Introduction
Platelet-derived growth factor (PDGF) is a potent
mitogen and chemoattractant that stimulates cell proliferation,
survival and migration in numerous types of tumors, such as
bladder, breast and cervical carcinoma (1). The PDGF family consists of five
disulfide-bonded dimers: PDGF-AA, -AB, -BB, -CC and -DD (2). The PDGF dimeric isoforms are
synthesized as precursor molecules. PDGF-AA, -AB and -BB are
cleaved and activated in secretory vesicles inside producer cells,
while PDGF-CC and -DD are secreted as inactive precursor molecules
that are converted into their active form by proteolytic cleavage
(3). PDGF isoforms exert their
cellular effects through structurally similar α- and β-tyrosine
kinase receptors (PDGFRα and PDGFRβ, respectively); PDGFRα can bind
all PDGF isoforms, except PDGF-DD, while PDGFRβ binds only PDGF-BB
and PDGF-DD with considerable affinity (4). The binding of PDGF polypeptide chains
to their receptors triggers the dimerization and
autophosphorylation of PDGFRs, which in turn activate several
downstream signaling pathways, such as the ERK and PI3K/AKT
signaling cascades (5).
Abnormal PDGF activity is frequently detected in a
number of human tumors (6–9). Tumor-derived PDGF ligands are
considered to act in either a paracrine or autocrine manner,
stimulating the phosphorylation of receptors on tumor and stromal
cells in the tumor microenvironment (10). Previous studies suggested that
tumor-derived PDGF may primarily promote tumor angiogenesis by
mediating the recruitment and growth of stromal fibroblasts,
perivascular cells and endothelial cells (11–14).
In this way, PDGF indirectly affects tumor growth, metastatic
dissemination and drug resistance. PDGF autocrine signaling may
also contribute to tumorigenesis. The tumor-promoting functions of
autocrine PDGF have been demonstrated in multiple non-epithelial
malignancies, including squamous cell carcinoma, glioblastoma and
osteosarcoma (15). Autocrine PDGF
signaling is capable of modulating the malignant phenotypes of
tumor cell proliferation, epithelial-to-mesenchymal transition
(EMT), energy metabolism, invasion, metastasis and colonization
(5). Targeting PDGF/PDGFR signaling
may therefore represent a therapeutic strategy in patients with
cancer (2,5).
Pancreatic cancer is one of the most lethal
malignancies worldwide, currently ranking as the fourth leading
cause of cancer-associated death in the USA and Europe, but is
expected to be the second leading cause of death by 2020 (16). The treatment of this disease is
currently problematic due to the difficulty of initial diagnosis,
strong aggressive features, primary and secondary resistance to
conventional chemotherapy, and high recurrence (17). Anoikis is a specialized type of
apoptosis in epithelial and endothelial cells that is triggered by
loss of contact with the extracellular matrix or adhesion to
inappropriate locations (18).
Anoikis resistance is a mechanism by which cancer cells avoid
undergoing apoptosis during tumor development and metastasis
(19).
Numerous studies have revealed that multiple
signaling pathways are involved in the progression of pancreatic
cancer, such as NF-κB, MAPK, TGFβ/Smad and Hedgehog signaling
pathways (20,21). Primary and metastatic malignant
endocrine pancreatic tumors express high levels of PDGFRβ compared
with normal endocrine pancreatic tissues (22). PDGFRβ has been identified as a
reliable prognostic marker of pancreatic adenocarcinoma, since
higher levels of PDGFRβ expression are associated with a poor
prognosis, as well as with lymphatic invasion and lymph node
metastasis (23). Additionally, in
SW1990 human pancreatic cancer xenograft models, PDGFRβ activation
is observed after radioimmunotherapy or chemotherapy with imatinib
(24). Transcriptional profiling
and functional screening have identified PDGFRβ as both necessary
and sufficient to mediate the proliferative and pro-metastatic
effects of mutant p53 (25). In
addition, Wnt-1/β-catenin signaling contributes to the autocrine
activation of PDGF/Src signaling in pancreatic cancer (26). PDGF-BB promotes the acquisition of
the EMT phenotype in pancreatic cancer AsPC-1 cells via the
induction of microRNA-221 (27) and
mimics the serum-induced dispersal of pancreatic epithelial cell
clusters (28). Furthermore,
dual-specificity phosphatase 28 and PDGF-A form an acquired
autonomous autocrine signaling pathway that promotes
chemoresistance and migration in pancreatic cancer (29). A neutralizing antibody directed
against PDGFRβ enhances the antitumor and anti-angiogenic activity
of a VEGF antagonist (30).
Despite the well-established association between
PDGF and tumor progression via ERK and AKT signaling cascades, the
precise mechanisms of autocrine PDGF signaling in pancreatic tumor
cells remain elusive. A previous study has revealed that PDGF can
affect tumorigenesis via ERK- and AKT-independent mechanisms
(31). The present study aimed to
study the roles of PDGF in pancreatic cancer biology, including
cell proliferation, anoikis resistance and invasion, as well as the
underlying mechanism through the transcriptional coactivator
Yes-associated protein (YAP)/Hippo pathway.
Materials and methods
Cell culture and drugs
Pancreatic adenocarcinoma BxPC-3 cells were
purchased from the Shanghai Institute of Cell Biology (Chinese
Academy of Sciences) and were cultured in RPMI 1640 medium
supplemented with 10% FBS (both Gibco; Thermo Fisher Scientific,
Inc.), penicillin (100 U/ml), streptomycin (100 µg/ml) and 2 mM
L-glutamine at 37°C in a humidified 5% CO2 incubator.
Regarding drugs, PDGF-BB (R&D Systems, Inc.) with
concentrations 0, 10, 25, 50 and 100 ng/ml was added and incubated
at 37°C in a 5% CO2 incubator for 24 h. Verteporfin, a
drug able to stop the formation of the YAP/TEAD complex in the
nucleus (cat. no. HY-B0146) was purchased from MedChemExpress.
Various concentrations (0.1, 0.5 and 1 µM) of Verteporfin were
added into the medium and incubated at 37°C in a 5% CO2
incubator for 24 h. CP-673451, a potent selective inhibitor of
PDGFR tyrosine kinase, was purchased from Selleck Chemicals (cat.
no. S1536). Cells were treated with 10 nM CP-673451 at 37°C for 24
h. Rhosin and calyculin A were purchased from MedChemExpress (cat.
nos. HY-12646 and HY-18983, respectively). The cells were treated
with 30 µM Rhosin and 30 µM calyculin A at 37°C for 24 h. PBS was
used as a control.
Transfection
Small interfering (si) RNA oligonucleotides for YAP1
and scrambled non-targeting negative control were purchased from
Thermo Fisher Scientific, Inc. (Stealth RNAi™ siRNA; cat. no.
AM16708). Cells (5.0×105) were transfected with siRNA
oligonucleotides (20 nM) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 24 h
according to the manufacturer's protocol. Cell culture medium was
replaced post-transfection and cells were allowed to grow for an
additional 24 h before subsequent experiments.
MTT assay
Briefly, cells (5×103 cells/well) were
seeded in 96-well plates in the absence of or at 10, 25, 50 and 100
ng/ml of PDGF-BB (R&D Systems, Inc.) for 24 h at 37°C. The
treatment was started 24 h after seeding. Subsequently, cells were
incubated with MTT solution at 37°C for 4 h, and formazan crystals
resulting from MTT reduction were dissolved by adding 100 µl DMSO
in each well and gently shaking for 15 min. The absorbance of
cultures was measured using a multiwell spectrophotometer at a
wavelength of 560 nm. Results were calculated as the percentage of
absorbance in control cultures.
