Introduction
Oral/oropharyngeal squamous cell carcinoma (OSCC) is
a common malignant tumor of the head and neck, and its overall
5-year relative survival rate is ~50%, urgently requiring new
directions in therapeutics (1).
Cancer stem-like cells (CSCs; also referred to as tumor-initiating
cells) are small subpopulations of tumor cells that have been
isolated from various primary tumors and cancer cell lines,
including OSCC (2). CSCs play a
crucial role in tumorigenicity, metastasis and recurrence and,
thus, are considered as the origin of the cancer (2). The stemness properties of CSCs are
supported by endogenous CSC signaling pathways, such as Notch,
Hedgehog, Wnt, Bmi1, phosphatase and tensin homolog, bone
morphogenetic protein and transforming growth factor-β, which are
frequently activated in human cancers (3). In addition, novel oral CSC molecular
determinants, such as histone demethylases (4), microRNA (5), human papilloma viruses (5) and calcium signalling (6) were recently identified. Therefore,
advancing our understanding of the molecular properties and
signaling pathways unique to oral CSCs is crucial for developing a
new generation of targeted and effective therapies for OSCC.
Nitrogen-containing bisphosphonates (N-BPs) are
commonly used anti-resorptive agents in the treatment of
bone-related diseases, such as osteoporosis and metastatic bone
tumors. Zoledronic acid (ZA) is the most potent N-BP available
clinically, and its antitumor effects have been demonstrated in a
number of solid tumors, including OSCC (7,8). ZA
can inhibit cancer cell growth by inducing apoptosis, cell cycle
arrest, and by stimulating autophagy (7,9–12),
which are crucial for tumor growth and survival. Moreover, the
antitumor effect of ZA was also demonstrated in various cancer
mouse models, such as breast (13)
and pancreas (14), indicating that
ZA is an effective agent for controlling human cancer. Indeed,
clinical trials of ZA as an adjuvant therapy have been conducted
for breast cancer (15,16). However, the effects of ZA on oral
CSCs and its underlying mechanism of action have not been fully
elucidated.
The aim of the present study was to demonstrate the
inhibitory effects of ZA on the CSC phenotype in OSCC cells, in
order to determine whether chemokine (C-C motif) ligand 3 (CCL3)
signaling may be of value as a novel downstream target of
ZA-induced CSC suppression.
Materials and methods
Cell culture and reagents
Six human OSCC cell lines, namely SCC4, UM17B, UM5,
UM2, BapT and SCC9/TNF (17), were
cultured in DMEM/Ham's F12 (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Gemini Bioproducts) and 0.4 µg/ml
hydrocortisone (Sigma-Aldrich; Merck KGaA). ZA (LKT Laboratories),
human CCL3 (Cell Signaling Technology, Inc.), etoposide
(Sigma-Aldrich; Merck KGaA), methotrexate (Sigma-Aldrich; Merck
KGaA) and Maraviroc (Sigma-Aldrich; Merck KGaA) were purchased and
used in the present study.
Anchorage-independent growth
Cells (10,000) were seeded in 60-mm dishes in
culture medium containing 0.3% agarose over a base layer of
serum-free medium containing 0.5% agarose, as described in our
prior studies (4–6,17).
After 3 weeks of incubation with the indicated ZA concentrations,
the cell colonies were counted. The experiment was performed in
triplicates.
Organotypic raft cultures
The detailed protocol of this assay was described in
our previous publications (5,17). The
mean epithelial thickness was obtained from 10 randomly selected
epithelial layers formed by OSCC cells. The thickness was measured
by Aperio's ImageScope v.12.4 (Aperio Technologies, Inc.).
Tumorsphere formation assay
Cells (3,000) were grown in 3 ml of tumorsphere
medium in Ultra-Low Attachment 6-well Plates (Corning, Inc.) for
6–10 days (4–6,17). The
medium contained serum-free DMEM/F12 supplemented with 1:50 B27
(Invitrogen; Thermo Fisher Scientific, Inc.), 20 ng/ml EGF, 20
ng/ml, 10 µg/ml insulin, penicillin, streptomycin and amphotericin
B.
Side population analysis
Cells (1,000,000) were incubated in culture medium
containing Hoechst 33342 (5 µg/ml; Thermo Fisher Scientific, Inc.)
for 90 min at 37°C with or without (1 or 2.5 µM ZA. The cells were
then spun down, resuspended in ice-cold PBS containing
7-aminoactinomycin D (2 µg/ml; Thermo Fisher Scientific, Inc.), and
subjected to fluorescence-activated cell sorting analysis. The
Hoechst dye was excited with the UV laser at 351 to 364 nm and its
fluorescence measured with a 515-nm side population filter (Hoechst
blue) and a 608 EFLP optical filter (Hoechst red). The assay was
performed and analyzed by the UCLA Flow Cytometry Core.
Migration assay
Cell migration was measured using 6.5-mm Transwell
chambers with 8.0-µm polycarbonate membranes (cat. no. 3422;
Corning, Inc.) as described in our previous publications (4–6,17).
Cells (20,000) were seeded with or without 1 µM ZA and incubated
for 2 days.
Chemo-radiosensitivity assays
Chemosensitivity was determined by measuring cell
viability using the tetrazolium salt (MTT) cell proliferation assay
kit (American Type Culture Collection). The detailed protocol is
described in our previous studies (4,17). The
cells were plated at a density of 2×103 cells per well
into 96-well plates and incubated in culture medium containing 40
µM etoposide or 40 µM methotrexate, with or without ZA, for 4 days.
Absorbance at 570 nm was determined using a microplate reader
(Synergy H1; BioTek Instruments, Inc.). For the radiosensitivity
assay, the cells were irradiated with 5 Gy using Mark I-30
Cesium-137 irradiator (JL Shepherd & Assoc.) with a delivery
rate of 4.86 Gy/min, and cultured in growth medium containing 0 or
1 µM ZA for 10 days. Subsequently, surviving colonies were stained
and counted. The detailed protocol was as previously described
(17).
Western blotting
Western blotting was performed as previously
described (4–6,17). The
following primary antibodies were used: Notch intracellular domain
(NICD; cat. no. 2421; 1:500; Val1744; Cell Signaling Technology,
Inc.), β-catenin (1:500; cat. no. 9562; Cell Signaling Technology,
Inc.), and GAPDH (1:1,000; cat. no. sc-25778; Santa Cruz
Biotechnology, Inc.). The secondary horseradish
peroxidase-conjugated antibody (cat. no. 7074; Cell Signaling
Technology, Inc.) dilution range was 1:2,000 or 1:4,000.
