Introduction
The number of deaths from lung cancer has been
increasing exponentially over the recent years worldwide (1). This malignant tumor is mainly caused
by environmental pollution and smoking (2). Up to 85% of all lung cancer cases are
of the NSCLC subtype, which is furthermore classified into
adenocarcinoma, squamous-cell carcinoma and large-cell carcinoma
(3). Therapeutic techniques for
NSCLC include surgery, targeted therapy, chemotherapy,
radiotherapy, immunotherapy, or their combination (2). In spite of recent developments in
diagnostic and therapeutic approaches of NSCLC, the prognosis of
NSCLC patients is still unsatisfactory (3,4).
Therefore, identifying the potential molecular targets and defining
the underlying molecular mechanisms involved in NSCLC progression
and metastasis are key to discovering the effective targets to
improve its prognosis.
Sirtuins consisting of sirtuin 1–7 (SIRT1-7) members
belong to the NAD+-dependent histone/protein deacetylase
family (5,6). Sirtuins bear a highly conserved
NAD+-binding and catalytic core domain but display
diverse deacetylation and ADP-ribosylation enzymatic activities,
substrate proteins and cellular localizations and functions
(5–7). Sirtuins have been previously reported
to participate in a number of cellular processes including
senescence, metabolism, proliferation, differentiation, apoptosis,
DNA repair, genomic stability, chromatin remodeling, gene
transcription and post-translational modification (5,6,8). Of
all the sirtuins, SIRT7 is a newly discovered member. SIRT7 can
promote ribosome biogenesis via inducing synthesis of ribosomal RNA
and transfer RNA (9–11). Notably, SIRT7 is a highly selective
acetylated histone H3 lysine 18 (H3K18Ac) deacetylase and
SIRT7-mediated deacetylation of H3K18Ac maintains oncogenic
transformation by transcriptionally suppressing expression of tumor
suppressor genes (12).
Accumulating evidence (13–22) has demonstrated that SIRT7 is
upregulated and functions as an oncogene in most cancers including
breast, thyroid, colorectal and gastric cancer, hepatocellular
carcinoma, cholangiocarcinoma, prostate cancer, bladder cancer and
angiosarcoma. Furthermore, high expression of SIRT7 has been
revealed to contribute to tumor cell resistance to chemotherapy and
radiotherapy (20,23,24).
Conversely, SIRT7 has been demonstrated to be downregulated in the
oral, neck and head squamous cell carcinoma and pancreatic cancer
(25–27). SIRT7 can attenuate tumor cell EMT
and metastasis through deacetylating mothers against
decapentaplegic homolog 4 (SMAD4) to antagonize transforming growth
factor-β (TGF-β) signaling (27,28).
These studies indicate that SIRT7 exerts tumor-promoting or
tumor-suppressive properties.
Several studies have revealed that SIRT7 plays key
roles in human lung cancer (29,30).
SIRT7 was revealed to be upregulated in human NSCLC cells and its
depletion could inhibit NSCLC cell growth by activating the
pro-apoptotic signaling pathway (29). miR-3666 suppressed NSCLC cell growth
by targeting SIRT7 (29). SIRT7
depletion could further sensitize NSCLC cells to gemcitabine
chemotherapy via inhibiting autophagy (30). However, the effects and the clinical
roles of SIRT7 in lung cancer are less understood. The present
study detected the expression of SIRT7 in clinical tissues of human
lung cancer and NSCLC cells, and analyzed the clinical association
between SIRT7 expression of lung cancer tissues and its
clinicopathological characteristics. The roles of SIRT7
overexpression or knockdown in growth and metastasis of NSCLC cells
were determined and the related molecular mechanism was
elucidated.
Materials and methods
Cellular and molecular biological
reagents
Dulbecco's modified Eagle's medium (DMEM) was
supplied by HyClone; GE Healthcare Life Sciences. Fetal bovine
serum (FBS) and puromycin were supplied by Gibco; Thermo Fisher
Scientific, Inc. Penicillin-streptomycin antibiotics, the
NheI and SgsI restriction enzymes and the reverse
transcription kit were supplied by Thermo Fisher Scientific, Inc.
Taq DNA polymerase, dNTPmix, T4 DNA ligase and the RNA extraction
kit were supplied by TaKaRa Biotechnology Co., Ltd. SYBR Green
Master was supplied from Roche Applied Science. Lipofectamine 2000
was supplied by Invitrogen; Thermo Fisher Scientific, Inc.
Blasticidin S was supplied by Yeasen Biotechnology Co., Ltd. Cell
Counting Kit-8 (CCK-8) was supplied by Beijing Solarbio Science
& Technology Co., Ltd. Propidium iodide (PI)/RNase Staining
Buffer Solution was supplied by BD Biosciences. The bicinchoninic
acid protein assay kit, the mammalian cell lysis buffer for western
blotting and the SuperEnhanced chemiluminescence detection (BeyoECL
Star) kit were supplied by Beyotime Institute of Biotechnology. The
diaminobenzidine substrate was supplied by Boster Biological
Technology. The primers were supplied by Sangon Biotech Co.,
Ltd.
Antibodies
Primary antibody rabbit anti-SIRT7 (cat. no.
12994-1-AP) was supplied by ProteinTech Group, Inc. Rabbit anti-AKT
(cat. no. AF6261), anti-ERK1/2 (cat. no. AF0155), anti-p-AKT
(Thr308) (T308) (cat. no. AF3262), anti-p-AKT (Ser473) (S473) (cat.
no. AF0016) and anti-p-ERK1/2 (Thr202/Tyr204) (cat. no. AF1015)
primary antibodies were supplied by Affinity Biosciences. Primary
antibodies including rabbit anti-β-actin (cat. no. YM3214),
anti-p21 (cat. no. YT3497), anti-p27 (cat. no. YT3502), anti-cyclin
D1 (cat. no. YT1173), anti-cyclin E1 (cat. no. YT1176), anti-CDK2
(cat. no. YT0832) and anti-CDK4 (cat. no. YT5198) were supplied by
ImmunoWay Biotechnology Company. Primary antibodies including
rabbit anti-E-cadherin (product no. 3195), anti-N-cadherin (product
no. 13116), anti-Vimentin (product no. 5741), anti-Snail (Snail1)
(product no. 3879) and anti-Slug (Snail2) (product no. 9585) for
western blotting were supplied by Cell Signaling Technology, Inc.
Primary antibody rabbit anti-Snail (cat. no. 101167-T10) for
immunohistochemistry was supplied by Sino Biological, Inc. Primary
antibody rabbit anti-Slug (cat. no. GTX128796) for
immunohistochemistry was supplied by GeneTex, Inc. Secondary
antibody horseradish peroxidase (HRP)-conjugated anti-rabbit IgG
(product no. 7074 for western blotting and product no. BM3894 for
immunohistochemistry) was supplied by Cell Signaling Technology,
Inc. and Boster Biological Technology, respectively.
Plasmid and lentiviral vectors
The pGEM/SIRT7 cloning plasmid carrying human SIRT7
coding sequence (CDS) (BC017305) was supplied by Sino Biological,
Inc. Plasmids including pLenti6.3/IRES/GFP lentiviral
plasmid-expressing blasticidin S deaminase and green fluorescent
protein (GFP) as well as pLP1, pLP2 and VSVG lentiviral packing
plasmids were supplied by Novobio Scientific, Inc. Lentiviruses
including control shRNA lentivirus (LV-shcontrol) and SIRT7 shRNA
(h) lentivirus (LV-shSIRT7) containing puromycin resistance gene
were supplied by Santa Cruz Biotechnology, Inc.
Cell culture
The human embryonic kidney cell line 293T, the
normal human bronchial epithelial cell line HBEpiC and the human
NSCLC cell lines including H292, A549, H1299 and H1975 were
supplied by the Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences. Cells were grown in culture medium (DMEM
containing 10% FBS and 100 U/ml of penicillin-streptomycin
antibiotics), identified by short tandem repeat (STR) profiling
(Genetic Testing Biotechnology) and confirmed to have no mycoplasma
contamination. 293T cells were cultured for 2 weeks before
transfected with plasmids for the construction of lentiviral
vectors, whereas HBEpiC, H292, A549, H1299 and H1975 cells were
cultured for 1 week for the following RT-qPCR, western blotting and
infection experiments. The culture medium supplemented with 10
µg/ml of blasticidin S was used to grow SIRT7-overexpressed
A549-SIRT7 and control A549-mock NSCLC cell lines. The culture
medium supplemented with 2 µg/ml of puromycin was used to grow
SIRT7-silenced H292-shSIRT7 and control H292-shcontrol NSCLC cell
lines. After antibiotic selection for about 4 weeks, stable
transgenic cell lines were established and frozen at once. For
in vitro assays, the frozen transgenic cells were thawed and
cultured for 2 weeks to be amplified. The cells were then used for
RT-qPCR and western blotting assays as well as in vitro
functional assays including CCK-8, colony formation, cell cycle,
wound healing and Transwell migration/invasion. The culture
duration of each in vitro functional assay was subsequently
specified. For in vivo animal experiments, the transgenic
cells were thawed and cultured for 3 weeks to be amplified before
being injected into nude mice. The aforementioned cells were
cultured in a humidified chamber at 37°C under 5% CO2 to
obtain enough cells for each of the following experiments.
Nude mice
Forty-eight four-week-old female athymic BALB/c nude
mice (average weight, 15 g) were supplied by Shanghai Laboratory
Animal Center. The mice were maintained in the animal facility at
Soochow University (Suzhou, China) under specific pathogen-free
conditions with a 12-h light/dark cycle at 22±2°C and 60±5%
humidity, provided with free access to food and water. All animal
experiments were approved by the Animal Research Ethics Committee
of Soochow University (IRB no. A201809059).
Lung cancer tissue specimens
We obtained 102 pairs of human lung cancer tumor
tissues and adjacent non-tumor lung tissues (collected at a
distance of more than 6 cm from the tumor site) derived from 102
lung cancer patients (age range, 39–83 years; 52 male and 50 female
patients) at the Department of Cardio-Thoracic Surgery of the First
Affiliated Hospital of Soochow University (Suzhou, China) from
January 2016 to April 2017. The patients had undergone lung cancer
surgery but had not received any neoadjuvant chemotherapy or
radiotherapy. The collected tissue samples were fixed in 10%
neutral formalin for 24 h at room temperature, and subsequently
embedded in paraffin. Two experienced pathologists independently
performed the pathological staging of tumors. The medical history
of patients was reviewed to describe their clinicopathological
features. The present study was conducted after approval by the
Ethics Committee of the First Affiliated Hospital of Soochow
University (IRB no. 2016128). Signed informed consent was obtained
from all the participants.