Anoikis assay
Anoikis assay was performed as previously described
(32). In order to induce anoikis
under anchorage-independent conditions, ~1×106 cells/ml
were plated in an ultra-low attachment 6-well plate (cat. no. 3471;
Corning, Inc.) with or without PDGF-BB addition. BxPC-3 cells were
cultured under suspension or adherent conditions for 48 h and then
all cells were harvested using centrifugation at 500 × g at room
temperature for 5 min for apoptosis analysis.
Apoptosis assay
For cell apoptosis analysis, apoptotic cells were
measured using an Annexin V-FITC Apoptosis Detection kit (Nanjing
KeyGen Biotech Co., Ltd.) according to the manufacturer's protocol.
Cells were harvested, washed twice with PBS and resuspended in 500
µl binding buffer. Cell suspensions were stained with 5 µl Annexin
V-FITC and 5 µl PI for 30 min at room temperature in the dark. The
cells were evaluated immediately via CytoFLEX LX Flow Cytometer
(Beckman Coulter, Inc.). A minimum of 10,000 cells per sample was
measured, and the analysis of apoptotic cells was performed using
BD CellQuest Pro software v3.3 (BD Biosciences).
Wound-healing assay
Wound-healing assays were performed to examine the
capacity of cell migration. Briefly, after BxPC-3 cells grew to
90–95% confluence in 6-well plates, a single scratch wound was
generated using a 200-ml disposable pipette tip. Cells were washed
to remove displaced and floating cells, and then incubated in fresh
serum-free RPMI 1640 medium for 24 h. Wound-healing was detected at
0 and 24 h within the scratched wounds, and representative fields
were photographed using an inverted light microscope with an
attached digital camera (Olympus Corporation; magnification, ×200),
and the distance between the borders of the wound were assessed for
quantification using Image Pro-Plus 7.0 (Media Cybernetics,
Inc.).
Transwell assay
Transwell chambers (EMD Millipore; 8-µm pore size)
were coated with Matrigel® at 37°C for 12 h (15
µg/filter). Cells (2.0×104) were plated in serum-free
DMEM into the upper chambers, while the lower chambers were filled
with complete medium. Cells treated with PDGF-BB or vehicle were
allowed to invade across the Matrigel-coated membrane at 37°C in 5%
CO2. After 24 h of incubation, the cells on the upper
surface of the insert were removed, and the cells on the lower
surface of the insert were fixed with 4% paraformaldehyde at room
temperature for 5 min and stained with 0.1% crystal violet for 3
min at room temperature. Images of migrated cells were captured,
and cell numbers in five randomly selected fields were counted
under light microscopy (magnification, ×200).
Luciferase assay
The 8×GTIIC-luc plasmid was obtained from Addgene,
Inc. Transient transfection was performed using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. For the luciferase reporter assay, BxPC-3 cells were
seeded in 12-well plates. The 8×GTIIC reporter plasmid containing
TEA-domain (TEAD)-binding elements together with Renilla-Luc
plasmids (Addgene, Inc.) were co-transfected into BxPC-3 cells.
After 4 h of transfection at 37°C, cells were exposed to 0, 10, 25,
50 or 100 ng/ml PDGF-BB. After treatment for 24 h at 37°C,
luciferase activities were measured using a Dual-Glo luciferase
assay kit (Promega Corporation) under a Victor3 1420 plate reader
(PerkinElmer, Inc.). Normalized luciferase signal was calculated by
dividing the firefly luciferase signal by the Renilla
luciferase signal.
Western blot analysis
Western blot analysis was performed using total cell
lysates. Total cell lysates from different experiments were
obtained by lysing the cells in RIPA buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors (100 µM
phenylmethylsulfonyl fluoride, 10 µM leupeptin, 10 µM pepstatin and
2 mM EDTA). The protein content was quantitated using a BCA Protein
Assay kit (Beyotime Institute of Biotechnology). Proteins (50
µg/lane) were separated via 4–20% SDS-PAGE and transferred to
nitrocellulose membranes (Pall Life Sciences). Membranes were
blocked with 5% non-fat milk for 1 h at room temperature and probed
overnight at 4°C with primary antibodies against YAP (cat. no.
4912), phospho- (p-)YAP (Ser127; cat. no. 4911S), Macrophage
Stimulating 1 (MST1; cat. no. 3682), p-MST1/2 (Thr183/180; cat. no.
3681), Large Tumor Suppressor Kinase 1 (LATS1; cat. no. 3477),
p-LATS1 (Ser909; cat. no. 9157), N-cadherin (cat. no. 13116),
E-cadherin (cat. no. 14472) and β-actin (cat. no. 4970), all at a
dilution of 1:1,000 (Cell Signaling Technology, Inc.). Membranes
were washed and incubated with horseradish peroxidase-conjugated
secondary antibodies (1:5,000) (cat. nos. AS014 and AS003; ABclonal
Biotech Co., Ltd.) at room temperature for 2 h, and were visualized
using an enhanced chemiluminescence system (EMD Millipore).
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was isolated from cultured cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The PrimeScript RT Master Mix kit (Takara Biotechnology Co.,
Ltd.) was used to synthesize cDNA for mRNA detection according to
the manufacturer's protocol. qPCR was performed with gene-specific
primers using the SYBR Green PCR kit (Takara Biotechnology Co.,
Ltd.) according to the manufacturer's protocol under the following
thermocycling conditions: 95°C for 5 min, followed by 40 cycles at
95°C for 10 sec for denaturation and 60°C for 30 sec for
annealing/extension. The expression levels of each target gene were
calculated using the 2−ΔΔCq method (33). Data were expressed as the
fold-change relative to GAPDH. The primer sequences were as
follows: c-MYC forward, 5′-CCCGCTTCTCTGAAAGGCTCTC-3′ and reverse,
5′-CTCTGCTGCTGCTGCTGCTGGTAG−3′; MCL-1 forward,
5′-CCAAGGCATGCTTCGGAAA-3′ and reverse, 5′-TCACAATCCTGCCCCAGTTT-3′;
GAPDH forward, 5′-TCACTGGCATGGCCTTCCGTG−3′ and reverse,
5′-GCCATGAGGTCCACCACCCTG−3′; N-cadherin forward,
5′-TGAAACGGCGGGATAAAGAG-3′ and reverse, 5′-GGCTCCACAGTATCTGGTTG-3′;
and E-cadherin forward, 5′-GGTTTTCTACAGCATCACCG-3′ and reverse,
5′-GCTTCCCCATTTGATGACAC-3′.
Immunofluorescence staining
After 100 ng/ml PDGF-BB treatment for 24 h at 37°C,
cells cultured on slides were rinsed with PBS, fixed in 3.7%
formaldehyde/PBS for 20 min at room temperature and permeabilized
using 0.1% Triton X-100/PBS for 10 min at room temperature. Slides
were blocked for 1 h with 1% BSA/PBS (Beyotime Institute of
Biotechnology), and were incubated with antibodies against YAP
(1:100; cat. no. sc-101199; Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Subsequently, cells were incubated with
FITC-conjugated secondary antibodies (1:1,000; cat. no. F0382;
Sigma-Aldrich; Merck KGaA) for 2 h at room temperature. Slides were
mounted with ProLong Gold antifade reagent with DAPI (Invitrogen;
Thermo Fisher Scientific, Inc.). Fluorescence images were collected
using a fluorescence microscope (IX70; Olympus Corporation;
magnification, ×600).