Reverse transcription-quantitative PCR
(RT-qPCR)
cDNA was synthesized from 2.5 µg total RNA using the
SuperScript First-Strand Synthesis system (Invitrogen; Thermo
Fisher Scientific, Inc.). Subsequently, qPCR was performed using
SYBR Green I Master mix (Roche Diagnostics) and LightCycler 480 II
(Roche Diagnostics) as described in our previous studies (4–6,17). All
primer sequences were obtained from the Universal Probe Library
(Roche Diagnostics), and may be made available upon request. The
second derivative Cq value determination method (Roche Diagnostics
GmbH) was used to compare fold-differences according to the
manufacturer's instructions.
Statistical analysis
The statistical analyses were performed using
GraphPad Prism 5 (GraphPad Software, Inc.). The data are expressed
as mean ± standard deviation. Data among multiple groups were
compared using one-way ANOVA with Newman-Keuls test, while data
between two groups were compared using t-tests. P<0.05 was
considered to indicate statistically significant differences.
Results
ZA suppresses the malignant growth
properties of OSCC cells
To investigate the effect of ZA on OSCC growth, 6
OSCC cell lines were exposed to increasing concentrations of ZA
(0–20 µM) for 4 days, and their growth was determined using the MTT
assay. ZA decreased the growth of 6 OSCC cell lines in a
dose-dependent manner; however, there was a significant difference
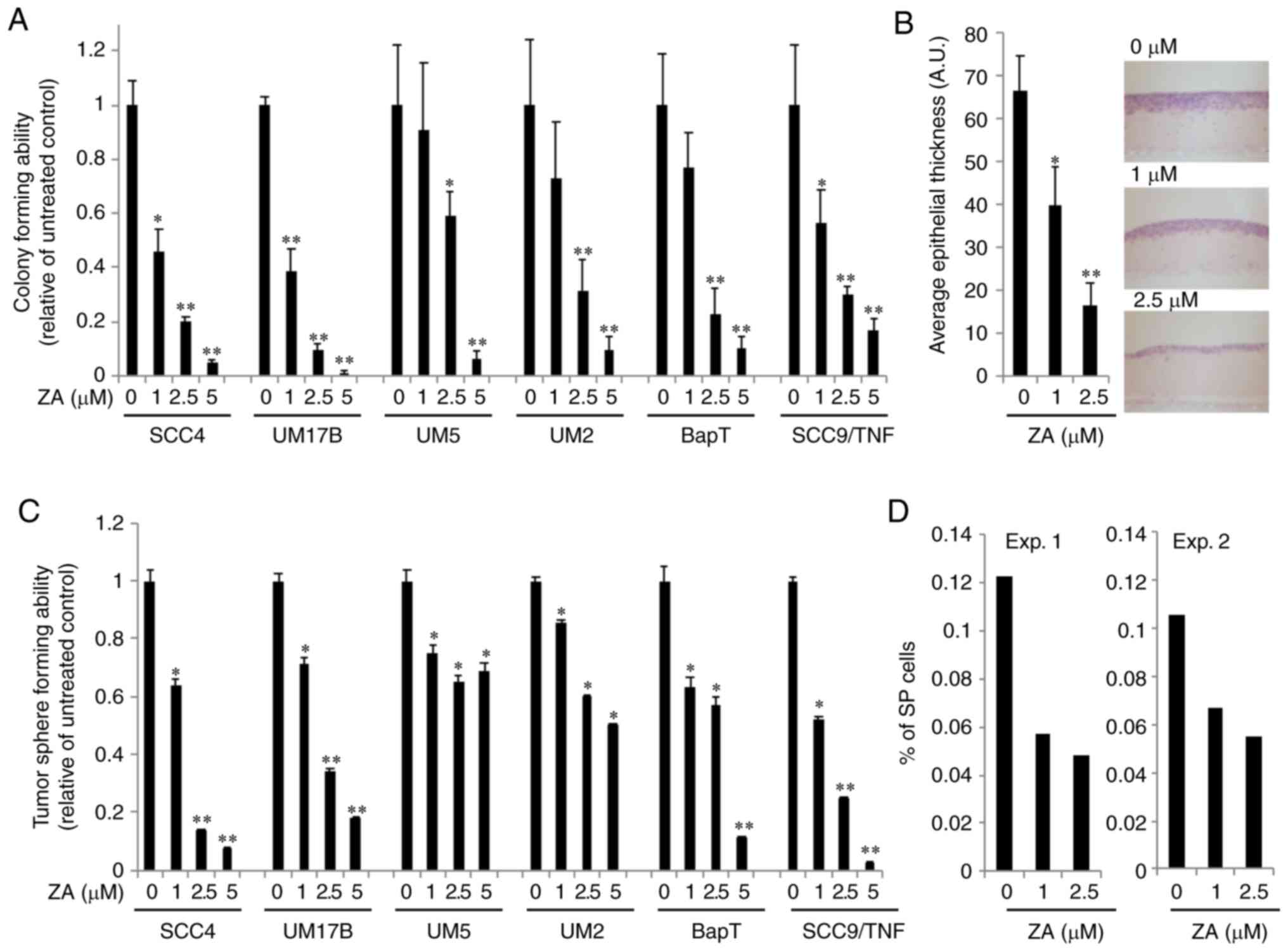
in the response to ZA among the tested OSCC cell lines (Fig. S1). To examine the effect of ZA on
malignant growth properties, its effect on anchorage-independent
growth ability of OSCC cells was investigated. Soft agar assay
revealed that OSCC cells exposed to ZA exhibited a significant
reduction in colony formation at minimal cytotoxic doses of ZA (1–5
µM; Fig. 1A). A 3D organotypic cell
culture system was also employed, whereby squamous epithelium was
reconstituted (17,18). As shown in Fig. 1B, OSCC cells exposed to ZA exhibited
significantly reduced epithelial thickness compared with the
unexposed control cells. Indeed, previous studies have demonstrated
successful inhibition of tumor growth by ZA in vivo
(9,11). These findings collectively suggest
that ZA suppresses malignant growth of OSCC cells.