Tissue microarray (TMA) and section
preparation
After tissue localization by hematoxylin and eosin
(H&E) analysis, the aforementioned paired lung cancer tumor
tissue and adjacent non-tumor lung tissue specimens fixed with
formalin and embedded in paraffin were used for preparation of
human lung cancer TMA with a sample diameter of 1.5 mm (102 cases,
102 pairs, 204 dots). The TMA was then cut into 3 µm-thick sections
for subsequent immunohistochemistry analysis.
Construction and titration of
lentiviral vectors
Amplification of CDS fragment (1203 bp) of human
SIRT7 was performed by polymerase chain reaction (PCR) with
pGEM/SIRT7 plasmid template and human full-length SIRT7
CDS-specific primer pair (SIRT7-F1:
5′-CTAGCTAGCGCCACCATGGCAGCCGGGGGTCTGAG-3′ and SIRT7-R1:
5′-TTGGCGCGCCTTACGTCACTTTCTTCCTTTTTGTGCG-3′). It was then inserted
into the lentiviral plasmid pLenti6.3/IRES/GFP between NheI
and SgsI sites to generate a recombinant lentiviral plasmid
pLenti6.3/SIRT7/IRES/GFP. The 293T cells were seeded in 10-cm
dishes at a density of 2×106 cells/10 ml medium/dish and
cultured overnight. Subsequently, the plasmid
pLenti6.3/SIRT7/IRES/GFP or pLenti6.3/IRES/GFP was co-transfected
into 293T cells with pLP1, pLP2 and VSVG by Lipofectamine 2000. The
cells were cultured for another 48 h at 37°C. The lentivirus
expressing SIRT7 (LV-SIRT7) and the blank lentivirus (LV) were
consequently produced and concentrated by ultracentrifugation. The
lentiviral biological titre (TU/ml) was determined based on the sum
of GFP-positive 293T cells as observed by fluorescence
microscopy.
Establishment of stable cell
lines
The A549 NSCLC cell line was infected with LV-SIRT7
or LV (control) at a multiplicity of infection (MOI) of 50 and
selected with blasticidin S (10 µg/ml), leading to generation of
A549-SIRT7 (SIRT7-overexpressed) or A549-mock (control) transgenic
cell line. The H292 NSCLC cell line was infected with LV-shSIRT7 or
LV-shcontrol (control) (50 MOI) and selected with puromycin (2
µg/ml), resulting in formation of H292-shSIRT7 (SIRT7-silenced) or
H292-shcontrol (control) transgenic cell line. The transgene
efficiency in A549 NSCLC cells was analyzed using the GFP reporter
by fluorescence microscopy and flow cytometry. The efficiency of
SIRT7 overexpression or knockdown in A549 or H292 NSCLC cells,
respectively, was analyzed by reverse transcription-quantitative
PCR (RT-qPCR) and western blotting.
RT-qPCR
The total RNAs of A549, H292, H1299, H1975, HBEpiC,
A549-SIRT7, A549-mock, H292-shSIRT7 and H292-shcontrol cells were
purified by a MiniBEST universal RNA extraction kit following the
supplier's instructions and reversely-transcribed to first-strand
cDNAs with a RevertAid RT Reverse Transcription kit. Subsequently,
qPCR analysis of human SIRT7 mRNA expression was performed by
SYBR-Green Master using human SIRT7-specific primer pair (SIRT7-F2:
5′-ACTTGGTCGTCTACACAGGC-3′ and SIRT7-R2:
5′-GGTGATGCTCATGTGGGTGA-3′, product size: 158 bp) (β-actin used as
an internal control) as previously described (31). RT was conducted at 42°C for 60 min,
followed by 70°C for 5 min. qPCR amplification was conducted at
95°C for 300 sec followed by 40 cycles at 95°C for 10 sec and 60°C
for 30 sec. SIRT7 mRNA expression level in A549, H292, H1299 and
H1975 NSCLC cells (HBEpiC used as a cell control);
SIRT7-overexpressed A549-SIRT7 NSCLC cells (A549-mock used as a
cell control); and SIRT7-silenced H292-shSIRT7 NSCLC cells
(H292-shcontrol used as a cell control) was standardized to β-actin
expression and computed via the 2−∆∆Cq method (32), respectively.
CCK-8 assay
In 96-well plates, the A549-SIRT7 vs. A549-mock and
H292-shSIRT7 vs. H292-shcontrol human NSCLC cells (1×104
cells/200 µl medium) were seeded per well. At the 1st, 2nd, 3rd and
4th day following cell culture, the tumor cell vitality was
evaluated by CCK-8 (10 µl/well). Using an automatic microplate
reader at 450 nm, the optical density (OD) was read. The growth
curve of tumor cells in vitro was plotted as the changes in
OD value over culture time.
Colony formation assay
In 6-well plates, the A549-SIRT7 vs. A549-mock and
H292-shSIRT7 vs. H292-shcontrol human NSCLC cells (200 cells/2 ml
medium) were seeded per well. After being incubated for two weeks,
the colonies of tumor cells were fixed with 4% paraformaldehyde for
15 min followed by staining in 0.5% crystal violet for 20 min at
room temperature. Colonies of >10 cells were counted under a
white light field using fluorescence microscope IX51 (Olympus
Corporation; magnification, ×200). Analysis of the relative
clonogenic ability of these tumor cells was then carried out by
GraphPad Prism 6 software (GraphPad, Inc.).
Cell cycle assay
After being cultured in 6-well plates, the
A549-SIRT7 vs. A549-mock and H292-shSIRT7 vs. H292-shcontrol human
NSCLC cells (0.5×106 cells) were collected and incubated
overnight at 4°C in ice-cold 70% ethanol. The PI/RNase Staining
Buffer Solution (500 µl per reaction) was then used to stain the
cells in the dark for 30 min. Finally, the cell cycle was detected
using CytoFLEX flow cytometer (Beckman Coulter, Inc.) and analyzed
with CXP analysis software 2.2 (Beckman Coulter, Inc.).
Wound healing assay
In 6-well plates, the A549-SIRT7 vs. A549-mock and
H292-shSIRT7 vs. H292-shcontrol human NSCLC cells (5×105
cells/5 ml medium) were seeded per well. Upon almost 100%
confluence of cells, scratches were generated and reference points
were marked on the outer bottom of 6-well plates nearby. Cellular
debris was removed from the wells by rinsing with fresh medium. To
maintain cell growth, the wells were supplemented with DMEM
containing 2% FBS. The migration of tumor cells was studied by
microscopic observation (white light field, fluorescence microscope
IX51; magnification, ×100) at 0, 24 and 48 h after wounding. The
capability of migration of tumor cells was quantitatively analyzed
by ImageJ 1.52v software (National Institutes of Health).
Transwell migration/invasion
assay
In 8 µm-pore size 24-well Transwell filters (EMD
Millipore), the A549-SIRT7 vs. A549-mock and H292-shSIRT7 vs.
H292-shcontrol human NSCLC cells with a density of 2×104
or 2×105 cells/100 µl serum-free medium were placed in
the upper chamber without or with 50 µl Matrigel (1:8 pre-diluted
with serum-free medium) (Corning, Inc.) for Transwell migration or
invasion assays, respectively. The lower chamber was supplemented
with culture medium. After 24 h, the tumor cells which had migrated
or invaded into bottom side were placed in 4% paraformaldehyde to
be fixed for 15 min and 0.5% crystal violet for 20 min at room
temperature to be stained. Microscopy (white light field,
fluorescence microscope IX51; magnification, ×200) was then used to
assess the migratory and invasive abilities of tumor cells.
Tumor xenograft mouse models
For establishment of a tumor subcutaneous xenograft
model, the A549-SIRT7 vs. A549-mock and H292-shSIRT7 vs.
H292-shcontrol human NSCLC cells (2×106 cells per mouse;
6 mice per group) were injected subcutaneously into the right
flanks of nude mice. The growth of tumors was tracked by monitoring
the tumor volume every week until 4 weeks after inoculation of
tumor cells and the weight 4 weeks after inoculation of tumor
cells. Tumor diameter was measured by a Vernier calliper every week
and the tumor volume was calculated using a formula: Volume
(mm3)=a2b/2 (a is the shortest diameter,
whereas b is the longest diameter). Four weeks after subcutaneous
injection, the tumor-bearing mice were humanely euthanized by
continuous inhalation with 30% CO2 for 5 min. Death was
confirmed when mice had no heartbeat for 30 sec accompanied by no
response to the toe pinch reflex. Subsequently, the xenograft
tumors were removed and weighed by an electronic analytical
balance. The xenograft tumor-derived sections (3 µm-thick) were
then prepared and used for subsequent immunohistochemical analysis.
For establishment of a tumor lung metastasis model, the
aforementioned tumor cells (2×106 cells per mouse; 6
mice per group) were injected intravenously into the tail veins of
nude mice. Six weeks after intravenous injection, the mice were
euthanized as aforementioned to harvest their lung tissues. The
lung tissue sections (3 µm-thick) were then prepared and used for
H&E analysis of lung metastatic nodules.
Western blotting
The protein expression of SIRT7 in A549, H292,
H1299, H1975 and HBEpiC cells as well as SIRT7, p-AKT
(T308/S473)/total AKT, p-ERK1/2/total ERK1/2, p21/27, cyclin D1/E1,
CDK2/4, E/N-cadherin, vimentin, Snail and Slug in A549-SIRT7,
A549-mock, H292-shSIRT7 and H292-shcontrol cells was analyzed by
western blotting (β-actin was used as a loading control) as
previously reported (31). The
primary antibodies and HRP-conjugated anti-rabbit IgG secondary
antibody were diluted at 1:1,000 and 1:3,000, respectively.
Membranes were incubated with the primary antibodies overnight at
4°C overnight and a secondary antibody for 1 h at room temperature.
Protein signals were determined by BeyoECL Star kit, scanned by Gel
Imaging System and quantified by ImageJ 1.52v software (National
Institutes of Health).