Statistical analysis
Data were expressed as the mean ± SD. Statistical
analysis was performed using unpaired Student's t-test or one-way
ANOVA followed by Tukey's post hoc test for comparisons among ≥3
groups using the statistical program SPSS 11.0 (SPSS, Inc.) for
Windows. P<0.05 was considered to indicate a statistically
significant difference.
Results
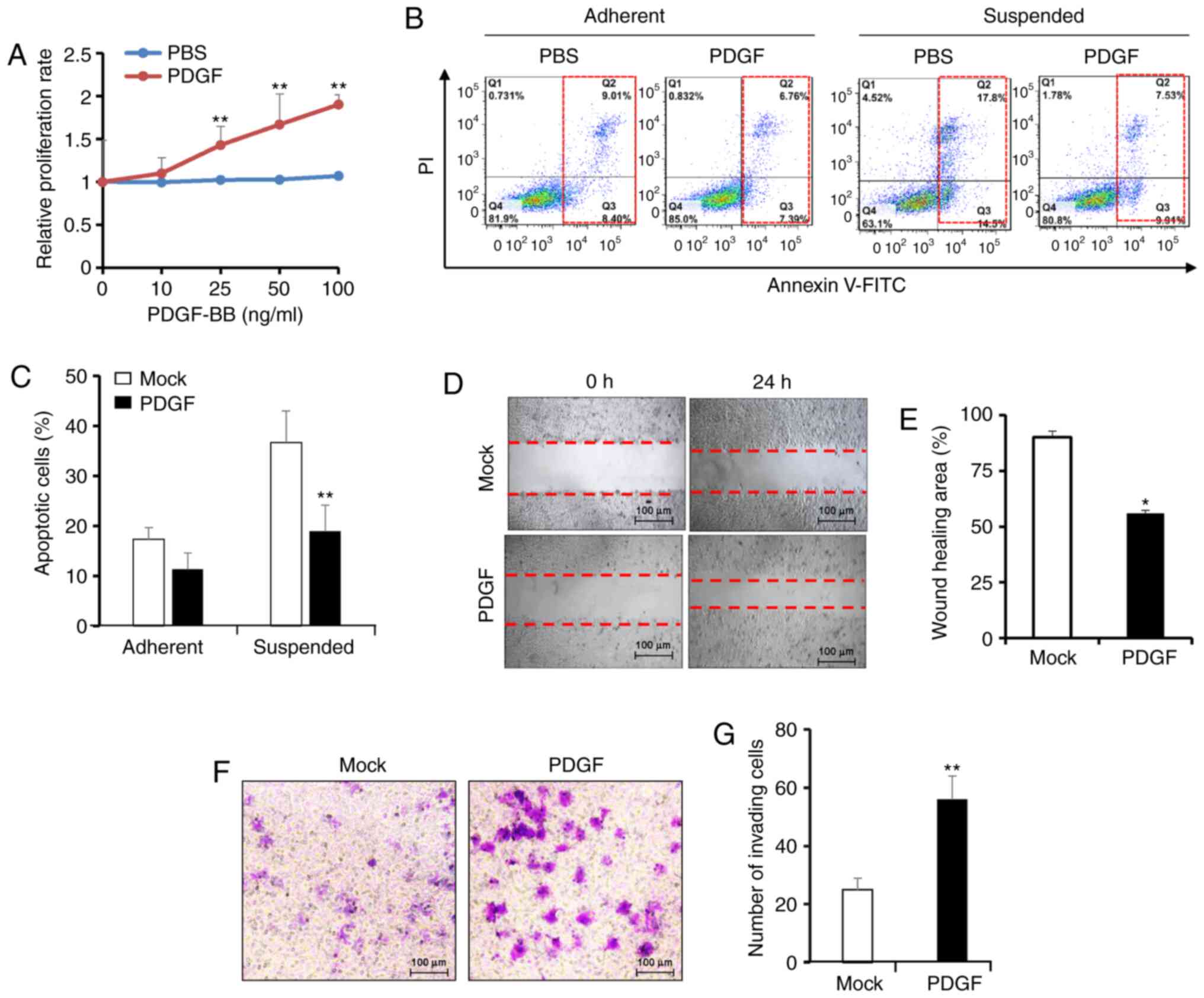
PDGF-BB promotes pancreatic cancer
malignancy
To determine the role of PDGF-BB in promoting tumor
growth, cell proliferation was determined in pancreatic
adenocarcinoma BxPC-3 cells using an MTT assay. After exposure to
PDGF-BB (0–100 ng/ml) for 24 h, BxPC-3 cells exhibited a
dose-dependent increase in proliferation (Fig. 1A). Cell proliferation was
significantly increased in BxPC-3 cells treated with concentrations
>25 ng/ml PDGF-BB. Anoikis is an anchorage-independent form of
cell death that is initiated after the disruption of the cell
matrix and cell-cell interactions; thus, anoikis resistance is an
initial step in the progression of metastatic cancer (34). Subsequently, the effects of PDGF-BB
on cell responsiveness to anoikis were evaluated. BxPC-3 cells were
cultured under suspension or adherent conditions for 48 h, after
which apoptotic cells were analyzed via flow cytometry. Compared
with adherent cells, suspended BxPC-3 cells exhibited a higher rate
of anoikis after culture without PDGF-BB for 48 h, with >30%
apoptotic cells. Treatment with 100 ng/ml PDGF-BB significantly
decreased cell apoptosis in the suspended cells, but had no
significance in adherent cells, indicating enhanced anoikis
resistance (Fig. 1B and C).
Subsequently, the migration of BxPC-3 cells in the presence or
absence of PDGF-BB was examined via wound-healing and Transwell
assays. PDGF-BB significantly accelerated wound closure of BxPC-3
cells compared with that of untreated cells (Fig. 1D and E). Consistently, the Transwell
assay revealed that PDGF-BB treatment increased the number of
invading BxPC-3 cells (Fig. 1F and
G). These results suggested that PDGF-BB may have a positive
effect on cell migration. Overall, the present data revealed that
PDGF-BB may promote pancreatic cancer malignancy via increasing
cell proliferation, anoikis resistance and cell migration.
