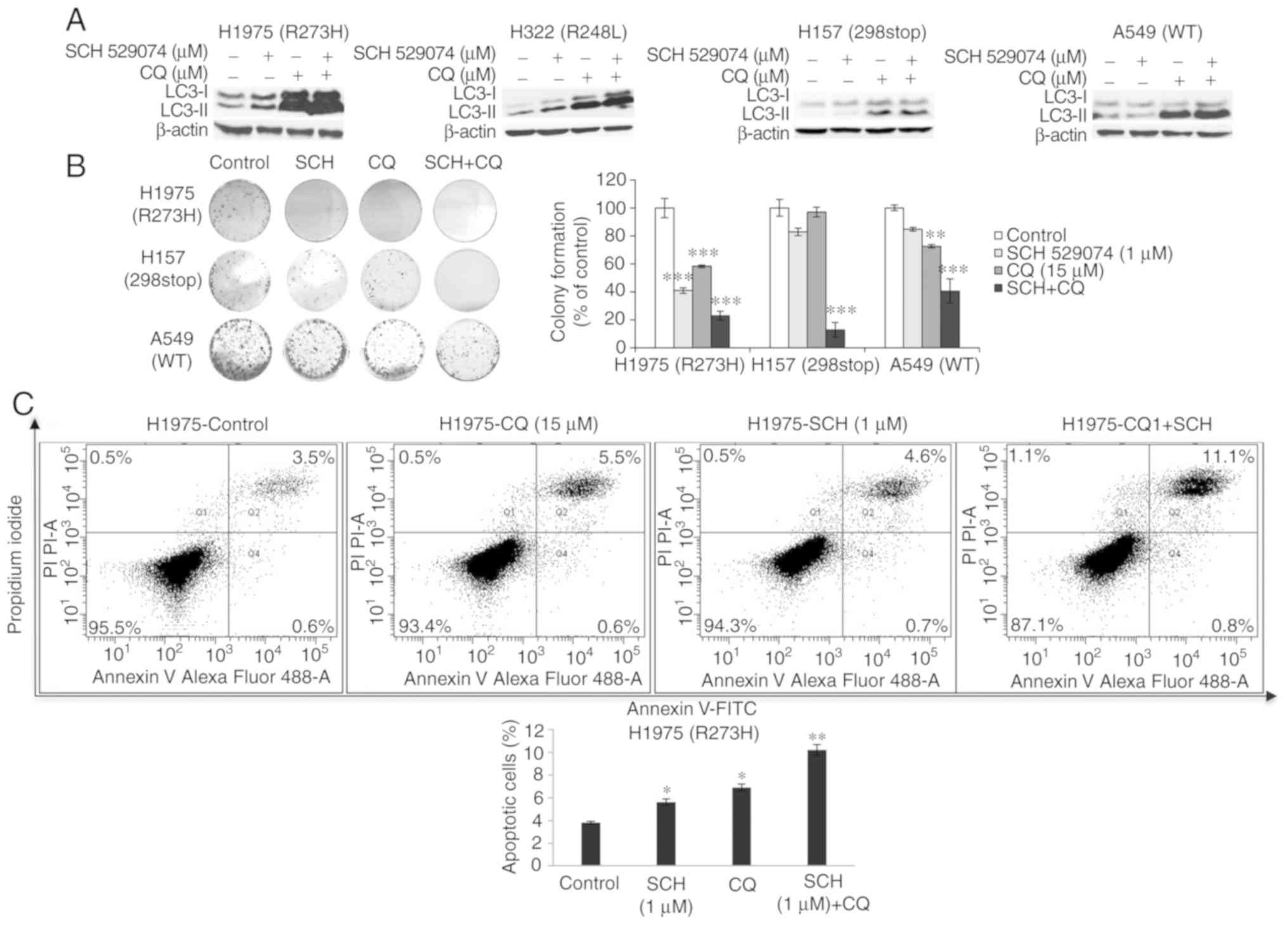
Combined treatment with CQ and SCH
529074 increases LC3-II levels, reduces cell survival and enhances
induction of apoptosis
A combination treatment of SCH 529074 and the
autophagy inhibitor CQ was performed. The cells were treated with a
low concentration of SCH 529074 (1 µM) and CQ (15 µM) for 72 h in
order to reduce the toxicity caused by the drugs. As revealed in
Fig. 6A, the levels of LC3-II were
increased in cells treated with SCH 529074 and CQ compared with
cells treated with SCH 529074 alone. However, LC3-II levels were
similar between the combined treatment and the CQ single treatment.