Immunohistochemistry (IHC)
The prepared 3 µm-thick human lung cancer TMA
section was subjected to IHC analysis of SIRT7 using rabbit
anti-SIRT7 (1:50) primary antibody and HRP-conjugated anti-rabbit
IgG (1:1,000) secondary antibody as previously described (31). SIRT7 expression level of each
specimen was evaluated by a weighted IHC score (0-1, -; 2–3, +;
4–5, ++; and 6–7, +++) (31). A
weighted score of ≥4 (++ or +++) was considered as SIRT7 high
expression in a tissue specimen. In addition, the sections derived
from A549-SIRT7, A549-mock, H292-shSIRT7 and H292-shcontrol
subcutaneous xenograft tumors were subjected to IHC analysis of
p-AKT (T308) (1:50), p-AKT (S473) (1:50), AKT (1:50), p-ERK1/2
(1:50), ERK1/2 (1:100), p21 (1:200), p27 (1:100), cyclin D1
(1:100), cyclin E1 (1:100), CDK2 (1:100), CDK4 (1:100), E-cadherin
(1:200), N-cadherin (1:125), vimentin (1:100), Snail (1:500) and
Slug (1:100), respectively. Briefly, the xenograft tumor-derived
sections (3 µm-thick) were fixed in 10% neutral formalin for 24 h
at room temperature and embedded in paraffin. After
deparaffinization, rehydration, rinse, antigen retrieval, quenching
of endogenous peroxidase activity with 3%
H2O2 and blocking with 5% bovine serum
albumin (BSA), the sections were incubated with primary antibodies
at 4°C overnight and a secondary antibody for 1 h at room
temperature. Finally, the sections were stained with
diaminobenzidine tetrahydrochloride (DAB) for 5 min and then
counterstained with H&E for 5 min at room temperature.
Following dehydration and mounting, observation was performed and
images were captured under a white light field using fluorescence
microscope IX51 (magnification, ×100). Protein expression was
analyzed by ImageJ 1.52v software (National Institutes of
Health).
AKT inhibition assay
The A549-SIRT7 human NSCLC cells were pre-treated
with an allosteric AKT inhibitor MK-2206 (APExBIO) (10 µM) or
dimethylsulfoxide (DMSO) without MK-2206 (vehicle control) for 1 h.
Then the MK-2206- and DMSO-treated A549-SIRT7 cells as well as the
untreated A549-SIRT7 and A549-mock cells were subjected to CCK-8,
wound healing and Transwell invasion assays. Additionally, the
aforementioned cells were cultured for another 24 h and then
subjected to western blot analysis of EMT markers.
Statistical analyses
The data of the IHC scoring of SIRT7 expression in
lung cancer tissues were presented as -, +, ++ or +++ and a
Mann-Whitney U test was used to assess the difference of the rank
data. The categorical data of high or low expression of SIRT7 in
lung cancer tissues were presented as the percentage of total cases
and a Pearson's χ2 test was used to analyze the
differences. The measurement data of the in vitro cell
studies and in vivo animal studies were analyzed by a normal
distribution test and presented as the mean ± standard deviation
(SD) when P>0.1. A Student's t-test was then used for
comparisons of differences between two independent groups. After
further testing homogeneity of variance (P>0.1 indicated
homogeneity of variance), one-way or two-way repeated measures
analysis of variance (ANOVA) with least significant difference
(LSD) post hoc multiple comparisons were used to analyze the
differences among groups. All aforementioned statistical tests were
performed with SPSS17.0 (SPSS, Inc.). A two-sided value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
SIRT7 is increased in human lung
cancer tissues and cell lines
For determination of the expression level of SIRT7
in human lung cancer clinical tissues, SIRT7 expression in human
lung cancer TMA (102 pairs of lung cancer tumor tissue and adjacent
non-tumor tissue specimens) was analyzed by IHC (Fig. 1A). Among these lung cancer tumor
tissues, high SIRT7 expression was observed in 71.6% of the cases
(73 cases; 35 cases scored ‘+++’ and 38 cases scored ‘++’) and low
SIRT7 expression was detected in 28.4% of the cases (29 cases; 18
cases scored ‘+’ and 11 cases scored ‘−’) (Fig. 1B and C). Whereas in the matched
adjacent non-tumor tissues, there were only 32.4% of cases (33
cases; 10 cases scored ‘+++’ and 23 cases scored ‘++’) exhibiting
high SIRT7 expression and 67.6% of cases (69 cases; 39 cases scored
‘+’ and 30 cases scored ‘−’) showing low SIRT7 expression (Fig. 1B and C). The data demonstrated that
SIRT7 was significantly increased in the human lung cancer tumor
tissues by comparison with the adjacent non-tumor control tissues
(P<0.05) (Fig. 1A-C).
Subsequently, the level of SIRT7 in human NSCLC cell lines
including A549, H292, H1299 and H1975 were detected by RT-qPCR
(Fig. 1D) and western blot
(Fig. 1E and F) analyses, revealing
that these 4 types of human NSCLC cell lines also expressed a
significantly higher level of SIRT7 than HBEpiC control cell line
(P<0.05). These results revealed that SIRT7 is elevated in human
lung cancer tissues and cell lines.
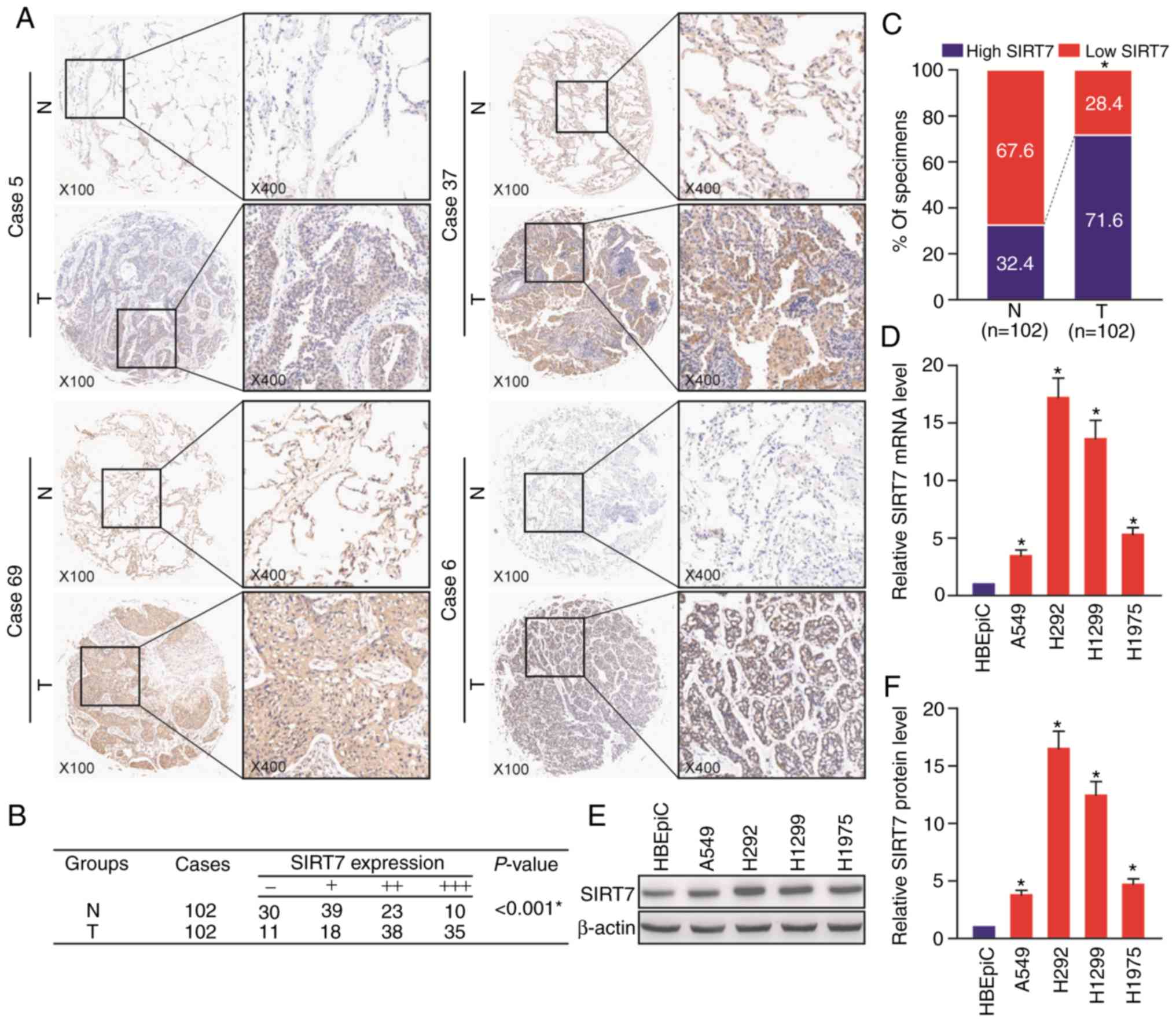 | Figure 1.SIRT7 is increased in human lung
cancer tissues and cell lines. (A-C) IHC analysis of SIRT7 in human
lung cancer TMA. (A) The representative IHC images of Case 5 (tumor
tissue IHC score: -), Case 37 (tumor tissue IHC score: +), Case 69
(tumor tissue IHC score: ++) and Case 6 (tumor tissue IHC score:
+++). (B) The number of IHC scoring -, +, ++ or +++ cases in 102
paired lung cancer tumor/adjacent non-tumor tissues. P<0.001,
Mann-Whitney U test. (C) The percentage of high/low SIRT7
expression in lung cancer tumor/adjacent non-tumor tissues.
*P<0.05, Pearson's χ2 test. T, tumor tissue; N,
adjacent non-tumor tissue. (D) The relative level of SIRT7 mRNA in
human NSCLC cells (HBEpiC served as a control) analyzed by RT-qPCR.