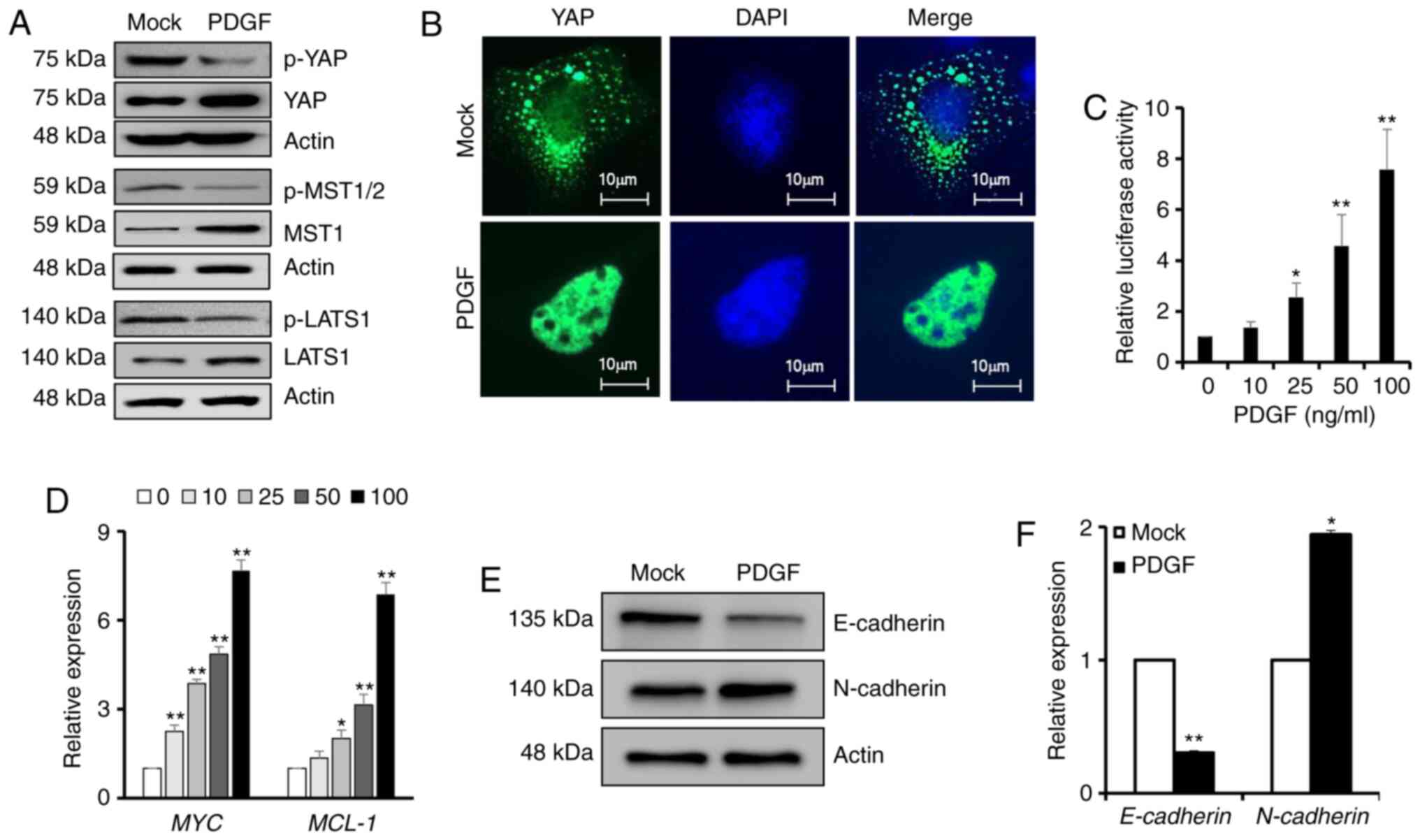
PDGF-BB activates YAP signaling
The transcriptional coactivator YAP, an effector of
the Hippo signaling pathway, can act as an oncogene to promote
tumor survival and metastasis if its activity is increased
abnormally (35). Therefore, the
present study estimated the effects of PDGF-BB on YAP activity.
Compared with the mock group, the total amount of YAP, MST1 and
LATS1 protein were all upregulated, while the phosphorylation of
YAP at Ser127, MST1 at Thr183, and LATS1 at Ser909 as the
degradation forms were markedly decreased in PDGF-BB-treated cells,
indicating the aberrant activation of YAP (Fig. 2A). Immunofluorescence staining
revealed the nuclear and cytoplasmic localization of YAP in BxPC-3
cells in the absence or presence of PDGF-BB, indicating that
PDGF-BB caused a redistribution of YAP and enhanced its nuclear
accumulation (Fig. 2B). In
addition, BxPC-3 cells were transfected with a luciferase reporter
plasmid (8×GTIIC-luc) containing TEAD-binding elements. PDGF-BB
administration at >25 ng/ml for 24 h significantly potentiated
YAP activity, as demonstrated by the luciferase assay (Fig. 2C). Accordingly, RT-qPCR revealed
that PDGF-BB treatment significantly upregulated the expression of
two YAP downstream genes, namely the MYC proto-oncogene and the
MCL-1 apoptosis regulator (Fig.
2D). MYC has a pivotal function in growth control, while MCL-1
promotes tumor survival by enabling cells to escape apoptosis
(36). Since EMT is closely
associated with tumor invasion and metastasis, and is also
regulated by Hippo/YAP signaling (37), the present study investigated the
effect of PDGF-BB on EMT by detecting the expression levels of
epithelial and mesenchymal markers. Western blot analysis suggested
that the expression levels of the epithelial marker E-cadherin were
decreased after PDGF-BB treatment, whereas the expression levels of
the mesenchymal marker N-cadherin were increased (Fig. 2E). Similar results were obtained via