ZA suppresses the CSC phenotype
Self-renewal capacity is a key feature of CSCs and
is considered to be pivotal for tumorigenic potential (2). Thus, the effect of ZA on the
self-renewal capacity of OSCC cells was investigated by performing
tumorsphere formation assay. OSCC cells exposed to minimal doses of
ZA demonstrated a significant reduction in tumorsphere formation
(Fig. 1C), indicating that ZA
suppresses the self-renewal capacity of OSCC cells. The effect of
ZA on side population (SP) cells, which are considered as a
stem-like cell population among a heterogeneous OSCC cell
population, was further investigated (19,20).
ZA diminished the number of SP cells (Fig. 1D). We further examined the effect of
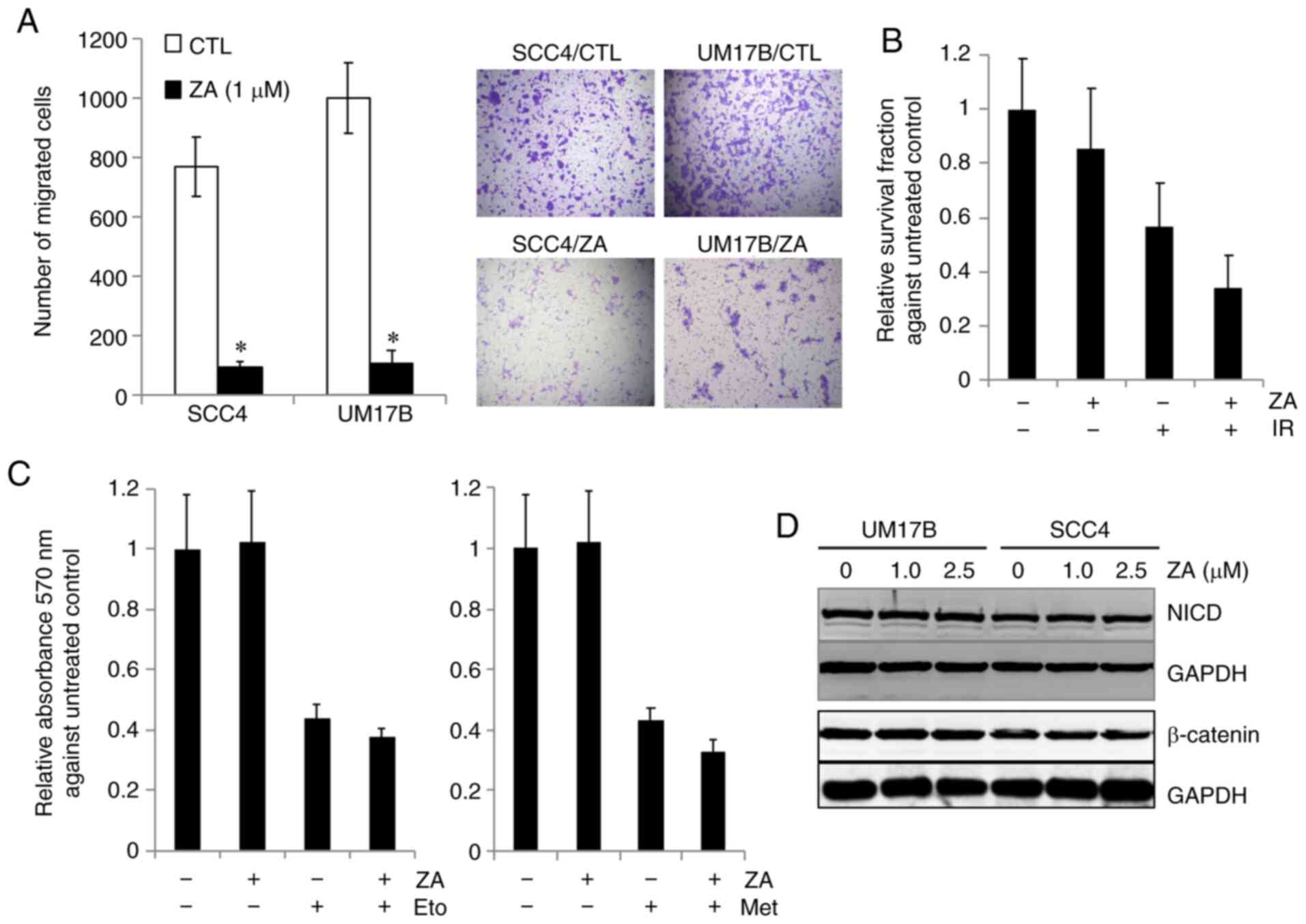
ZA on other CSC properties, i.e., migration and sensitivity to
chemoradiotherapy (2). As
demonstrated by a Transwell migration assay (Fig. 2A), ZA strongly inhibited the
motility in OSCC cells. ZA sensitized OSCC cells to radiation
(Fig. 2B) and to chemotherapeutic
drugs, i.e., etoposide and methotrexate (Fig. 2C). As expected, 1.0 µM ZA alone did
not inhibit OSCC cell growth. ZA slightly potentiated the cytotoxic
effect of chemotherapeutic drugs and radiation on OSCC cells. Taken
together, these findings indicate that ZA suppresses the CSC
phenotype in OSCC cells.
The CSC phenotype can be maintained by several
endogenous signaling pathways, including Notch and Wnt (2). Thus, we sought to test whether ZA
suppresses the CSC phenotype through targeting these signaling
pathways. The effect of ZA on Notch signaling was examined by
measuring the expression of proteolytic release of NICD, indicative
of Notch activation, in the control and ZA-treated cells. The
expression of NICD was not altered by ZA in SCC4 and UM17B cells
(Fig. 2D). The effect of ZA on Wnt
signaling was also determined by measuring the expression of
β-catenin, the key transcription factor in Wnt signaling. ZA
exerted no effect on the expression of β-catenin (Fig. 2D) and its downstream targets (data
not shown). Taken together, these results suggest the involvement
of other CSC-related signaling pathways in the ZA-mediated CSC
inhibition.
ZA reduces the CCL3 expression
required to support the CSC phenotype in OSCC cells
The role of cytokines in the CSC phenotype has been
demonstrated in various cancers, including OSCC (4,17).
Therefore, to determine whether there is a possible functional
association between cytokines and the ZA-mediated inhibition of the
CSC phenotype, we first profiled the expression of 19 cytokines in
control and ZA-treated SCC4 cells (Fig. S2). qPCR analysis demonstrated that
IL7, IL8, IL36RN, CCL3, CCL5 and VEGF were markedly reduced by ZA
(Figs. S2 and 3A). Among those, the inhibitory effect of
ZA on the expression of IL36RN and CCL3 was further confirmed in
other OSCC cell lines (Fig. 3A).