(E and F) Analysis of SIRT7 protein in NSCLC cells by western
blotting. (E) The representative western blot images. (F) The
relative level of SIRT7 protein in NSCLC cells (HBEpiC served as a
control). *P<0.05, one-way repeated measures ANOVA with LSD post
hoc multiple comparisons, n=6 per group. SIRT7, sirtuin 7; IHC,
immunohistochemistry; TMS, tissue microarray; NSCLC, non-small cell
lung cancer; ANOVA, analysis of variance; LSD, least significant
difference. |
Upregulation of SIRT7 is correlated
with malignant clinicopathological features in human lung
cancer
Based on SIRT7 expression level of lung cancer tumor
tissues, 29 patients with low SIRT7 expression (‘+’ and ‘−’) in
tumor tissues were classified as the SIRT7-low expression group,
and 73 patients with high SIRT7 expression (‘+++’ and ‘++’) in
tumor tissues were classified as the SIRT7-high expression group.
The association between high or low SIRT7 expression in lung cancer
tissues and clinicopathological variables including age, sex,
histology, differentiation, tumor size, pathologic stage and lymph
node metastasis was then assessed (Table I). The data revealed that the SIRT7
expression level was positively associated with malignant
clinicopathological characteristics. A high level of SIRT7 was
associated with large tumor size, high pathologic stage and
presence of lymph node metastasis (P<0.05), indicating that
increased expression SIRT7 may be involved in the progression and
metastasis of human lung cancer.
 | Table I.The association of SIRT7 expression
with lung cancer clinicopathological features. |
Table I.
The association of SIRT7 expression
with lung cancer clinicopathological features.
| Variables | Total cases (n=102)
(%) | SIRT7-low
expression (n=29; 28.4%) | SIRT7-high
expression (n=73; 71.6%) | P-value |
|---|
| Age (years) |
|
|
| 0.433 |
|
≤65 | 52 (51.0) | 13 | 39 |
|
|
>65 | 50 (49.0) | 16 | 34 |
|
| Sex |
|
|
| 0.925 |
|
Male | 52 (51.0) | 15 | 37 |
|
|
Female | 50 (49.0) | 14 | 36 |
|
| Histology |
|
|
| 0.483 |
|
Adenocarcinoma | 76 (74.5) | 23 | 53 |
|
|
SCC/Other | 26 (25.5) | 6 | 20 |
|
|
Differentiation |
|
|
| 0.069 |
|
Well | 8 (7.8) | 5 | 3 |
|
|
Moderate/Poor | 94 (92.2) | 24 | 70 |
|
| Tumor size |
|
|
|
<0.001a |
| T1 | 44 (43.1) | 21 | 23 |
|
|
T2/T3/T4 | 58 (56.9) | 8 | 50 |
|
| Pathologic
stage |
|
|
| 0.036a |
|
I/II | 77 (75.5) | 26 | 51 |
|
|
III/IV | 25 (24.5) | 3 | 22 |
|
| Lymph node
metastasis |
|
|
| 0.012a |
| N0 | 69 (67.6) | 25 | 44 |
|
|
N1/N2 | 33 (32.4) | 4 | 29 |
|
Forced expression or knockdown of
SIRT7 in human NSCLC cells
To generate a SIRT7-overexpressed lung cancer cell
line, LV-SIRT7 or LV (control) lentivirus infected into the A549
NSCLC cell line (relative low expression of SIRT7) and stable cell
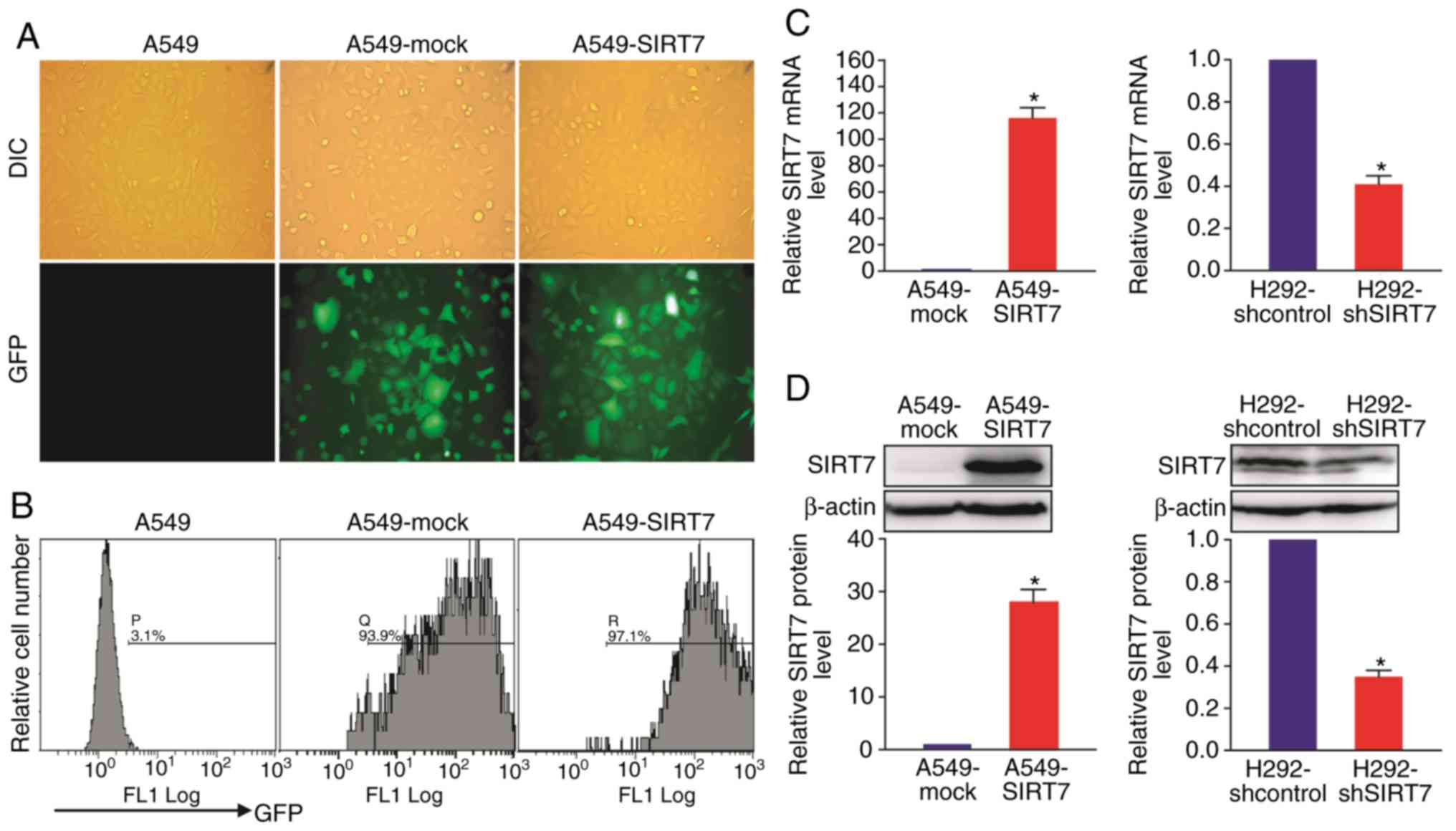
lines were selected by blasticidin S. According to the fluorescence
microscopic (Fig. 2A) and flow
cytometric (Fig. 2B) detection,
nearly all (more than 90%) of the LV-SIRT7- or LV-infected A549
(A549-SIRT7 or A549-mock) NSCLC cells expressed GFP, demonstrating
a high efficiency of transgene expression. The lentivirus-directed
SIRT7 overexpression efficiency in A549 cells was then determined
using RT-qPCR and western blot analyses, and it was revealed that
both the mRNA (Fig. 2C) and protein
(Fig. 2D) levels of SIRT7 in
A549-SIRT7 cells were increased compared with A549-mock control
cells (P<0.05). To establish a SIRT7-silenced lung cancer cell
line, the H292 NSCLC cell line (relative high expression of SIRT7)
was infected with LV-shSIRT7 or LV-shcontrol (control) lentivirus
and selected by puromycin. Results of RT-qPCR (Fig. 2C) and western blotting (Fig. 2D) further demonstrated that compared
with LV-shcontrol-infected H292 (H292-shcontrol) control cells,
LV-shSIRT7-infected H292 (H292-shSIRT7) cells exhibited a reduction
in mRNA and protein expression of SIRT7 (P<0.05). These data
revealed that SIRT7-overexpressed A549 and SIRT7-silenced H292
human NSCLC cell lines were successfully obtained.
SIRT7 promotes human NSCLC cell growth
and G1 to S phase transition
To address whether SIRT7 influences human NSCLC cell
growth, a CCK-8 assay was used to assess the proliferation and
viability of A549-SIRT7 vs. A549-mock and H292-shSIRT7 vs.
H292-shcontrol human NSCLC cells in vitro. As revealed in
Fig. 3A, overexpression of SIRT7
significantly promoted A549 NSCLC cell growth in vitro,
whereas knockdown of SIRT7 suppressed H292 NSCLC cell growth in
vitro (P<0.05). Moreover, colony formation assay (Fig. 3B and C) demonstrated that A549-SIRT7
NSCLC cells formed larger and more colonies than A549-mock control
cells, whereas H292-shSIRT7 NSCLC cells generated smaller and less
colonies than H292-shcontrol control cells (P<0.05). The results
indicated that SIRT7 can enhance clonogenicity of human NSCLC
cells. To further determine whether SIRT7 could accelerate growth
of human NSCLC cells in vivo, a human NSCLC subcutaneous
xenograft model was established by injecting A549-SIRT7 vs.
A549-mock or H292-shSIRT7 vs. H292-shcontrol cells into athymic
BALB/c nude mice. The tumor growth including tumor volume and
weight of the aforementioned cells was monitored. The in
vivo data (Fig. 3D-F) also
demonstrated that overexpression of SIRT7 significantly promoted
A549 subcutaneous xenograft tumor growth, whereas knockdown of
SIRT7 significantly inhibited H292 subcutaneous xenograft tumor
growth (P<0.05). To examine the cellular mechanism by which
SIRT7 promotes NSCLC cell growth, a cell cycle assay was performed
and A549-SIRT7 vs. A549-mock and H292-shSIRT7 vs. H292-shcontrol
NSCLC cells were analyzed by flow cytometry. According to Fig. 3G and H, in A549 cells,
overexpression of SIRT7 favored transition of the cell cycle phase
from G1 to S and accumulation of S phase (P<0.05). In contrast,
in H292 cells, knockdown of SIRT7 induced G1-phase arrest and
S-phase reduction (P<0.05). Therefore, the aforementioned
results demonstrated the roles of SIRT7 in promoting human NSCLC
cell proliferation and growth possibly by facilitating G1 to
S-phase transition.