RT-qPCR, indicating that PDGF-BB treatment significantly suppressed
E-cadherin expression and promoted N-cadherin expression (Fig. 2F). In summary, the current results
strongly suggested that PDGF-BB may stimulate YAP activity and the
expression of its downstream genes.
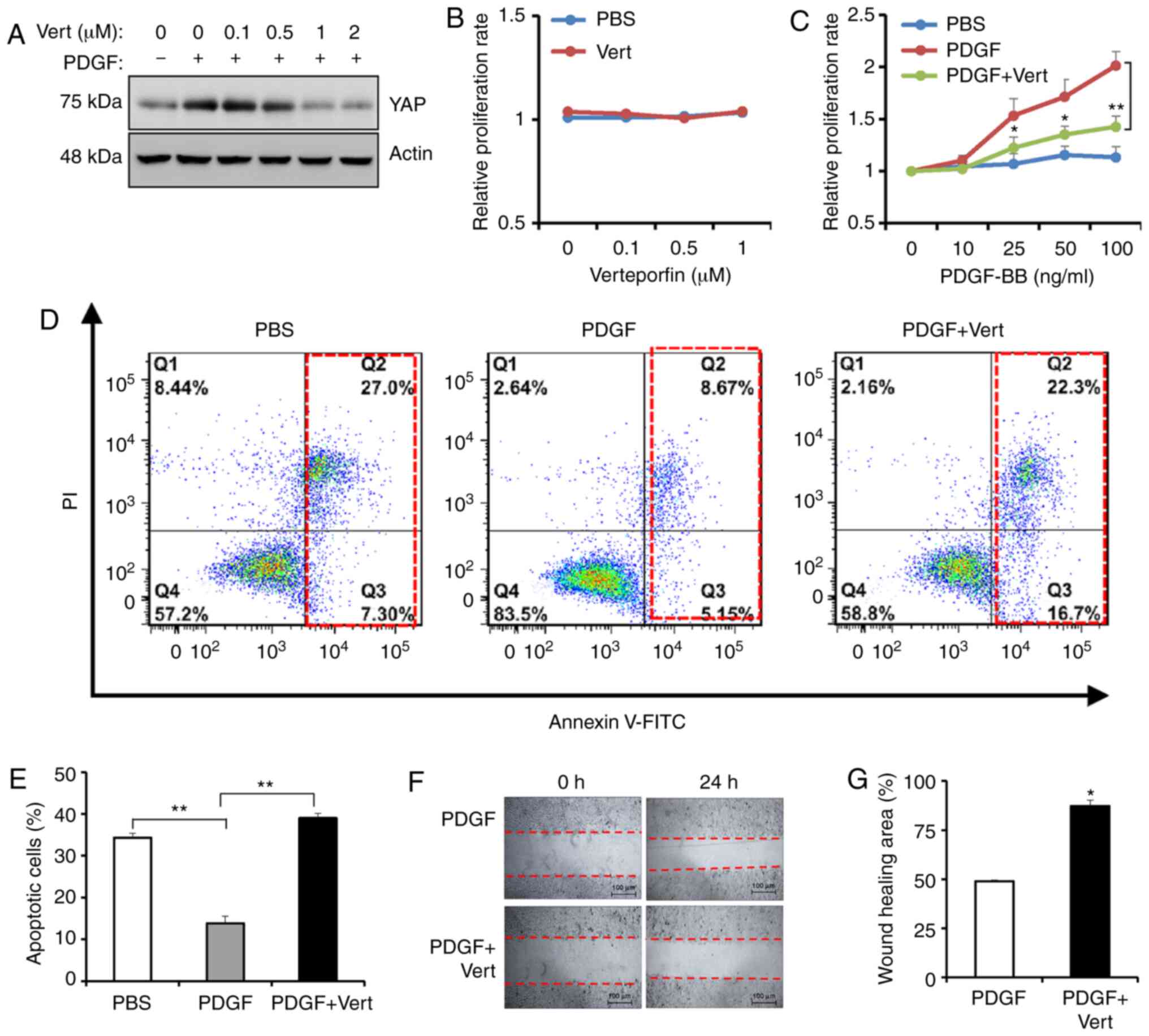
YAP activation contributes to
PDGF-BB-enhanced pancreatic cancer malignancy
To ascertain the effects of YAP activity in
PDGF-BB-induced cancer malignancy, BxPC-3 cells were treated with
verteporfin, a YAP inhibitor, and PDGF-BB. Increasing
concentrations of verteporfin (0.1, 0.5 and 1 µM) gradually
decreased the levels of YAP protein (Fig. 3A). Treatment with verteporfin for 24
h had no effect on cell proliferation, as measured via MTT assay
(Fig. 3B). Subsequently, the
combined effects of 0.1 µM verteporfin (the minimum effective dose)
and PDGF-BB on cell proliferation, anoikis resistance and cell
migration were assessed. Notably, 0.1 µM verteporfin partially
reversed the promoting effects of PDGF-BB on cell proliferation
(Fig. 3C). Additionally,
verteporfin increased anoikis in the presence of PDGF-BB (Fig. 3D and E). A wound-healing assay
revealed that verteporfin attenuated the migration of tumor cells
enhanced by PDGF-BB (Fig. 3F and
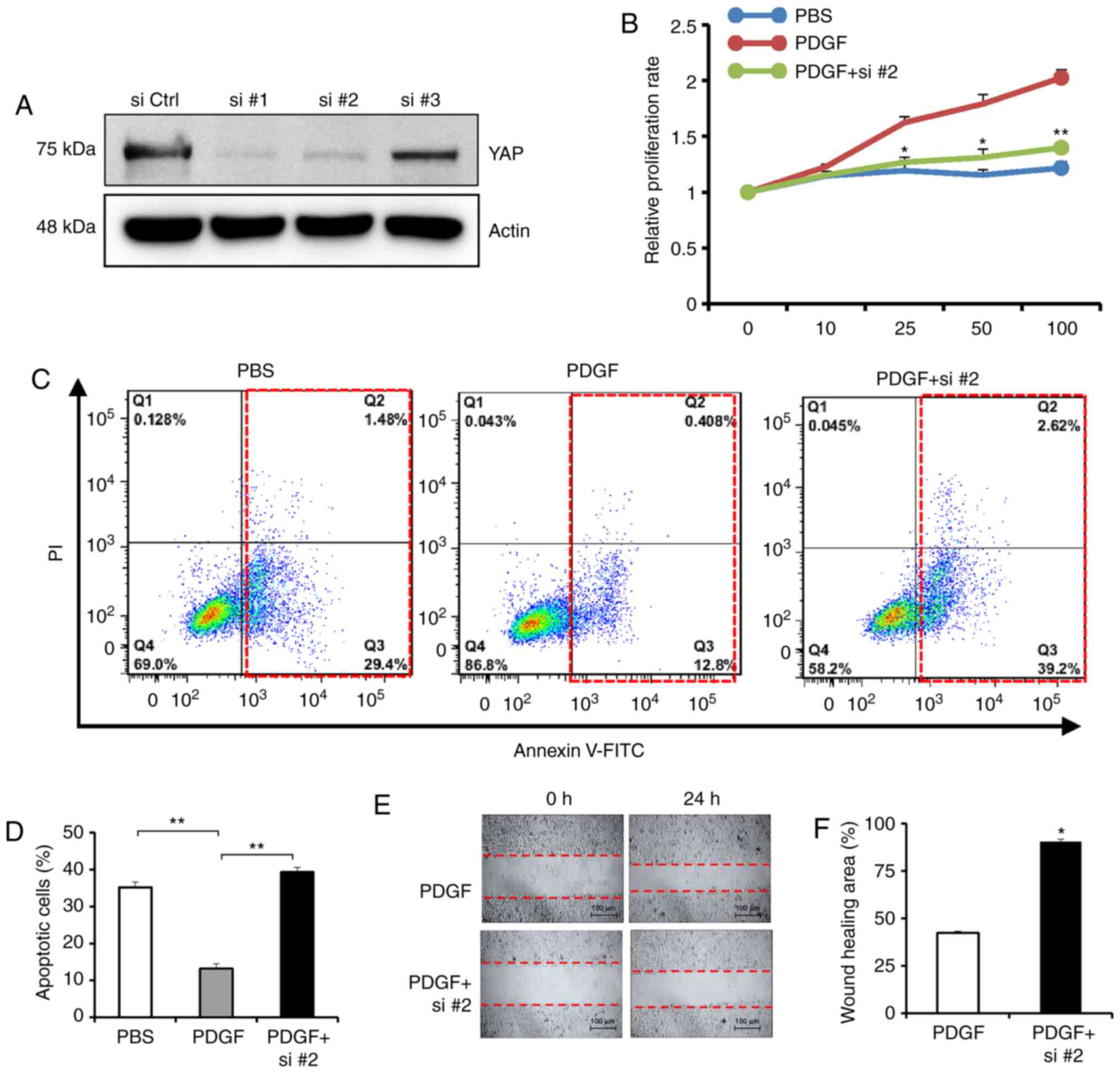
G). Subsequently, YAP expression was knocked down using three
siRNAs, and siRNA2 was chosen for further experiments (Fig. 4A). Similarly, YAP siRNA abrogated
the cell proliferation promoting effects of PDGF-BB (Fig. 4B) and promoted anoikis in the
presence of PDGF-BB (Fig. 4C and
D). A wound-healing assay indicated that YAP siRNA reversed the
PDGF-BB-enhanced migration of tumor cells (Fig. 4E and F). These results revealed that
blockade of YAP activity with a pharmacological inhibitor partially
abrogated the effects of PDGF-BB on cancer malignancy, suggesting
that YAP activation may be a causal mechanism for PDGF-BB-induced
tumor progression.