The present study focused on CCL3, as its role in CSC was
demonstrated in human cancer (21).
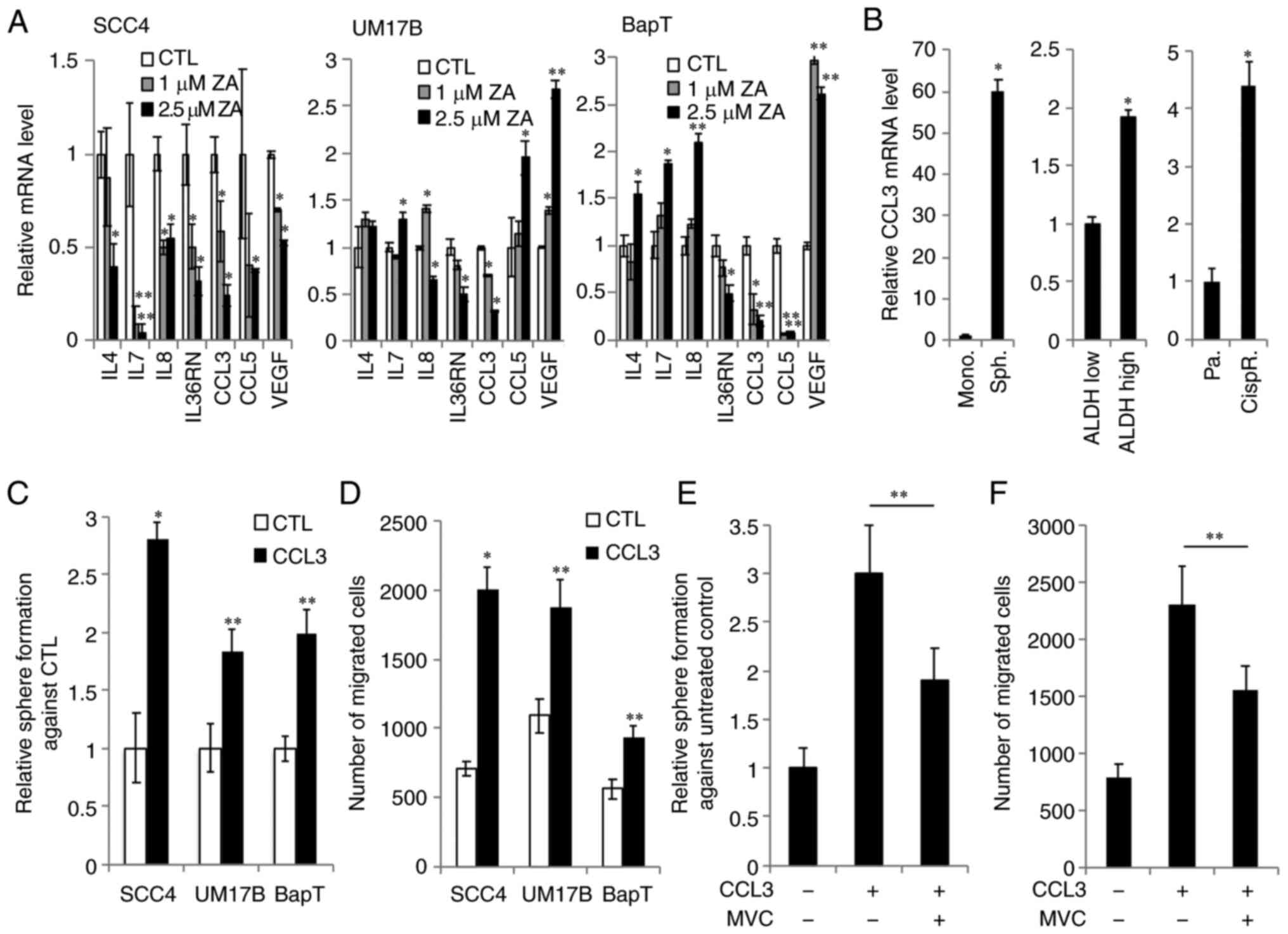 | Figure 3.ZA reduces CCL3 expression required
to support the CSC phenotype in OSCC. (A) The effect of ZA on CCL3
expression in OSCC was determined by quantitative PCR. Decreased
expression of CCL3 was confirmed in 3 OSCC cell lines (SCC4, UM17B
and BapT). The cells were treated with the indicated ZA
concentrations for 2 days. *P<0.05 and **P<0.01 vs. control
(CTL) in one-way ANOVA. (B) The level of CCL3 expression was
determined in CSC-enriched population and non-CSC population
derived from SCC4 cells. Monolayer adherent cells (Mono., non-CSC
population) vs. spheres (Sph., CSC-enriched population);
ALDHlow (non-CSC population) vs. ALDHhigh
(CSC-enriched population); parental cells (Pa., non-CSC population)
vs. cisplatin-resistant cells (CispR., CSC-enriched population).
*P<0.01 vs. corresponding non-CSC populations by t-test. (C) The
effect of CCL3 on self-renewal capacity was assessed in multiple
OSCC cell lines by tumorsphere formation assay. Tumorsphere
formation assay was performed without CCL3 (CTL) or with 10 ng/ml
CCL3 (CCL3). *P<0.01 and **P<0.05 compared to CTL by t-test.
(D) The effect of CCL3 on the migration ability of OSCC cells was
determined by Transwell assay. *P<0.01 and **P<0.05 compared
to CTL by t-test. (E) The effect of maraviroc (MVC) on CCL3-induced
self-renewal capacity of SCC4 cells was assessed by tumorsphere
formation assay. The assay was performed in medium containing no
treatment (−), 10 ng/ml CCL3 (+), or 100 µM MVC (+) for 7 days.
**P<0.05 in one-way ANOVA. (F) The effect of MVC on CCL3-induced
migration in SCC4 cells was assessed by Transwell assay. The assay
was performed with no treatment (−), 10 ng/ml CCL3 (+), or 100 µM
MVC (+) for 48 h. **P<0.05 in one-way ANOVA. ZA, zoledronic
acid; CSC, cancer stem-like cells; OSCC, oral/oropharyngeal
squamous cell carcinoma; CCL3, chemokine (C-C motif) ligand 3;
ALDH, aldehyde dehydrogenase; IL, interleukin; VEGF, vascular
endothelial growth factor. |
The expression level of CCL3 was first compared
between CSC-enriched and corresponding non-CSC populations. CCL3
was found to be highly enriched in self-renewing CSC populations,
i.e., tumorspheres compared with their corresponding non-CSC
populations, i.e., adherent monolayer cells derived from SCC4 cells
(Fig. 3B). The activity of aldehyde
dehydrogenase 1 (ALDH1) has been used as a CSC marker for human
cancers, including OSCC (2).