Moreover, combination treatment significantly reduced the ability
of NSCLC cells (H1975, H157 and A549) to form colonies compared
with that noted by single drug treatment (Fig. 6B). Furthermore, flow cytometric
analysis was performed in order to investigate if the combination
treatment could synergistically increase the apoptotic rates. SCH
529074 (1 µM) and CQ treatment produced a significantly increased
apoptotic rate in H1975 cells (for both P<0.05), whereas the
combination treatment resulted in a significantly higher apoptotic
rate compared with that observed following single drug treatment of
the cells (P<0.01, Fig. 6C).
Discussion
SCH 529074 is a small molecular weight compound that
can bind to the p53-DNA binding domain, restore growth-suppressive
activity of mutant p53 and interrupt Mdm2-mediated ubiquitination
of WT mutant p53 (10). In the
present study, the role of SCH 529074 was investigated in NSCLC
cells with different p53 mutational status and two colorectal
cancer cell lines HCT116 and HCT116 p53−/− were included
as controls, since these paired cell lines have been widely applied
to analyze the function of p53 (19,20).
Initially, the mutation status and protein
expression levels of p53 were assessed in 4 lung cancer and 2 colon
cancer cell lines. A high protein level of p53 was detected in
H1975 (R273H) and H322 (R248L) cells. Mutant p53 fails to induce
transcription of its negative regulator Mdm2, leading to loss of
ubiquitination and reduced degradation by Mdm2 (21). This results to an accumulation of
p53 in these cells (21). In line
with these findings, it was revealed that the two p53 WT cell lines
(A549 and HCT116) exhibited low levels of p53, which could be
attributed to its degradation by Mdm2. Notably, p53 expression was
not detectable in H157 cells containing a heterogeneous mutant
(298stop) form of p53. This phenomenon may be caused by epigenetic
modification of p53 and/or p53 regulatory genes. Regardless of the
p53 mutation status, suppressed cell survival was observed in all
these cell lines including HCT116 p53−/−, which was
associated with a reduced colony formation activity following drug
treatment, suggesting that SCH 529074 inhibited cell survival in a
p53-independent manner.
p53 is involved in regulating the cell cycle and in
the induction of apoptosis (5). SCH
529074 treatment resulted in G0/G1 cell cycle arrest irrespective
of the p53 status of the cells. The cell cycle is strictly
regulated by cell cycle regulators, such as cyclins, CDKs
(cyclin-dependent kinase), and CDKIs (cyclin-dependent kinase
inhibitors) (22). Following SCH
529074 treatment, one of the cyclin-dependent kinase inhibitors p21
was upregulated at both the mRNA and protein level in all assessed
NSCLC cell lines. The p21 gene was initially reported to
induce cell cycle arrest by mediating p53-dependent G1 growth
arrest (23). It has also been
revealed that p21 is an effector of multiple pathways required for
promoting p53-independent anti-proliferative activities (23). The p53-independent induction of p21
could be achieved through downregulation of c-Myc (24). In contrast to other cyclins, which
play roles in cell cycle progression, G cyclins negatively regulate
cell proliferation (25). The data
indicated that NSCLC cells expressed significantly higher levels of
CCNG2 mRNA following drug treatment and that its expression
was notably high in H157 (298stop) and A549 (p53 WT) cells.
Decreased expression of CCNG2 was detected in various tumors
including lung cancer, and it was correlated with lymph node
metastasis and poor overall survival (26). Notably, SCH 529074 treatment did not
alter the expression levels of p21 and CCNG2 in colon cancer cells,
indicating a tissue specific effect/mechanism. The data revealed
that SCH 529074 treatment induced a p53-independent cell cycle
arrest and an altered expression of cell cycle-associated genes in
lung cancer cells.
In addition to the effects of SCH 529074 on cell
cycle arrest, induction of apoptosis was observed in all NSCLC
cells via a p53-independent manner. A dose-response effect was
observed with regard to SCH 529074 treatment and the induction of
apoptosis, which was accompanied by increased caspase-3/7 activity.