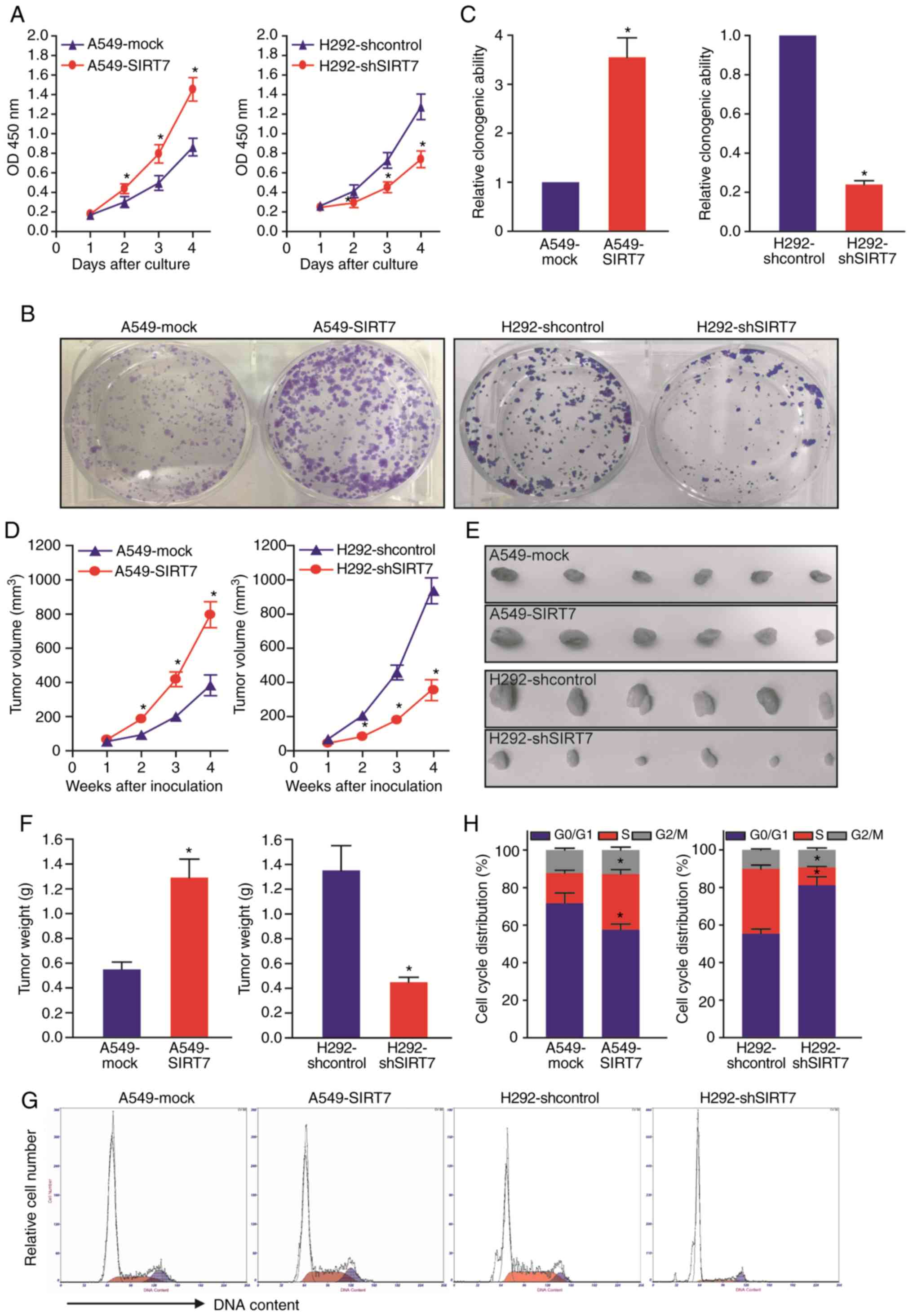 | Figure 3.SIRT7 promotes growth and G1 to
S-phase transition of human NSCLC cells. (A) CCK-8 analysis of
NSCLC cell proliferation/growth. *P<0.05, two-way repeated
measures ANOVA with LSD post hoc multiple comparisons, n=6 per
group. (B and C) Colony formation assay. (B) The representative
images. (C) The relative clonogenic ability of A549-SIRT7
(A549-mock served as a control) and H292-shSIRT7 (H292-shcontrol
served as a control) NSCLC cells. *P<0.05, Student
t-test, n=6 per group. (D-F) Tumor subcutaneous xenograft
mouse model. (D) Tumor volume. *P<0.05, two-way repeated
measures ANOVA with LSD post hoc multiple comparisons, n=6 per
group. (E) The xenograft tumor images. (F) Tumor weight.
*P<0.05, Student's t-test, n=6 per group. (G and H) Cell
cycle analysis by flow cytometry. (G) The representative images.
(H) The percentage of each cell cycle phase. *P<0.05, Student's
t-test, n=6 per group. SIRT7, sirtuin 7; NSCLC, non-small
cell lung cancer; CCK-8, Cell Counting Kit-8; ANOVA, analysis of
variance; LSD, least significant difference. |
SIRT7 promotes human NSCLC cell
migration and invasion in vitro as well as distant lung metastasis
in vivo
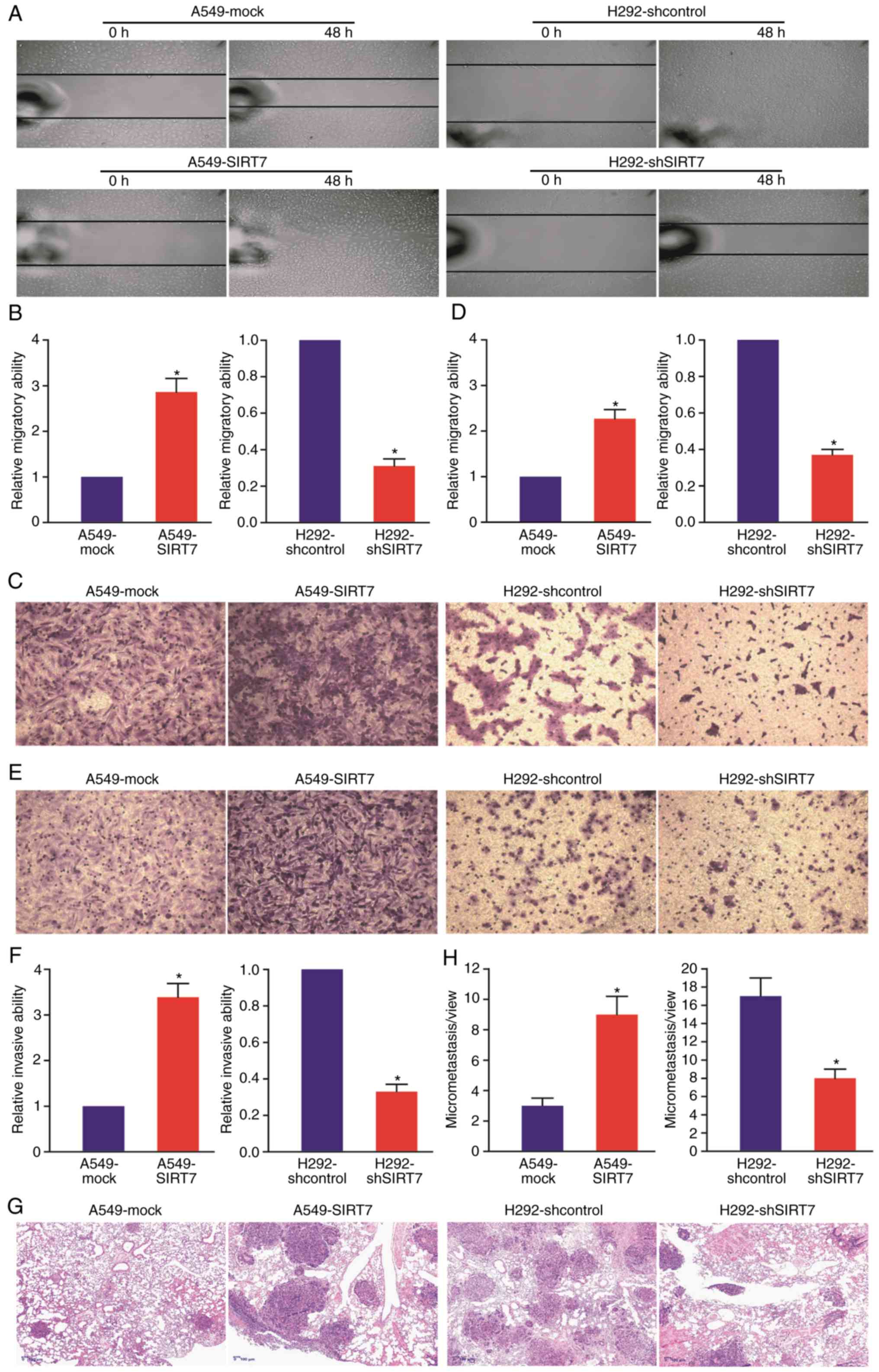
To explore the roles of SIRT7 in NSCLC cell
metastasis, wound healing assay and Transwell migration and
invasion assays were respectively carried out to determine the
migratory and invasive abilities of A549-SIRT7 vs. A549-mock and
H292-shSIRT7 vs. H292-shcontrol human NSCLC cells. As revealed in
Fig. 4A-D, compared with A549-mock
or H292-shcontrol control cells, the migratory capability of
A549-SIRT7 NSCLC cells was significantly enhanced and the migratory
capability of H292-shSIRT7 NSCLC cells was weakened (P<0.05).
Additionally, overexpression of SIRT7 in A549 NSCLC cells promoted
A549 cell invasion, whereas knockdown of SIRT7 in H292 NSCLC cells
impeded H292 cell invasion (P<0.05) (Fig. 4E and F). To further determine
whether the association of SIRT7 with human NSCLC metastatic
ability obtained in vitro could be replicated in
vivo, the A549-SIRT7 vs. A549-mock or H292-shSIRT7 vs.
H292-shcontrol cells were intravenously injected into nude mice.