ALDH1High cancer cells display enhanced CSC properties
compared with ALDH1Low cells (2). We observed that sorted
ALDH1High SCC4 cells expressed higher CCL3 mRNA levels
compared with sorted ALDH1Low SCC4 cells (Fig. 3B). The CSC population can be
enriched after treatment with chemotherapeutic drugs (2). Thus, the CCL3 expression was also
measured in cisplatin-resistant SCC4 cells that were isolated from
SCC4 cells treated with 25 µM cisplatin for 2 days. CCL3 mRNA was
highly expressed in cisplatin-resistant SCC4 cells compared with
their parental control SCC4 cells (Fig.
3B). Moreover, the functional role of CCL3 in CSC properties
was investigated. Tumorsphere formation assay revealed that CCL3
promoted self-renewal capacity in multiple OSCC cell lines
(Fig. 3C). CCL3 also significantly
increased the migration of OSCC cells (Fig. 3D). Collectively, these findings
indicate that CCL3 is an important regulator of CSCs in OSCC.
To further confirm the importance of CCL3-mediated
signaling for the CSC phenotype, the role of CCL3 receptor, CCR5,
in the CCL3-mediated CSC regulation was examined. CCR5 binds to
CCL3 with a high affinity (22). We
examined the effect of the CCR5 antagonist maraviroc on the
CCL3-induced CSC phenotype, and found that maraviroc attenuated the
promoting effect of CCL3 on CSC properties, such as self-renewal
capacity (Fig. 3E) and migration
ability of OSCC cells (Fig. 3F).
Maraviroc at 100 µM was used, as this concentration exerts no
cytotoxic effects on OSCC cells for 7 days (data not shown). Taken
together, these findings indicate that the CCL3/CCR5 axis plays an
important role in regulating CSCs, further suggesting a functional
involvement of CCL3 in ZA-mediated CSC inhibition.
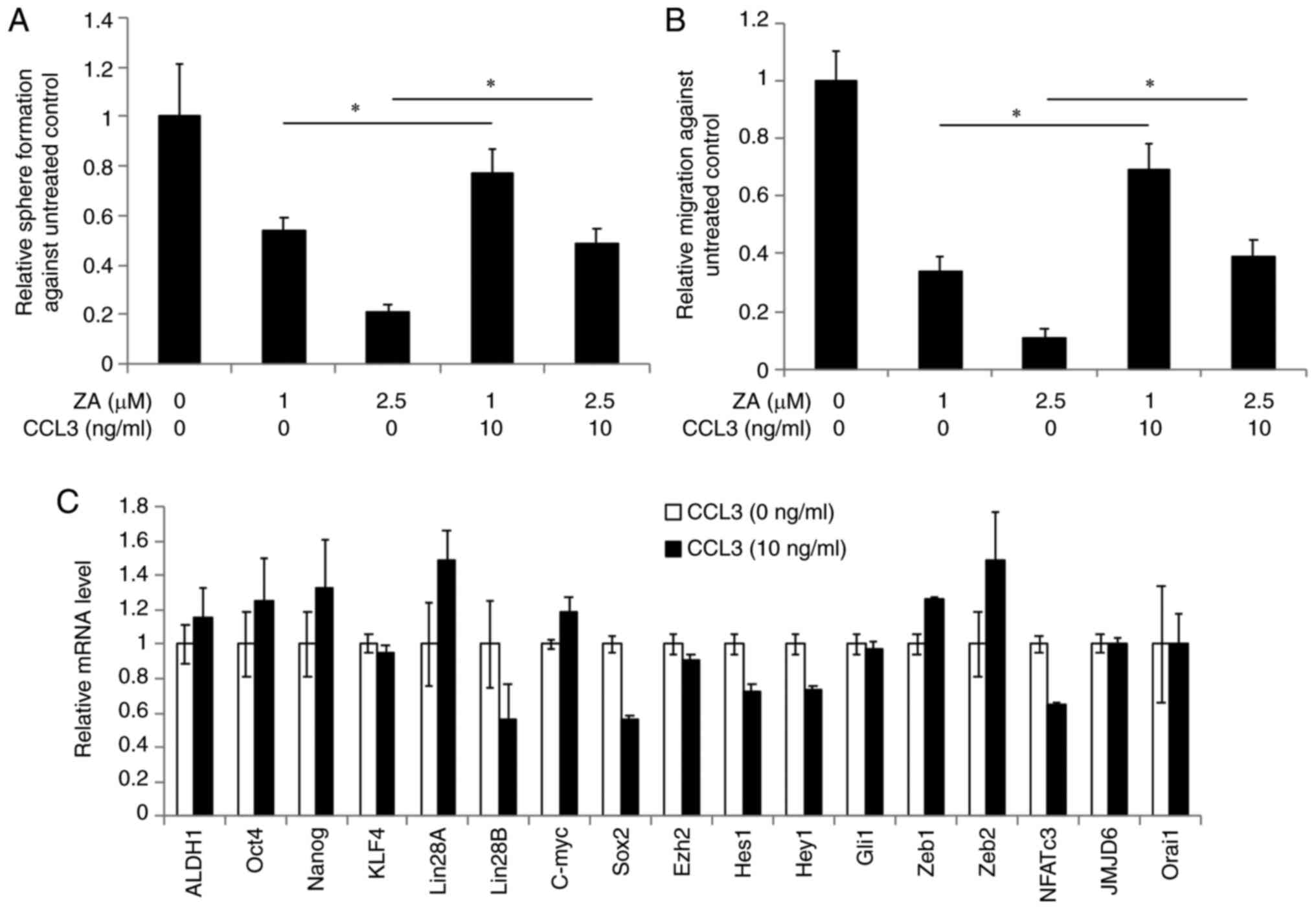
Exogenous CCL3 rescues CSC properties
in ZA-treated OSCC cells
We hypothesized that ZA suppresses the CSC phenotype
via reduction of CCL3 expression. To test this hypothesis, we
investigated whether CCL3 can rescue the CSC properties suppressed
by ZA. Tumorsphere formation assays revealed that addition of
recombinant human CCL3 rescued self-renewal capacity in the
ZA-treated OSCC cells (Fig. 4A).