Although p53 is the one of the key players in the apoptotic
pathway, other supplementary pathways independent of p53 exist
(27). However, the cell lines
(H157 and A549) that exhibited the highest apoptotic rate did not
exhibit the highest caspase-3/7 activity, suggesting that SCH
529074-induced apoptosis may be mediated by both caspase-dependent
and -independent pathways in these cells. The induction of
apoptosis is activated when the expression of the pro-apoptotic
proteins (PUMA, NOXA, BAX, BIM, BAD and BID) overcomes the
expression of the anti-apoptotic proteins (BCL-2, BCL-xl, MCl-1 and
BCL-W) (28). PUMA is a BH3-only
protein and a critical mediator of the p53-dependent and
-independent apoptotic pathways (29). For example, Wu et al revealed
that p53 independent induction of PUMA mediated intestinal
apoptosis in response to ischemia-reperfusion (30). Upon treatment of SCH 529074, PUMA
mRNA and protein levels were upregulated in all cancer cell lines
regardless of their p53 status, indicating that SCH 529074 induced
a p53-independent expression of PUMA. In HCT116 p53−/−
cells treated with 2 µM of SCH 529074, no apoptosis was present,
but when these cells were treated with 4 µM of SCH 529074, a
significantly higher early apoptotic rate was detected, in line
with the increased expression level of PUMA in this cell line. It
is reported that PUMA alone was not sufficient to induce apoptosis.
This may be due to a change in the subcellular localization of the
PUMA protein. Ambroise et al observed that neither PUMA
upregulation in normal activated human B lymphocytes, nor high
levels of PUMA in Burkitt's lymphoma cells were associated with
cell death (31). This was due to
the subcellular localization of PUMA occurring in the cytosol and
not in the mitochondria (31).
Increased PUMA levels potently induce apoptosis by antagonizing the
expression levels of the anti-apoptotic Bcl-2 family members and/or
by directly activating the pro-apoptotic protein members, leading
to mitochondrial dysfunction and initiation of the caspase
activation cascade (32). In
contrast to the enhanced expression of PUMA observed in all
assessed cell lines, no significant alteration of BAX expression
was observed. Moreover, NOXA was induced only in p53 mutant H1975
cells, which may be associated with the genetic feature of the cell
line including mutation status of p53, p53-regulatory genes and
genes interacting with p53 (33).
The expression levels of p53 were not markedly altered in mutant
cell lines, although they did change in the WT cell lines A549 and
HCT116, possibly due to the ability of the compound to interrupt
Mdm2-mediated ubiquitination of WT p53 (10). Collectively, the data indicated that
SCH 529074 may induce both p53-dependent and -independent
apoptosis. Similarly, a pre-clinical study also reported that
NJ-26854165 (Serdemetan), a p53 activating tryptamine derivative,
had growth inhibitory activities in both wild-type and mutant p53
tumors, indicating that the mechanism involves both p53-dependent
and -independent functions (34).
Several studies have reported that genotoxic stimuli
could trigger autophagy in cancer cells. Cordani et al
reported that mutant p53 interacted with the induction of autophagy
during cancer progression (35).
The present study revealed that SCH 529074 treatment caused
increased LC3-II levels regardless of the p53 mutational status,
indicating its potential involvement in autophagy. However, the
LC3-II levels in the cells treated by SCH 529074 and the autophagy
inhibitor CQ were similar to those observed in the cells treated
with CQ alone, which was possibly due to the fact that the
autophagy inhibitor may have caused the inhibition of the
autophagosome-lysosome fusion, leading to accumulation of LC3-II
(36,37). Nevertheless, combination treatment
with low concentration of SCH 529074 and the autophagy inhibitor CQ
led to reduced tumor cell survival rates compared with those
observed following single drug treatment, indicating the efficacy
of this combination therapy in NSCLC cells. It has been previously
revealed that the application of combination therapy can maximize
sensitivity and minimize the toxicity observed in the treatment of
cancer cells (38). The precise
role of SCH 529074 in autophagy requires further investigation by
the analysis of additional autophagic markers. Furthermore, the
effects of SCH 529074 should also be assessed in normal bronchial
epithelial cells and in in vivo animal models in future
studies.