Six weeks later, the lung tissues of mice were harvested for
H&E analysis of lung metastatic nodules. As revealed in
Fig. 4G and H, tumor metastatic
nodules in the A549-SIRT7-injected mouse-derived lungs displayed an
increase compared with the A549-mock control model, whereas tumor
metastatic nodules in the H292-shSIRT7-injected mouse-derived lungs
exhibited a decrease compared with the H292-shcontrol control model
(P<0.05), indicating that SIRT7 significantly strengthens the
distant metastatic capacity of human NSCLC cells in vivo as
well. Thus, the results demonstrated that SIRT7 can promote the
metastatic potential of human NSCLC cells.
SIRT7 activates AKT and ERK1/2
signaling as well as regulates the expression of G1-phase
checkpoint molecules and EMT-associated molecules in human NSCLC
cells
To elucidate the molecular mechanism for
SIRT7-induced promoting effects on human NSCLC cell growth and
metastasis, the protein expression levels of AKT/ERK1/2 signaling
molecules including p-AKT (T308/S473)/total AKT and p-ERK1/2/total
ERK1/2, G1-phase checkpoint molecules including p21/27, cyclin
D1/E1 and CDK2/4, and EMT-associated molecules including
E/N-cadherin, vimentin, Snail and Slug in A549-SIRT7 vs. A549-mock
and H292-shSIRT7 vs. H292-shcontrol human NSCLC cells were
determined by western blotting. It was revealed that compared with
A549-mock control cells, the expression levels of p-AKT (T308),
p-AKT (S473), p-ERK1/2, cyclin D1, cyclin E1, CDK2, CDK4,
N-cadherin, vimentin, Snail and Slug in A549-SIRT7 cells were
significantly higher, while the expression levels of p21, p27 and
E-cadherin in A549-SIRT7 cells were significantly lower (P<0.05)
(Fig. 5A and B). The data
demonstrated that overexpression of SIRT7 activated AKT and ERK1/2
signaling, regulated the expression of G1-phase checkpoint
molecules to induce G1 to S-phase transition, and promoted EMT in
A549 NSCLC cells. Notably, knockdown of SIRT7 in H292 NSCLC cells
exerted the opposite effects in regulating the expression of the
aforementioned molecules (P<0.05) (Fig. 5A and B). The in vivo
modulatory influence of SIRT7 on the expression of these molecules
was further confirmed by IHC analysis of A549-SIRT7 vs. A549-mock
and H292-shSIRT7 vs. H292-shcontrol NSCLC xenograft tumors
(Fig. 5C). To further confirm the
role of AKT signaling in SIRT7-mediated effects on the biological
behavior of NSCLC cells and expression of EMT markers, AKT
inhibition assays using MK-2206 in A549-SIRT7 human NSCLC cells was
carried out. As revealed in Fig.
6A-C, inhibition of AKT signaling impaired the promoting roles
of SIRT7 in NSCLC cell proliferation, migration and invasion
(P<0.05). Moreover, AKT inhibition attenuated the regulating
effects of SIRT7 on the expression of EMT markers such as
E-cadherin, N-cadherin, vimentin, Snail and Slug in NSCLC cells
(Fig. 6D). The findings indicated
that SIRT7 can promote human NSCLC cell growth and metastasis
probably by activating AKT and ERK1/2 signaling as well as
regulating the expression of G1-phase checkpoint molecules to
promote G1 to S-phase transition and EMT-associated molecules to
facilitate EMT.
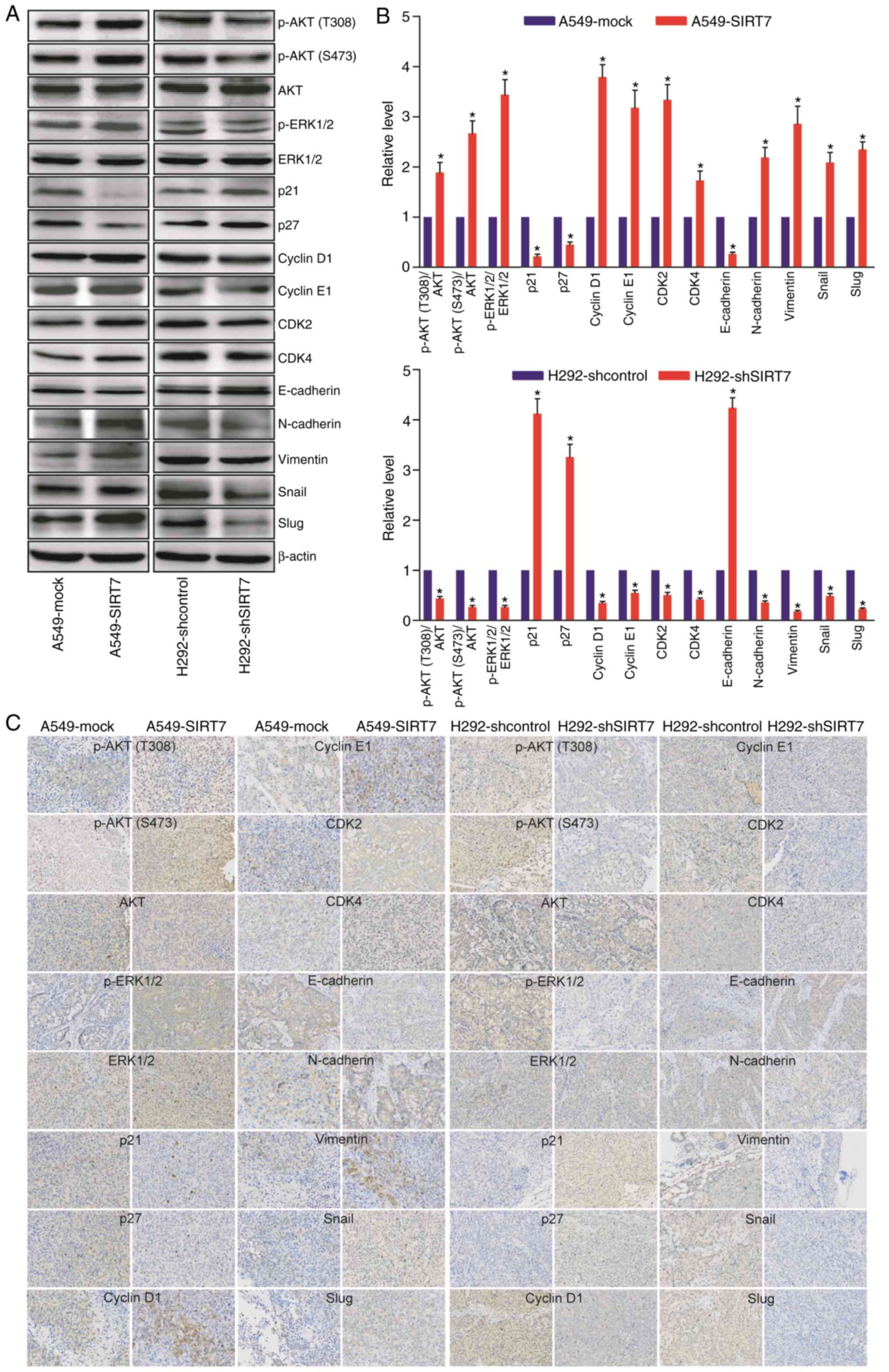 | Figure 5.SIRT7 activates AKT/ERK1/2 signaling
and regulates the expression of G1-phase checkpoint molecules for
G1 to S transition as well as EMT molecules for EMT induction in
NSCLC cells. (A and B) Western blot analysis of AKT/ERK1/2,
G1-phase checkpoint and EMT molecules. (A) The representative
western blot images. (B) The relative level of p-AKT (T308)/AKT,
p-AKT (S473)/AKT and p-ERK1/2/ERK1/2 as well as p21, p27, cyclin
D1, cyclin E1, CDK2, CDK4, E-cadherin, N-cadherin, vimentin, Snail
and Slug in A549-SIRT7 (A549-mock served as a control) or
H292-shSIRT7 (H292-shcontrol served as a control) NSCLC cells.
*P<0.05, Student's t-test, n=6 per group. (C) IHC
analysis of AKT/ERK1/2, G1-phase checkpoint and EMT molecules in
xenograft tumor tissues. The representative IHC images are
presented. SIRT7, sirtuin 7; EMT, epithelial-mesenchymal
transition; NSCLC, non-small cell lung cancer; AKT, protein kinase;
ERK1/2, extracellular signal-regulated kinase 1/2; CDK,
cyclin-dependent kinase; IHC, immunohistochemistry. |
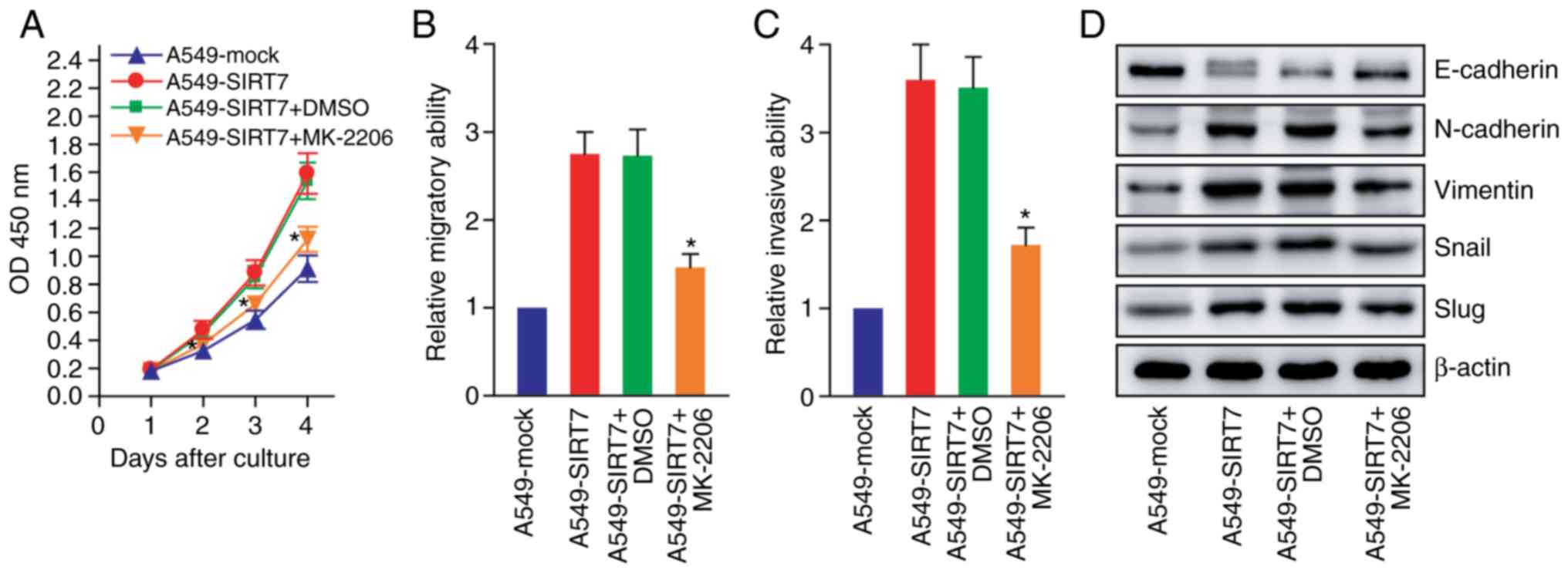 | Figure 6.SIRT7 promotes NSCLC cell
proliferation and EMT progression by activating AKT signaling. (A)
CCK-8 assay after treatment with an AKT inhibitor (MK-2206).