Similarly, CCL3 also rescued migration ability in the ZA-treated
OSCC cells (Fig. 4B). These
findings indicate that addition of recombinant human CCL3 can
rescue the CSC properties suppressed by ZA. Finally, we examined
whether CCL3 upregulates CSC-promoting factors, and observed no
significant changes in their expression by CCL3 (Fig. 4C). Collectively, these data indicate
that decreased CCL3 expression is likely a major cause of the
ZA-mediated inhibition of the CSC phenotype, suggesting a novel CSC
inhibitory mechanism by ZA.
Discussion
The present study demonstrated the antitumor effects
of ZA on OSCC. ZA inhibited the malignant growth properties of OSCC
cells, such as anchorage-independent growth and squamous epithelium
formation in organotypic raft cultures. Moreover, ZA treatment led
to suppression of self-renewal capacity, which is considered as a
key feature of CSCs. ZA also inhibited important CSC properties,
including migration and chemo-radioresistance. Interestingly, CCL3,
a regulator of the CSC phenotype, was inhibited by ZA and
successfully rescued the suppressed CSC properties in ZA-treated
OSCC cells. Therefore, ZA may be an effective therapeutic agent for
oral cancer via suppressing cancer cell stemness.
CSCs have been identified in a broad spectrum of
solid tumors, including OSCC. Similar to normal stem cells, CSCs
retain their self-renewal capacity, and are thus responsible for
tumorigenicity (2,3). They are also considered as key
contributors to manifestations of tumor aggressiveness, such as
metastasis and drug resistance. Therefore, targeting CSCs appears
to be an effective therapeutic intervention. The present study
suggested that ZA inhibits tumor aggressiveness by suppressing
CSCs. ZA suppressed not only the number of CSCs, but also important
CSC properties. Treatment of OSCC cells with ZA resulted in
decreased self-renewing CSCs and SP cells. SP cells comprise a
stem-like cell population and have been identified in different
types of cancer, including OSCC (19,20).
SP cancer cells display a high self-renewal potential (19,20).
It was clearly demonstrated that ZA suppressed self-renewal
capacity in multiple OSCC cell lines. Moreover, ZA also inhibited
important CSC properties, such as migration and
chemo-radioresistance. The finding of the present study are
consistent with previous reports showing the inhibitory effect of
ZA on the migration ability of breast cancer cells (23). In the present study, ZA reversed
epithelial-to-mesenchymal transition (EMT), the key cellular
process in metastasis, by inactivating nuclear factor-κB (23). Furthermore, a recent study
demonstrated that ZA reduced the motility of CSCs isolated from
breast cancer cells (24). Thus,
the effect of ZA on EMT and EMT-related gene expression in OSCC
should be further investigated. It was also reported that ZA
sensitized OSCC cells to cisplatin and radiation. This observation
is consistent with previous reports (25,26).
Since CSCs play a key role in tumor growth and aggressiveness, the
findings of the present study are of paramount importance for the
development of more effective oral cancer therapies by using ZA.
However, the underlying mechanism through which ZA suppresses OSCC
CSC phenotype has not been fully elucidated.
CCL3 has been shown to play an important role in
carcinogenesis. Elevated CCL3 expression was found not only in OSCC
cells (27), but also in a
carcinogen-induced animal model of oral carcinogenesis (28). CCL3 overexpression has also been
associated with poor survival rates of OSCC patients (27). CCL3 can promote cancer cell growth
and migration (28–30), whereas inhibition of CCL3 suppressed
tumor growth and angiogenesis (30)
and increased cellular sensitivity to therapeutic drugs (31), suggesting the important role of CCL3
in CSC properties. Indeed, CCL3 was found to be highly expressed in
CSC-enriched OSCC cell populations compared with non-CSC
populations, and promoted CSC properties, including self-renewal
capacity, migration and chemoresistance. Interestingly, our study
demonstrated that ZA treatment resulted in decreased CCL3
expression in OSCC cells, suggesting that ZA inhibits OSCC cell CSC
phenotype by reducing CCL3. It is well known that CCL3 exerts its
biological effects by activating downstream signaling cascades
after binding to its receptors, i.e., CCR5 (32). Ectopic CCR5 expression can increase
CSC population and phenotype in breast cancer (33). Conversely, inhibition of CCR5 by its
antagonist, maraviroc, reduced tumor growth (34) and CSC properties (35), such as metastasis and
chemoresistance. Indeed, the CCL3/CCR5 axis has demonstrated
pro-tumorigenic activity in oral carcinogenesis (28). Similarly, it was observed that the
CCR5 antagonist attenuated CCL3-induced self-renewal capacity and
migration of OSCC cells, suggesting a novel role of the CCL3/CCR5
axis in the regulation of CSC properties. However, the
CSC-promoting downstream signaling pathway activated by the
CCL3/CCR5 axis remains elusive. Interestingly, a recent study
demonstrated that CCL3 treatment resulted in activation of the
mitogen-activated protein kinase (MAPK) signaling pathway in cancer
cells (30). The MAPK signaling
pathway, including extracellular signal-regulated kinase (ERK),
plays an important role in malignant tumor growth. Maehara et
al demonstrated that an activated ERK signaling pathway is
crucial for the maintenance of the CSC numbers and properties in
esophageal squamous cell carcinoma (36). Thus, future studies should
investigate whether ZA suppresses the CSC phenotype by inhibiting
the CCL3/ERK signaling axis. Of note, ZA also inhibited IL36RN
expression in multiple OSCC cell lines (Fig. 3A). Since IL36RN expression on cancer
cells and its role in CSCs remain unknown, further investigation is
required to elucidate the role of IL36RN in OSCC.
In summary, the present study demonstrated the
inhibitory effect of ZA on the CSC phenotype in OSCC cells.
Identification of CCL3 signaling as a novel downstream target of
ZA-induced CSC suppression provides new insight into the mechanism
through which ZA exerts its antitumor effects. Since CSCs are
considered as key to tumor aggressiveness, targeting CSCs by ZA may
be an effective therapeutic approach to the treatment of oral
cancer.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank all members of The
Shapiro Family Laboratory of Viral Oncology and Aging Research for
discussing data interpretation and sharing their knowledge. Part of
this study was presented at the annual meeting of the International
Journal of Dental Research, June 2019 and was published as Abstract
no. 3394.