In summary, the data from the present study
indicated that SCH 529074 exerts growth inhibitory effects on NSCLC
cells by inducing cell cycle arrest, apoptosis and possibly
autophagy. These results were in agreement with those reported by
Demma et al (10). However,
it was demonstrated that this compound further exhibited effects on
p53-deficient cells, indicating a p53-independent mechanism of
action. Additionally, the ability of SCH 529074 to induce autophagy
is a novel finding. SCH 529074 bears therapeutic potential and may
be used in combination with a low concentration of other drugs for
the treatment of patients with lung cancer.
Supplementary Material
Supporting Data
Acknowledgements
We thank Professor Berit Jungnickel for kindly
providing us with the colon cancer cell lines HCT116 and HCT116
p53−/−. We thank Claudia Seliger for technical
support.
Funding
The present study was supported by University
Hospital Jena.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, IP and GL conceived and designed the study. MN,
DH, ZZ, YL and YM performed the experiments. YM was responsible for
data analysis and preparation of the figures. MN and YC wrote the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar
|
|
2
|
Oser MG, Niederst MJ, Sequist LV and
Engelman JA: Transformation from non-small-cell lung cancer to
small-cell lung cancer: Molecular drivers and cells of origin.
Lancet Oncol. 16:e165–e172. 2015. View Article : Google Scholar :
|
|
3
|
Mascaux C, Tomasini P, Greillier L and
Barlesi F: Personalised medicine for nonsmall cell lung cancer. Eur
Respir Rev. 26:1700662017. View Article : Google Scholar
|
|
4
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.
|
|
5
|
Green DR and Chipuk JE: p53 and
metabolism: Inside the TIGAR. Cell. 126:30–32. 2006. View Article : Google Scholar
|
|
6
|
Mogi A and Kuwano H: TP53 mutations in
nonsmall cell lung cancer. J Biomed Biotechnol. 2011:5839292011.
View Article : Google Scholar :
|
|
7
|
Brosh R and Rotter V: When mutants gain
new powers: News from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009. View
Article : Google Scholar
|
|
8
|
Steels E, Paesmans M, Berghmans T, Branle
F, Lemaitre F, Mascaux C, Meert AP, Vallot F, Lafitte JJ and
Sculier JP: Role of p53 as a prognostic factor for survival in lung
cancer: A systematic review of the literature with a meta-analysis.
Eur Respir J. 18:705–719. 2001. View Article : Google Scholar
|
|
9
|
Yue X, Zhao Y, Xu Y, Zheng M, Feng Z and
Hu W: Mutant p53 in cancer: Accumulation, gain-of-function and
therapy. J Mol Bio. 429:1595–1606. 2017. View Article : Google Scholar
|
|
10
|
Demma M, Maxwell E, Ramos R, Liang L, Li
C, Hesk D, Rossman R, Mallams A, Doll R, Liu M, et al: SCH529074, a
small molecule activator of mutant p53, which binds p53 DNA binding
domain (DBD), restores growth-suppressive function to mutant p53
and interrupts HDM2-mediated ubiquitination of wild type p53. J Bio
Chem. 285:10198–10212. 2010. View Article : Google Scholar
|
|
11
|
Parrales A and Iwakuma T: Targeting
oncogenic mutant p53 for cancer therapy. Front Oncol. 5:2882015.
View Article : Google Scholar :
|
|
12
|
Cui T, Chen Y, Yang L, Knösel T, Zöller K,
Huber O and Petersen I: DSC3 expression is regulated by p53, and
methylation of DSC3 DNA is a prognostic marker in human colorectal
cancer. Br J Cancer. 15:1013–1019. 2011. View Article : Google Scholar
|
|
13
|
Li Y, Chen Y, Ma Y, Nenkov M, Haase D and
Petersen I: Collagen prolyl hydroxylase 3 has a tumor suppressive
activity in human lung cancer. Exp Cell Res. 363:121–128. 2018.
View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
16
|
Weinberg RA: p53 and apoptosis: Master
guardian and executioner. The Biology of Cancer. Garland Science,
Taylor & Francis Group; New York, NY: pp. 331–389. 2014
|
|
17
|
Levine B and Abrams J: p53: The Janus of
autophagy? Nat Cell Biol. 10:637–639. 2008. View Article : Google Scholar :
|
|
18
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar
|
|
19
|
Zhou X, Hai Q and Lu H: Mutant p53 in
cancer therapy-the barrier or the path. J Mol Cell Biol.