*P<0.05 compared with A549-SIRT7 or A549-SIRT7+DMSO, two-way
repeated measures ANOVA with LSD post hoc multiple comparisons, n=6
per group. (B) Wound healing assay after treatment with the AKT
inhibitor (MK-2206). *P<0.05 compared with A549-SIRT7 or
A549-SIRT7+DMSO, one-way repeated measures ANOVA with LSD post hoc
multiple comparisons, n=6 per group. (C) Transwell invasion assay
after treatment with the AKT inhibitor. *P<0.05 compared with
A549-SIRT7 or A549-SIRT7+DMSO, one-way repeated measures ANOVA with
LSD post hoc multiple comparisons, n=6 per group. (D) Western blot
analysis of EMT markers after treatment with the AKT inhibitor. The
representative western blot images are presented. SIRT7, sirtuin 7;
EMT, epithelial-mesenchymal transition; NSCLC, non-small cell lung
cancer; EMT, epithelial-mesenchymal transition; AKT, protein kinase
B; CCK-8, Cell Counting Kit-8; DMSO, dimethylsulfoxide; ANOVA,
analysis of variance; LSD, least significant difference. |
Discussion
As the newest member of sirtuin family, SIRT7 plays
essential roles and exhibits dual functions in tumorigenesis and
progression (13,33). The present study demonstrated that
SIRT7 was increased in human lung cancer tissues and NSCLC cell
lines, and there was a positive correlation between SIRT7
expression level in tumor tissues and malignant clinicopathological
characteristics (large tumor size, high pathologic stage and
positive lymph node metastasis) of lung cancer patients. Using
human NSCLC cell and xenograft mouse models, it was revealed that
SIRT7 overexpression promoted NSCLC cell in vitro and in
vivo proliferation/growth, G1 to S-phase transition, migration
and invasion as well as in vivo distant lung metastasis,
whereas SIRT7 knockdown suppressed these processes. Furthermore,
SIRT7 overexpression strengthened activation of AKT/ERK1/2
signaling as well as regulated the expression of G1-phase
checkpoint molecules to promote G1 to S-phase transition and
EMT-associated molecules to facilitate EMT in NSCLC cells.
The abnormal activation of the phosphatidylinositol
3-kinase (PI3K)-AKT and mitogen-activated protein kinase
(MAPK)-ERK1/2 pathway is considered to contribute to carcinogenesis
and cancer progression (34–36). A
high level of phosphorylated AKT and ERK1/2 has also been revealed
to frequently occur in human NSCLC, affirming them as potential
therapeutic targets for NSCLC (37). Previous studies have revealed the
role of SIRT7 in modulating AKT and ERK1/2 signaling. Vakhrusheva
et al (38) revealed the
increase of phosphorylated AKT in the heart of SIRT7−/−
mice. Under energy stress conditions, SIRT7 can suppress AKT
activation via deacetylation of FKBP51 and subsequent enhanced
interaction between PHLPP and AKT, thereby sensitizing breast
cancer cells to cytotoxic agents (39). Controversially, SIRT7 has been
revealed to promote activation of AKT and p70S6K1 in thyroid cancer
via the DBC1/SIRT1 axis (40). In
addition, SIRT7 was able to activate the MAPK-ERK1/2 pathway as
well as increase ERK1/2 phosphorylation in colorectal cancer
(16) and hepatocellular carcinoma
(41). In accordance with these
findings (16,40,41),
the present study revealed that AKT and ERK1/2 phosphorylation were
increased in SIRT7-overexpressed NSCLC cells, whereas their levels
were reduced in SIRT7-knocked down NSCLC cells. The data indicated
that SIRT7 was capable of activating AKT/ERK1/2 signaling in NSCLC
cells, which may contribute to SIRT7-induced promoting effects on
NSCLC cell growth and metastasis.
Cell cycle dysregulation control may result in
tumorigenesis and cancer development (42). Proteins involved in cell cycle
regulation include cyclins, cyclin-dependent kinases (CDKs) and CDK
inhibitors (42). The main G1-phase
checkpoint molecules include cyclin A, cyclin D and cyclin E
cyclins, CDK2, CDK4 and CDK6 CDKs, and p21 and p27 CDK inhibitors
(42). Among these elements, p21
can suppress the activity of the cyclin E/A-CDK2 complex, whereas
p27 can suppress the activity of the cyclin D-CDK4/6 complex,
leading to cell cycle arrest in the G1 phase (42). In order to explore the cellular
mechanism for SIRT7-induced NSCLC cell growth promotion, cell cycle
analysis in SIRT7-overexpressed and SIRT7-knocked down NSCLC cells
was performed by flow cytometry. It was demonstrated that SIRT7
overexpression accelerated cell cycle progression by favoring cell
cycle phase transition from G1 to S in NSCLC cells, while SIRT7
knockdown induced G1-phase arrest. The effects of SIRT7 on the
expression of G1-phase checkpoint molecules including p21, p27,
cyclin D1, cyclin E1, CDK2 and CDK4 were then detected,
demonstrating that overexpression of SIRT7 upregulated cyclin D1,
cyclin E1, CDK2 and CDK4, and downregulated p21 and p27, whereas
knockdown of SIRT7 exhibited the opposite regulatory effects.
Previous studies (18,19) have revealed that SIRT7 can promote
tumor cell cycle progression via downregulation of p21 and
upregulation of cyclin D1 and CDK2, which are consistent with the
present results. Thus, it is concluded that SIRT7 promotes human
NSCLC proliferation and growth to a great extent via accelerating
G1 to S-phase transition by regulating the expression of G1-phase
checkpoint molecules.
EMT endows epithelial-derived tumor cells with
enhanced migratory, invasive and metastatic ability and is involved
in cancer metastasis (43,44). Main steps can be observed in EMT
progression. For example, the epithelial cell-cell junctions are
disintegrated, the apical-basal polarity is lost, the front-rear
polarity is acquired, the cytoskeletal architecture is reorganized
and the cell shape is altered. It can also be detected that the
expression of E-cadherin epithelial marker is downregulated and the
expression of N-cadherin and vimentin mesenchymal markers is
upregulated (43,44). EMT can be driven by Snail/Slug,
Twist (Twist1)/Twist2 and Zeb1/Zeb2 transcription factors with the
ability to suppress E-cadherin and activate N-cadherin and vimentin
directly or indirectly (43,44).
Previous studies have revealed the role of SIRT7 in activation of
EMT in several cancers (16,20,45,46).
To further clarify the association between SIRT7 and EMT in NSCLC
cells, the present study determined the effects of SIRT7
overexpression or knockdown on the expression of EMT-related
molecules in human NSCLC cells. As anticipated, overexpression of
SIRT7 downregulated E-cadherin as well as upregulated Snail, Slug,
N-cadherin and vimentin. However, knockdown of SIRT7 exhibited the
opposite effects. The present findings indicated that SIRT7
promoted EMT of NSCLC cells, which may be involved in
SIRT7-stimulated NSCLC metastasis. Although the exact mechanism by
which SIRT7 regulates EMT-associated molecules in NSCLC cells
remains elusive, the SIRT7-induced upregulation of Snail and Slug
may be responsible for SIRT7-mediated E-cadherin reduction or
N-cadherin and vimentin increase. Active glycogen synthase kinase
3β (GSK3β) has been revealed to be capable of phosphorylating
Snail/Slug and increasing β-transducin repeat-containing protein
(βTrCP)- or carboxyl terminus of Hsc70-interacting protein
(CHIP)-induced Snail or Slug ubiquitylation, leading to proteasomal
degradation of Snail and Slug (44,47–49).
It has also been demonstrated that active AKT and ERK1/2 can
phosphorylate GSK3β at Ser9 residue and inactivate activity of
GSK3β (49,50). Therefore, SIRT7-induced activation
of AKT/ERK1/2 signaling may participate in SIRT7-induced
upregulation of Snail and Slug in NSCLC cells by positively
modulating their stability through the AKT/ERK1/2-GSK3β-Snail/Slug
axis. Notably, emerging evidence has revealed that SIRT7 can
directly bind to the promoter of E-cadherin to promote H3K18Ac
deacetylation, resulting in inhibition of E-cadherin transcription
(45,46), which prompted us to infer that SIRT7
may also directly suppress E-cadherin expression of NSCLC cells in
a chromatin-remodeling manner.
Collectively, the present study demonstrated that
SIRT7 was capable of promoting human NSCLC cell growth and
metastasis possibly by activating AKT/ERK1/2 signaling as well as
favoring cell cycle phase transition from G1 to S via regulation of
G1-phase checkpoint molecules and inducing EMT. SIRT7 functions as
an oncogene and could be a novel therapeutic target in human
NSCLC.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (NNSFC) (nos.
81772645, 81572992, 81602704 and 81372443), the Science and
Technology Department of Jiangsu Province (nos. BY2015039-03 and
BL2014039), the Health and Family Planning Commission of Jiangsu
Province (no. LGY2016030), the Suzhou Institute of Biomedical
Engineering and Technology of the Chinese Academy of Sciences (no.