Funding
The present study was supported in part by the UCLA
School of Dentistry faculty seed grant and the NIH/NIDCR grant (no.
R01DE023348).
Availability of data and materials
The datasets generated and analyzed in the present
study are included in this published article. Data not shown in the
manuscript are available from the corresponding author upon
reasonable request.
Authors' contributions
KS and RHK contributed to the study conception,
design and manuscript preparation. SHL and NR performed the cell
line experiments and biochemical assays. CEM and AN contributed to
the acquisition of gene expression data. MKK and NP contributed to
data evaluation and writing of the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocols were registered and
approved by the UCLA Institutional Biosafety committee
(BUA-2016-302-001).
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interest.
References
|
1
|
Warnakulasuriya S: Global epidemiology of
oral and oropharyngeal cancer. Oral Oncol. 45:309–316. 2009.
View Article : Google Scholar
|
|
2
|
Shin KH and Kim RH: An updated review of
oral cancer stem cells and their stemness regulation. Crit Rev
Oncog. 23:189–200. 2018. View Article : Google Scholar
|
|
3
|
O'Brien CA, Kreso A and Jamieson CH:
Cancer stem cells and self-renewal. Clin Cancer Res. 16:3113–3120.
2010. View Article : Google Scholar
|
|
4
|
Lee CR, Lee SH, Rigas NK, Kim RH, Kang MK,
Park NH and Shin KH: Elevated expression of JMJD6 is associated
with oral carcinogenesis and maintains cancer stemness properties.
Carcinogenesis. 37:119–128. 2016. View Article : Google Scholar
|
|
5
|
Lee SH, Lee CR, Rigas NK, Kim RH, Kang MK,
Park NH and Shin KH: Human papillomavirus 16 (HPV16) enhances tumor
growth and cancer stemness of HPV-negative oral/oropharyngeal
squamous cell carcinoma cells via miR-181 regulation.
Papillomavirus Res. 1:116–125. 2015. View Article : Google Scholar
|
|
6
|
Lee SH, Rigas NK, Lee CR, Bang A, Srikanth
S, Gwack Y, Kang MK, Kim RH, Park NH and Shin KH: Orai1 promotes
tumor progression by enhancing cancer stemness via NFAT signaling
in oral/oropharyngeal squamous cell carcinoma. Oncotarget.
7:43239–43255. 2016. View Article : Google Scholar
|
|
7
|
Tamura T, Shomori K, Nakabayashi M, Fujii
N, Ryoke K and Ito H: Zoledronic acid, a third-generation
bisphosphonate, inhibits cellular growth and induces apoptosis in
oral carcinoma cell lines. Oncol Rep. 25:1139–1143. 2011.
View Article : Google Scholar
|
|
8
|
Martin CK, Werbeck JL, Thudi NK, Lanigan
LG, Wolfe TD, Toribio RE and Rosol TJ: Zoledronic acid reduces bone
loss and tumor growth in an orthotopic xenograft model of
osteolytic oral squamous cell carcinoma. Cancer Res. 70:8607–8616.
2010. View Article : Google Scholar
|
|
9
|
Okamoto S, Kawamura K, Li Q, Yamanaka M,
Yang S, Fukamachi T, Tada Y, Tatsumi K, Shimada H, Hiroshima K, et
al: Zoledronic acid produces antitumor effects on mesothelioma
through apoptosis and S-phase arrest in p53-independent and Ras
prenylation-independent manners. J Thorac Oncol. 7:873–882. 2012.
View Article : Google Scholar
|
|
10
|
Corey E, Brown LG, Quinn JE, Poot M,
Roudier MP, Higano CS and Vessella RL: Zoledronic acid exhibits
inhibitory effects on osteoblastic and osteolytic metastases of
prostate cancer. Clin Cancer Res. 9:295–306. 2003.
|
|
11
|
Jiang P, Zhang P, Mukthavaram R, Nomura N,
Pingle SC, Teng D, Chien S, Guo F and Kesari S: Anti-cancer effects
of nitrogen-containing bisphosphonates on human cancer cells.
Oncotarget. 7:57932–57942. 2016. View Article : Google Scholar
|
|
12
|
Sewing L, Steinberg F, Schmidt H and Göke
R: The bisphosphonate zoledronic acid inhibits the growth of
HCT-116 colon carcinoma cells and induces tumor cell apoptosis.
Apoptosis. 13:782–789. 2008. View Article : Google Scholar
|
|
13
|
Li Y, Du Y, Sun T, Xue H, Jin Z and Tian
J: PD-1 blockade in combination with zoledronic acid to enhance the
antitumor efficacy in the breast cancer mouse model. BMC Cancer.
18:6692018. View Article : Google Scholar
|
|
14
|
Vitellius C, Fizanne L, Menager-Tabourel
E, Nader J, Baize N, Laly M, Lermite E, Bertrais S and Caroli-Bosc
FX: The combination of everolimus and zoledronic acid increase the
efficacy of gemcitabine in a mouse model of pancreatic
adenocarcinoma. Oncotarget. 9:28069–28082. 2018. View Article : Google Scholar
|
|
15
|
Barrett-Lee P, Casbard A, Abraham J, Hood
K, Coleman R, Simmonds P, Timmins H, Wheatley D, Grieve R,
Griffiths G and Murray N: Oral ibandronic acid vs. intravenous
zoledronic acid in treatment of bone metastases from breast cancer:
A randomised, open label, non-inferiority phase 3 trial. Lancet
Oncol. 15:114–122. 2014. View Article : Google Scholar
|
|
16
|
Coleman R, Cameron D, Dodwell D, Bell R,
Wilson C, Rathbone E, Keane M, Gil M, Burkinshaw R, Grieve R, et
al: Adjuvant zoledronic acid in patients with early breast cancer:
Final efficacy analysis of the AZURE (BIG 01/04) randomised
open-label phase 3 trial. Lancet Oncol. 15:997–1006. 2014.
View Article : Google Scholar
|
|
17
|
Lee SH, Hong HS, Liu ZX, Kim RH, Kang MK,
Park NH and Shin KH: TNFα enhances cancer stem cell-like phenotype
via Notch-Hes1 activation in oral squamous cell carcinoma cells.