PDGFR/RhoA/protein phosphatase-1
(PP-1) cascade participates in PDGF-BB-induced YAP activation
There is a complicated network regulating YAP
activity. A previous study revealed that platelets mediate YAP
dephosphorylation and promote its nuclear translocation via the
RhoA/MYPT1/PP-1 cascade (32).
Therefore, the present study explored the mechanism associated with
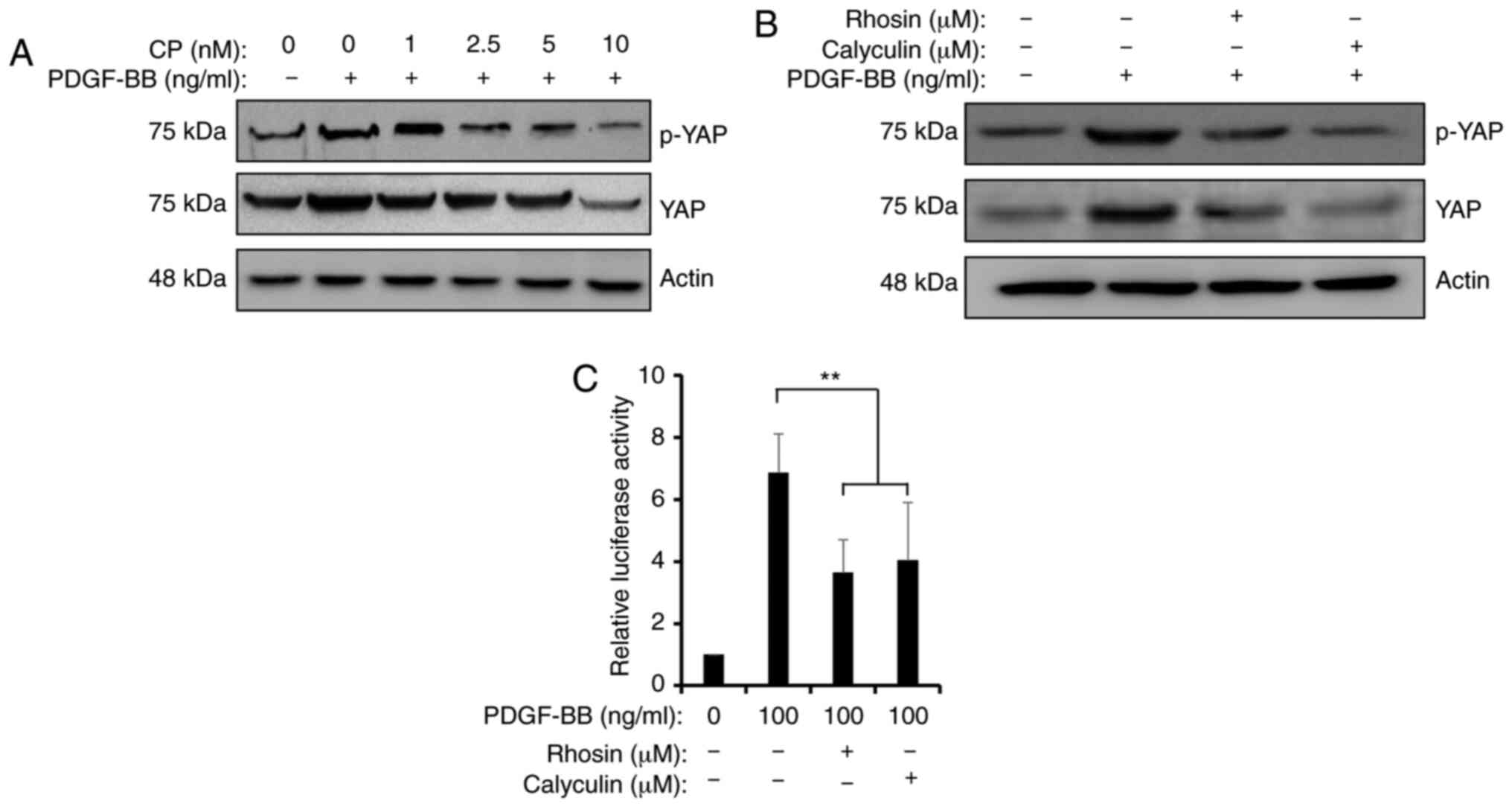
PDGF-induced YAP activation. Firstly, CP-673451, a potent selective
inhibitor of PDGFR tyrosine kinase, was used to block PDGF
downstream signaling. Treatment with 10 nM CP-673451 completely
abolished PDGF-BB-induced YAP stabilization and phosphorylation
(Fig. 5A). Secondly, Rhosin or
calyculin A were used to inhibit RhoA or PP-1 in the presence of
PDGF-BB, respectively. Treatment with 30 µM Rhosin or 30 nM
calyculin A partially attenuated PDGF-BB-induced YAP accumulation
and dephosphorylation to different extents (Fig. 5B). Additionally, a luciferase assay
demonstrated that Rhosin and calyculin A repressed the
PDGF-BB-induced activity of the 8×GTIIC-luc reporter (Fig. 5C). Therefore, the current data
revealed that the PDGFR/RhoA/PP-1 cascade may be involved in
PDGF-BB-induced YAP activation.
PDGFR inhibition decreases YAP
activity and cancer malignancy
PDGF is the principal mitogen in serum and is
produced by platelets and macrophages (3). In tumors, PDGF can be expressed by
tumor or adjacent stroma cells, thereby acting as either a
paracrine or autocrine factor (10). Therefore, the effects of CP-673451
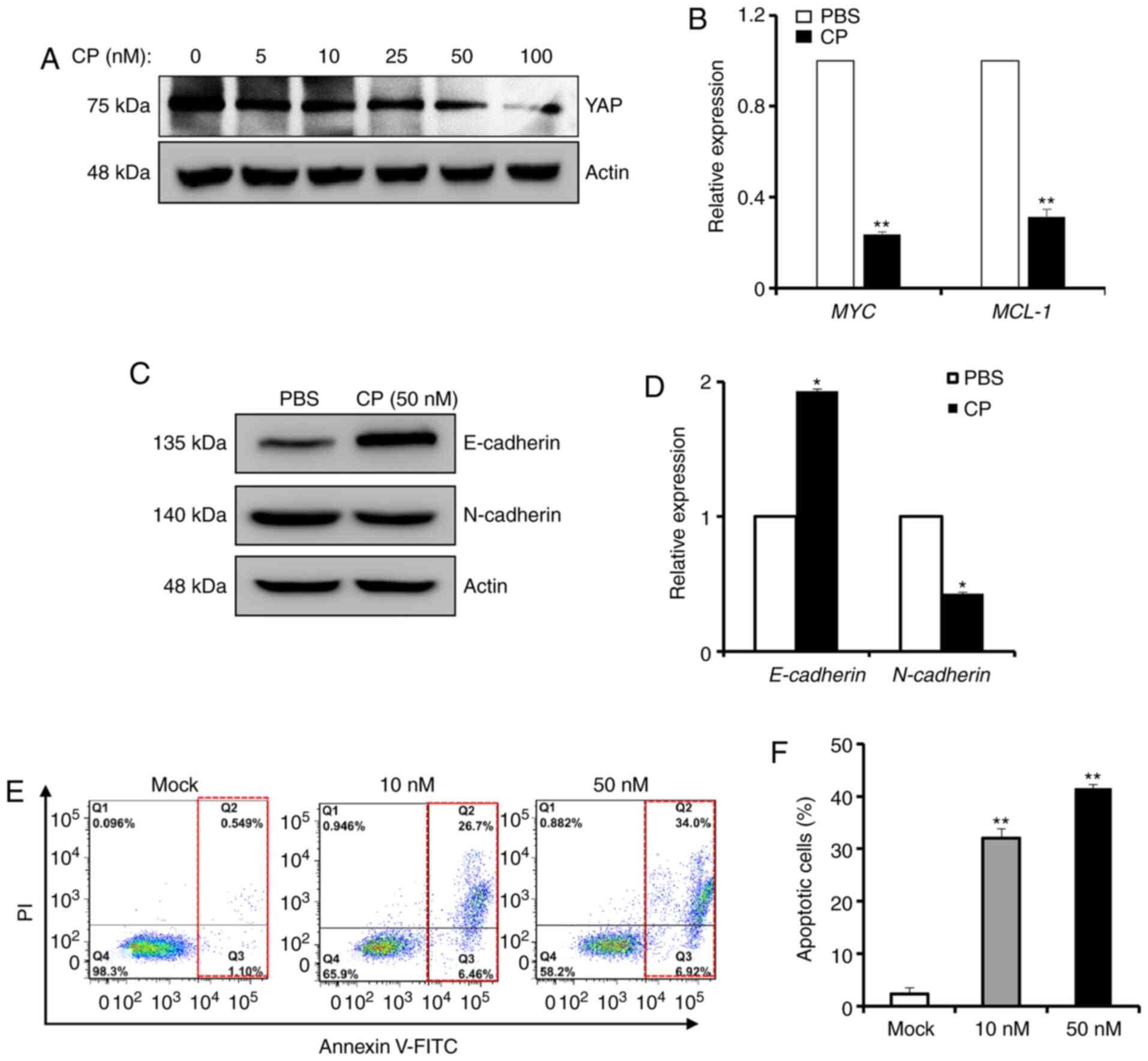
on YAP activity and cancer malignancy were investigated. A gradual
decrease in the total amount of YAP protein was observed during
PDGFR inhibition with up to 100 nM CP-673451 (Fig. 6A). As a result, the expression
levels of two YAP downstream genes (MYC and MCL-1) were
significantly decreased after treatment with 50 µM CP-673451 for 24
h (Fig. 6B). Additionally, western
blot analysis suggested that E-cadherin expression was increased
after CP-673451 treatment, while N-cadherin expression was
decreased (Fig. 6C). qPCR analysis
indicated that E-cadherin and N-cadherin expression was regulated
by CP-673451 (Fig. 6D).
Furthermore, treatment with CP-673451 for 24 h alone induced BxPC-3
cells to undergo apoptosis in a dose-dependent manner, as
demonstrated by flow cytometric Annexin V apoptosis analysis
(Fig. 6E and F). Therefore, the
present results indicated that PDGF inhibition may inhibit cancer
malignancy by mediating YAP inactivation.
Discussion
Several studies have revealed the positive
association between abnormal YAP activity and tumorigenesis
(38–41). The findings of the present study
suggested that PDGF-BB signaling promoted the malignancy of
pancreatic cancer via YAP activation, that the RhoA/PP-1 cascade
was involved in the PDGF-BB-induced dephosphorylation of YAP and
that targeting PDGFR repressed YAP activity and induced tumor
apoptosis.
The transcriptional coactivators YAP and its paralog
TAZ are vital downstream effectors of the Hippo signaling cascade
and serve versatile roles in the control of developmental
transitions, organ size, cell fate and tumorigenesis (42–45).
When the Hippo-signaling pathway becomes active, the MST1/2 and