11:293–305. 2019. View Article : Google Scholar
|
|
20
|
Lee YS, Yoon S, Park MS, Kim JH, Lee JH
and Sonog CW: Influence of p53 expression on sensitivity of cancer
cells to bleomycin. J Biochem Mol Toxicol. 24:260–269. 2010.
View Article : Google Scholar
|
|
21
|
Bykov VJ, Issaeva N, Shilov A, Hultcrantz
M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002. View Article : Google Scholar
|
|
22
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar :
|
|
23
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar
|
|
24
|
Jeong JH, Kang SS, Park KK, Chang HW,
Magae J and Chang YC: p53-independent induction of G1 arrest and
p21WAF1/CIP1 expression by ascofuranone, an isoprenoid antibiotic,
through downregulation of c-Myc. Mol Cancer Ther. 9:2102–2113.
2010. View Article : Google Scholar
|
|
25
|
Arachchige Don AS, Dallapiazza RF, Bennin
DA, Brake T, Cowan CE and Horne MC: Cyclin G2 is a
centrosome-associated nucleocytoplasmic shuttling protein that
influences microtubule stability and induces a p53-dependent. Exp
Cell Res. 312:4181–4204. 2006. View Article : Google Scholar :
|
|
26
|
Sun GG, Zhang J and Hu WN: CCNG2
expression is down-regulated in colorectal carcinoma and its
clinical significance. Tumour Biol. 35:3339–3346. 2014. View Article : Google Scholar
|
|
27
|
Strasser A, Harris AW, Jacks T and Cory S:
DNA damage can induce apoptosis in proliferating lymphoid cells via
p53-independent mechanisms inhibitable by Bcl-2. Cell. 79:329–339.
1994. View Article : Google Scholar
|
|
28
|
Montero J and Letai A: Why do BCL-2
inhibitors work and where should we use them in the clinic? Cell
Death Differ. 25:56–64. 2018. View Article : Google Scholar
|
|
29
|
Yu J and Zhang L: PUMA, a potent killer
with or without p53. Oncogene. 27 (Suppl 1):S71–S83. 2008.
View Article : Google Scholar :
|
|
30
|
Wu B, Qiu W, Wang P, Yu H, Cheng T,
Zambetti GP, Zhang L and Yu J: p53 independent induction of PUMA
mediates intestinal apoptosis in response to ischaemia-reperfusion.
Gut. 56:645–654. 2007. View Article : Google Scholar
|
|
31
|
Ambroise G, Portier A, Roders N, Arnoult D
and Vazquez A: Subcellular localization of PUMA regulates its
pro-apoptotic activity in Burkitt's lymphoma B cells. Oncotarget.
6:3818–3894. 2015. View Article : Google Scholar
|
|
32
|
Zheng X, He K, Zhang L and Yu J:
Crizotinib induces PUMA-dependent apoptosis in colon cancer cells.
Mol Cancer Ther. 12:777–786. 2013. View Article : Google Scholar :
|
|
33
|
Hafner A, Bulyk ML, Jambhekar A and Lahav
G: The multiple mechanisms that regulate p53 activity and cell
fate. Nat Rev Mol Cell Biol. 20:199–210. 2019. View Article : Google Scholar
|
|
34
|
Smith MA, Gorlick R, Kolb EA, Lock R,
Carol H, Maris JM, Keir ST, Morton CL, Reynolds CP, Kang MH, et al:
Initial testing of JNJ-26854165 (Serdemetan) by the pediatric
preclinical testing program. Pediatr Blood Cancer. 59:329–332.
2012. View Article : Google Scholar
|
|
35
|
Cordani M, Butera G, Pacchiana R and
Donadelli M: Molecular interplay between mutant p53 proteins and
autophagy in cancer cells. Biochim Biophys Acta Rev Cancer.
1867:19–28. 2017. View Article : Google Scholar
|
|
36
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar
|
|
37
|
Huang S and Sinicrope FA:
Celecoxib-induced apoptosis is enhanced by ABT-737 and by
inhibition of autophagy in human colorectal cancer cells.
Autophagy. 6:256–269. 2010. View Article : Google Scholar :
|
|
38
|
Matlock K, Berlow N, Keller C and Pal R:
Combination therapy design for maximizing sensitivity and
minimizing toxicity. BMC Bioinformatics. 18 (Suppl 4):S1162017.
View Article : Google Scholar
|