Biochem Biophys Res Commun. 424:58–64. 2012. View Article : Google Scholar
|
|
18
|
Shin KH, Bae SD, Hong HS, Kim RH, Kang MK
and Park NH: miR-181a shows tumor suppressive effect against oral
squamous cell carcinoma cells by downregulating K-ras. Biochem
Biophys Res Commun. 404:896–902. 2011. View Article : Google Scholar
|
|
19
|
Yanamoto S, Kawasaki G, Yamada S,
Yoshitomi I, Kawano T, Yonezawa H, Rokutanda S, Naruse T and Umeda
M: Isolation and characterization of cancer stem-like side
population cells in human oral cancer cells. Oral Oncol.
47:855–860. 2011. View Article : Google Scholar
|
|
20
|
Zhang P, Zhang Y, Mao L, Zhang Z and Chen
W: Side population in oral squamous cell carcinoma possesses tumor
stem cell phenotypes. Cancer Lett. 277:227–234. 2009. View Article : Google Scholar
|
|
21
|
Baba T, Naka K, Morishita S, Komatsu N,
Hirao A and Mukaida N: MIP-1α/CCL3-mediated maintenance of
leukemia-initiating cells in the initiation process of chronic
myeloid leukemia. J Exp Med. 210:2661–2673. 2013. View Article : Google Scholar
|
|
22
|
Blanpain C, Buser R, Power CA, Edgerton M,
Buchanan C, Mack M, Simmons G, Clapham PR, Parmentier M and
Proudfoot AE: A chimeric MIP-1 alpha/RANTES protein demonstrates
the use of different regions of the RANTES protein to bind and
activate its receptors. J Leukocyte Biol. 69:977–985. 2001.
|
|
23
|
Schech AJ, Kazi AA, Gilani RA and Brodie
AH: Zoledronic acid reverses the epithelial-mesenchymal transition
and inhibits self-renewal of breast cancer cells through
inactivation of NF-κB. Mol Cancer Ther. 12:1356–1366. 2013.
View Article : Google Scholar
|
|
24
|
Buhler H, Hoberg C, Fakhrian K and
Adamietz IA: Zoledronic acid inhibits the motility of cancer
stem-like cells from the human breast cancer cell line MDA-MB 231.
In Vivo. 30:761–768. 2016. View Article : Google Scholar
|
|
25
|
Rouhrazi H, Turgan N and Oktem G:
Zoledronic acid overcomes chemoresistance by sensitizing cancer
stem cells to apoptosis. Biotech Histochem. 93:77–88. 2018.
View Article : Google Scholar
|
|
26
|
Kijima T, Koga F, Fujii Y, Yoshida S,
Tatokoro M and Kihara K: Zoledronic acid sensitizes renal cell
carcinoma cells to radiation by downregulating STAT1. PLoS One.
8:e646152013. View Article : Google Scholar
|
|
27
|
Silva TA, Ribeiro FL, Oliveira-Neto HH,
Watanabe S, Alencar Rde C, Fukada SY, Cunha FQ, Leles CR, Mendonça
EF and Batista AC: Dual role of CCL3/CCR1 in oral squamous cell
carcinoma: Implications in tumor metastasis and local host defense.
Oncol Rep. 18:1107–1113. 2007.
|
|
28
|
da Silva JM, Moreira Dos Santos TP, Sobral
LM, Queiroz-Junior CM, Rachid MA, Proudfoot AEI, Garlet GP, Batista
AC, Teixeira MM, Leopoldino AM, et al: Relevance of CCL3/CCR5 axis
in oral carcinogenesis. Oncotarget. 8:51024–51036. 2017. View Article : Google Scholar
|
|
29
|
Hsu CJ, Wu MH, Chen CY, Tsai CH, Hsu HC
and Tang CH: AMP-activated protein kinase activation mediates
CCL3-induced cell migration and matrix metalloproteinase-2
expression in human chondrosarcoma. Cell Commun Signal. 11:682013.
View Article : Google Scholar
|
|
30
|
Liao YY, Tsai HC, Chou PY, Wang SW, Chen
HT, Lin YM, Chiang IP, Chang TM, Hsu SK, Chou MC, et al: CCL3
promotes angiogenesis by dysregulation of miR-374b/ VEGF-A axis in
human osteosarcoma cells. Oncotarget. 7:4310–4325. 2016. View Article : Google Scholar
|
|
31
|
Kim JH, Kim WS, Hong JY, Ryu KJ, Kim SJ
and Park C: Epstein-Barr virus EBNA2 directs doxorubicin resistance
of B cell lymphoma through CCL3 and CCL4-mediated activation of
NF-kappaB and Btk. Oncotarget. 8:5361–5370. 2017. View Article : Google Scholar
|
|
32
|
Jin J, Colin P, Staropoli I,
Lima-Fernandes E, Ferret C, Demir A, Rogée S, Hartley O,
Randriamampita C, Scott MG, et al: Targeting spare CC chemokine
receptor 5 (CCR5) as a principle to inhibit HIV-1 entry. J Biol
Chem. 289:19042–19052. 2014. View Article : Google Scholar
|
|
33
|
Jiao X, Velasco-Velázquez MA, Wang M, Li
Z, Rui H, Peck AR, Korkola JE, Chen X, Xu S, DuHadaway JB, et al:
CCR5 governs DNA damage repair and breast cancer Stem cell
expansion. Cancer Res. 78:1657–1671. 2018. View Article : Google Scholar
|
|
34
|
Mencarelli A, Graziosi L, Renga B,
Cipriani S, D'Amore C, Francisci D, Bruno A, Baldelli F, Donini A
and Fiorucci S: CCR5 antagonism by maraviroc reduces the potential
for gastric cancer cell dissemination. Transl Oncol. 6:784–793.
2013. View Article : Google Scholar
|
|
35
|
Velasco-Velázquez M, Jiao X, De La Fuente
M, Pestell TG, Ertel A, Lisanti MP and Pestell RG: CCR5 antagonist
blocks metastasis of basal breast cancer cells. Cancer Res.
72:3839–3850. 2012. View Article : Google Scholar
|
|
36
|
Maehara O, Suda G, Natsuizaka M, Ohnishi
S, Komatsu Y, Sato F, Nakai M, Sho T, Morikawa K, Ogawa K, et al:
Fibroblast growth factor-2-mediated FGFR/Erk signaling supports
maintenance of cancer stem-like cells in esophageal squamous cell
carcinoma. Carcinogenesis. 38:1073–1083. 2017. View Article : Google Scholar
|