Y851411105), the Natural Science Foundation of the Jiangsu Higher
Education Institutions of China (no. 16KJB320012), the Beijing
Xisike Clinical Oncology Research Foundation (no. Y-MX2016-017),
the Pushin HK Jiangsu Medical Technology Ltd., Inc. (no.
P112200315) and the Wu Jieping Medical Foundation (no.
P112200914).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
This study was conceived and designed by YX, MT, CX
and YZ. The clinical tissue specimens were collected by CX, DZ and
CL. The experiments and the data analyses were completed by YZ and
XY. Other authors including RC, QG, DZ and YQ also participated in
the data analyses of this study. The manuscript was written by YZ
and XY, and supervised by YX and MT. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The First Affiliated Hospital of Soochow University
(IRB no. 2016128). All patients had provided written informed
consent prior to obtaining the samples. All animal experiments were
approved by the Animal Research Ethics Committee of Soochow
University (IRB no. A201809059).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zappa C and Mousa SA: Non-small cell lung
cancer: Current treatment and future advances. Transl Lung Cancer
Res. 5:288–300. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Allemani C, Matsuda T, Di Carlo V,
Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ,
Estève J, et al: Global surveillance of trends in cancer survival
2000-14 (CONCORD-3): Analysis of individual records for 37 513 025
patients diagnosed with one of 18 cancers from 322 population-based
registries in 71 countries. Lancet. 391:1023–1075. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Finkel T, Deng CX and Mostoslavsky R:
Recent progress in the biology and physiology of sirtuins. Nature.
460:587–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chalkiadaki A and Guarente L: The
multifaceted functions of sirtuins in cancer. Nat Rev Cancer.
15:608–624. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Michishita E, Park JY, Burneskis JM,
Barrett JC and Horikawa I: Evolutionarily conserved and
nonconserved cellular localizations and functions of human SIRT
proteins. Mol Biol Cell. 16:4623–4635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Houtkooper RH, Pirinen E and Auwerx J:
Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol
Cell Biol. 13:225–238. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ford E, Voit R, Liszt G, Magin C, Grummt I
and Guarente L: Mammalian Sir2 homolog SIRT7 is an activator of RNA
polymerase I transcription. Genes Dev. 20:1075–1080. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen S, Blank MF, Iyer A, Huang B, Wang L,
Grummt I and Voit R: SIRT7-dependent deacetylation of the U3-55k
protein controls pre-rRNA processing. Nat Commun. 7:107342016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tsai YC, Greco TM and Cristea IM: Sirtuin
7 plays a role in ribosome biogenesis and protein synthesis. Mol
Cell Proteomics. 13:73–83. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barber MF, Michishita-Kioi E, Xi Y,
Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL,
Chen K, et al: SIRT7 links H3K18 deacetylation to maintenance of
oncogenic transformation. Nature. 487:114–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paredes S, Villanova L and Chua KF:
Molecular pathways: Emerging roles of mammalian Sirtuin SIRT7 in
cancer. Clin Cancer Res. 20:1741–1746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Geng Q, Peng H, Chen F, Luo R and Li R:
High expression of Sirt7 served as a predictor of adverse outcome
in breast cancer. Int J Clin Exp Pathol. 8:1938–1945.
2015.PubMed/NCBI
|
|
15
|
de Nigris F, Cerutti J, Morelli C,
Califano D, Chiariotti L, Viglietto G, Santelli G and Fusco A:
Isolation of a SIR-like gene, SIR-T8, that is overexpressed in
thyroid carcinoma cell lines and tissues. Br J Cancer. 86:917–923.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X,
Zhou Y, Wang H, Pan C and Huang W: Overexpression of sirt7 exhibits
oncogenic property and serves as a prognostic factor in colorectal
cancer. Clin Cancer Res. 20:3434–3445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen X, Li P, Xu Y, Chen X, Sun H, Zhao Y,
Liu M and Zhang W: Association of sirtuins with clinicopathological
parameters and overall survival in gastric cancer. Oncotarget.
8:74359–74370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ,
Kim MG, Chang YG, Shen Q, Park WS, Lee JY, et al: Sirtuin7
oncogenic potential in human hepatocellular carcinoma and its
regulation by the tumor suppressors MiR-125a-5p and MiR-125b.
Hepatology. 57:1055–1067. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li W, Sun Z, Chen C, Wang L, Geng Z and
Tao J: Sirtuin7 has an oncogenic potential via promoting the growth
of cholangiocarcinoma cells. Biomed Pharmacother. 100:257–266.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haider R, Massa F, Kaminski L, Clavel S,
Djabari Z, Robert G, Laurent K, Michiels JF, Durand M, Ricci JE, et
al: Sirtuin 7: A new marker of aggressiveness in prostate cancer.
Oncotarget. 8:77309–77316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han Y, Liu Y, Zhang H, Wang T, Diao R,
Jiang Z, Gui Y and Cai Z: Hsa-miR-125b suppresses bladder cancer
development by down-regulating oncogene SIRT7 and oncogenic long
non-coding RNA MALAT1. FEBS Lett. 587:3875–3882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X and Song Y: MicroRNA-340 inhibits
the growth and invasion of angiosarcoma cells by targeting SIRT7.
Biomed Pharmacother. 103:1061–1068. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kiran S, Oddi V and Ramakrishna G: Sirtuin
7 promotes cellular survival following genomic stress by
attenuation of DNA damage, SAPK activation and p53 response. Exp
Cell Res. 331:123–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang M, Lu X, Zhang C, Du C, Cao L, Hou T,
Li Z, Tu B, Cao Z, Li Y, et al: Downregulation of SIRT7 by
5-fluorouracil induces radiosensitivity in human colorectal cancer.
Theranostics. 7:1346–1359. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lai CC, Lin PM, Lin SF, Hsu CH, Lin HC, Hu
ML, Hsu CM and Yang MY: Altered expression of SIRT gene family in
head and neck squamous cell carcinoma. Tumour Biol. 34:1847–1854.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McGlynn LM, McCluney S, Jamieson NB,
Thomson J, MacDonald AI, Oien K, Dickson EJ, Carter CR, McKay CJ
and Shiels PG: SIRT3 & SIRT7: Potential novel biomarkers for
determining outcome in pancreatic cancer patients. PLoS One.
10:e01313442015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Zhu D and Qin S: SIRT7 suppresses
the epithelial-to-mesenchymal transition in oral squamous cell
carcinoma metastasis by promoting SMAD4 deacetylation. J Exp Clin
Cancer Res. 37:1482018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang X, Shi L, Xie N, Liu Z, Qian M, Meng
F, Xu Q, Zhou M, Cao X, Zhu WG and Liu B: SIRT7 antagonizes TGF-β
signaling and inhibits breast cancer metastasis. Nat Commun.
8:3182017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi H, Ji Y, Zhang D, Liu Y and Fang P:
MicroRNA-3666-induced suppression of SIRT7 inhibits the growth of
non-small cell lung cancer cells. Oncol Rep. 36:3051–3057. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang Y, Han Z, Wang Y and Hao W:
Depletion of SIRT7 sensitizes human non-small cell lung cancer
cells to gemcitabine therapy by inhibiting autophagy. Biochem
Biophys Res Commun. 506:266–271. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qian F, Hu Q, Tian Y, Wu J, Li D, Tao M,
Qin L, Shen B and Xie Y: ING4 suppresses hepatocellular carcinoma
via a NF-κB/miR-155/FOXO3a signaling axis. Int J Biol Sci.
15:369–385. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li L and Bhatia R: The controversial role
of Sirtuins in tumorigenesis-SIRT7 joins the debate. Cell Res.
23:10–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S,
Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell.
170:605–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ciuffreda L, Incani UC, Steelman LS,
Abrams SL, Falcone I, Curatolo AD, Chappell WH, Franklin RA, Vari
S, Cognetti F, et al: Signaling intermediates (MAPK and PI3K) as
therapeutic targets in NSCLC. Curr Pharm Des. 20:3944–3957. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vakhrusheva O, Smolka C, Gajawada P,
Kostin S, Boettger T, Kubin T, Braun T and Bober E: Sirt7 increases
stress resistance of cardiomyocytes and prevents apoptosis and
inflammatory cardiomyopathy in mice. Circ Res. 102:703–710. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu J, Qin B, Wu F, Qin S, Nowsheen S, Shan
S, Zayas J, Pei H, Lou Z and Wang L: Regulation of serine-threonine
kinase Akt activation by NAD+-dependent deacetylase SIRT7. Cell
Rep. 18:1229–1240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li H, Tian Z, Qu Y, Yang Q, Guan H, Shi B,
Ji M and Hou P: SIRT7 promotes thyroid tumorigenesis through
phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1
axis. Oncogene. 38:345–359. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu X, Yang L, Tu J, Cai W, Zhang M, Shou
Z, Yao Y and Xu Q: microRNA-526b servers as a prognostic factor and
exhibits tumor suppressive property by targeting Sirtuin 7 in
hepatocellular carcinoma. Oncotarget. 8:87737–87749. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Deng Z, Wang X, Long X, Liu W, Xiang C,
Bao F and Wang D: Sirtuin 7 promotes colorectal carcinoma
proliferation and invasion through the inhibition of E-cadherin.
Exp Ther Med. 15:2333–2342. 2018.PubMed/NCBI
|
|
46
|
Malik S, Villanova L, Tanaka S, Aonuma M,
Roy N, Berber E, Pollack JR, Michishita-Kioi E and Chua KF: SIRT7
inactivation reverses metastatic phenotypes in epithelial and
mesenchymal tumors. Sci Rep. 5:98412015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schlessinger K and Hall A: GSK-3beta sets
Snail's pace. Nat Cell Biol. 6:913–915. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kao SH, Wang WL, Chen CY, Chang YL, Wu YY,
Wang YT, Wang SP, Nesvizhskii AI, Chen YJ, Hong TM and Yang PC:
GSK3β controls epithelial-mesenchymal transition and tumor
metastasis by CHIP-mediated degradation of Slug. Oncogene.
33:3172–3182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu P, Lv J, Yang Z, Guo L, Zhang L, Li M,
Han W, Chen X, Zhuang H and Lu F: Protocadherin 9 inhibits
epithelial-mesenchymal transition and cell migration through
activating GSK-3β in hepatocellular carcinoma. Biochem Biophys Res
Commun. 452:567–574. 2014. View Article : Google Scholar : PubMed/NCBI
|