LATS1/2 kinases are activated by phosphorylation (46). LATS1/2 kinases phosphorylate and
inhibit YAP, thereby sequestering YAP in the cytosol and limiting
its transcriptional activity (46).
In addition, YAP phosphorylated at Ser127 can be ubiquitinated by
β-TrCP ubiquitin ligase and subsequently targeted for proteasomal
degradation (47). In addition, YAP
is a substrate of autophagy, and autophagy accelerates the
degradation of YAP (48). Thus, the
total protein forms and the phosphorylated forms of LATS1/2, MST1/2
and YAP are in a balance to maintain the activation of the Hippo
signaling pathway (45). Upon
silencing of Hippo signaling, YAP is dephosphorylated and active
YAP is translocated to the nucleus (45). Within the nucleus, YAP functions as
a transcriptional coactivator of TEAD transcription factors
(49). Furthermore, YAP can
interact with Smad family members and other transcription factors
to regulate the expression of target genes (50). In this way, YAP is implicated in
cell proliferation, apoptosis, migration, chemoresistance and
angiogenesis (51,52).
YAP induces EMT and promotes the progression of
cholangiocarcinoma (53). In
addition, YAP participates in the development, progression and
recurrence of pancreatic cancer (49). Furthermore, YAP is associated with
chemoresistance and poor prognosis in pancreatic cancer (49). Verteporfin, an agent that disrupts
YAP-TEAD complexes, suppresses the survival of pancreatic ductal
adenocarcinoma cells (54).
Verteporfin inhibits tumor angiogenesis by downregulating
angiopoietin-2 and suppresses vasculogenic mimicry by decreasing
the expression levels of matrix metallopeptidase 2, vascular
endothelial cadherin and α-smooth muscle actin (55). YAP activation through
cyclin-dependent kinase 1-mediated mitotic phosphorylation promotes
pancreatic cancer cell motility, invasion and tumorigenesis
(56). Considering the role of YAP
in pancreatic malignancy and clinical outcome, it is reasonable to
develop drugs targeting YAP for future interventions.
The results of the present study indicated that
PDGF-BB induced YAP activation, contributing to cancer malignancy
in pancreatic cancer cells. The YAP inhibitor verteporfin partially
attenuated the effects of PDGF-BB on cell proliferation, apoptosis
and migration. Additionally, PDGF-BB upregulated MYC expression, an
oncogene that promotes cell division. Furthermore, PDGF-BB enhanced
MCL-1 expression, which is a member of the BCL-2 family and
prevents cells from undergoing apoptosis (57). Knockdown of MCL-1 by RNA
interference renders B-RAF melanoma cells sensitive to anoikis,
which is a unique anchorage-independent form of apoptotic cell
death that occurs as a result of insufficient cell-matrix
interactions (58). Anoikis
resistance is a critical contributor to tumor invasion and
metastasis, and malignant cells take advantage of several
mechanisms to resist anoikis and thereby maintain survival
(59). In addition, the present
study revealed that PDGF-BB treatment altered the expression levels
of E-cadherin and N-cadherin, two master regulators of EMT.
Consistently, PDGF-BB increased the aggressive capability of
pancreatic cancer cells, as demonstrated by Transwell and
wound-healing assays. Finally, YAP inhibition effectively reversed
the oncogenic effects of PDGF-BB.
Meanwhile, CP-673451, an inhibitor of PDGFR
signaling, was used to block the effects of PDGF. CP-673451
downregulated YAP activation and repressed tumor malignancy. Its
effects suggested that PDGF signaling may activate YAP to affect
cell proliferation, survival and migration. Furthermore, the
current results revealed that PDGF-BB resulted in YAP
dephosphorylation and transactivation via the RhoA/PP-1 cascade, as
RhoA or PP-1 inhibition abolished YAP activation. A previous study
revealed that platelets can promote YAP dephosphorylation and
nuclear translocation via the RhoA/MYPT1/PP-1 cascade to induce a
pro-survival gene expression signature (32). Small GTPases, such as RhoA, Rac and
Cdc42, can activate YAP by inhibiting its phosphorylation (60). PP-1 is a mediator of PDGF signaling
in primary cultures of vascular smooth muscle cells (61). RhoA is one of the determinants of
the PDGF-BB-induced migration of rat hepatic stellate cells
(62). Notably, YAP is regulated by
complicated mechanisms. A previous study revealed that PDGF can
regulate YAP transcriptional activity via Src family
kinase-dependent tyrosine phosphorylation (63). Conversely, a study on genome-wide
profiling of highly aggressive Schwann cell lineage-derived
sarcomas revealed that TAZ/YAP-TEAD complexes can directly activate
PDGFR signaling and other oncogenic programs (64).
In conclusion, the present study suggested that
there may be a convergence between the Hippo/YAP and PDGF-PDGFR
signaling pathways in the malignant progression of pancreatic
cancer. Therefore, the concomitant manipulation of the YAP and PDGF
signaling pathways may improve the efficacy of therapy against
malignant tumors.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81972758), the
Interdisciplinary Medicine Seed Fund of Peking University (grant
no. BMU2018MX018) and the Science Foundation of Peking University
Cancer Hospital (grant no. 2017-23).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TL and TG performed the experiments, HL and HJ
analyzed and interpreted the data, YW designed the study and
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hainan Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PDGF-BB
|
platelet-derived growth factor-BB
|
|
YAP
|
Yes-associated protein
|
|
PP-1
|
protein phosphatase-1
|
|
PDGFR
|
PDGF receptor
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
References
|
1
|
Bartoschek M and Pietras K: PDGF family
function and prognostic value in tumor biology. Biochem Biophys Res
Commun. 503:984–990. 2018. View Article : Google Scholar
|
|
2
|
Heldin CH: Targeting the PDGF signaling
pathway in tumor treatment. Cell Commun Signal. 11:972013.
View Article : Google Scholar
|
|
3
|
Fredriksson L, Li H and Eriksson U: The
PDGF family: Four gene products form five dimeric isoforms.
Cytokine Growth Factor Rev. 15:197–204. 2004. View Article : Google Scholar
|
|
4
|
Roskoski R Jr: The role of small molecule
platelet-derived growth factor receptor (PDGFR) inhibitors in the
treatment of neoplastic disorders. Pharmacol Res. 129:65–83. 2018.
View Article : Google Scholar
|
|
5
|
Heldin CH, Lennartsson J and Westermark B:
Involvement of platelet-derived growth factor ligands and receptors
in tumorigenesis. J Intern Med. 283:16–44. 2018. View Article : Google Scholar
|
|
6
|
Cao Y: Multifarious functions of PDGFs and
PDGFRs in tumor growth and metastasis. Trends Mol Med. 19:460–473.
2013. View Article : Google Scholar
|
|
7
|
Martinho O, Longatto-Filho A, Lambros MB,
Martins A, Pinheiro C, Silva A, Pardal F, Amorim J, Mackay A,
Milanezi F, et al: Expression, mutation and copy number analysis of
platelet-derived growth factor receptor A (PDGFRA) and its ligand
PDGFA in gliomas. Br J Cancer. 101:973–982. 2009. View Article : Google Scholar
|
|
8
|
Nazarenko I, Hede SM, He X, Hedrén A,
Thompson J, Lindström MS and Nistér M: PDGF and PDGF receptors in
glioma. Ups J Med Sci. 117:99–112. 2012. View Article : Google Scholar
|
|
9
|
Saito Y, Haendeler J, Hojo Y, Yamamoto K
and Berk BC: Receptor heterodimerization: Essential mechanism for
platelet-derived growth factor-induced epidermal growth factor
receptor transactivation. Mol Cell Biol. 21:6387–6394. 2001.
View Article : Google Scholar
|
|
10
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008. View Article : Google Scholar
|
|
11
|
Paulsson J, Sjöblom T, Micke P, Pontén F,
Landberg G, Heldin CH, Bergh J, Brennan DJ, Jirström K and Ostman
A: Prognostic significance of stromal platelet-derived growth
factor beta-receptor expression in human breast cancer. Am J
Pathol. 175:334–341. 2009. View Article : Google Scholar
|
|
12
|
Dhar K, Dhar G, Majumder M, Haque I, Mehta
S, Van Veldhuizen PJ, Banerjee SK and Banerjee S: Tumor
cell-derived PDGF-B potentiates mouse mesenchymal stem
cells-pericytes transition and recruitment through an interaction
with NRP-1. Mol Cancer. 9:2092010. View Article : Google Scholar
|
|
13
|
Krenzlin H, Behera P, Lorenz V, Passaro C,
Zdioruk M, Nowicki MO, Grauwet K, Zhang H, Skubal M, Ito H, et al:
Cytomegalovirus promotes murine glioblastoma growth via pericyte
recruitment and angiogenesis. J Clin Invest. 129:1671–1683. 2019.
View Article : Google Scholar
|
|
14
|
Ostman A: PDGF receptors-mediators of
autocrine tumor growth and regulators of tumor vasculature and
stroma. Cytokine Growth Factor Rev. 15:275–286. 2004. View Article : Google Scholar
|
|
15
|
Heldin CH: Autocrine PDGF stimulation in
malignancies. Ups J Med Sci. 117:83–91. 2012. View Article : Google Scholar
|
|
16
|
Malvezzi M, Bertuccio P, Rosso T, Rota M,
Levi F, La Vecchia C and Negri E: European cancer mortality
predictions for the year 2015: Does lung cancer have the highest
death rate in EU women? Ann Oncol. 26:779–786. 2015. View Article : Google Scholar
|
|
17
|
Ansari D, Tingstedt B, Andersson B,
Holmquist F, Sturesson C, Williamsson C, Sasor A, Borg D, Bauden M
and Andersson R: Pancreatic cancer: Yesterday, today and tomorrow.
Future Oncol. 12:1929–1946. 2016. View Article : Google Scholar
|
|
18
|
Frisch SM and Screaton RA: Anoikis
mechanisms. Curr Opin Cell Biol. 13:555–562. 2001. View Article : Google Scholar
|
|
19
|
Gupta P, Gupta N, Fofaria NM, Ranjan A and
Srivastava SK: HER2-mediated GLI2 stabilization promotes anoikis
resistance and metastasis of breast cancer cells. Cancer Lett.
442:68–81. 2019. View Article : Google Scholar
|
|
20
|
Khader S, Thyagarajan A and Sahu RP:
Exploring signaling pathways and pancreatic cancer treatment
approaches using genetic models. Mini Rev Med Chem. 19:1112–1125.
2019. View Article : Google Scholar
|
|
21
|
Bai Y, Bai Y, Dong J, Li Q, Jin Y, Chen B
and Zhou M: Hedgehog signaling in pancreatic fibrosis and cancer.
Medicine (Baltimore). 95:e29962016. View Article : Google Scholar
|
|
22
|
Fjällskog ML, Hessman O, Eriksson B and
Janson ET: Upregulated expression of PDGF receptor beta in
endocrine pancreatic tumors and metastases compared to normal
endocrine pancreas. Acta Oncol. 46:741–746. 2007. View Article : Google Scholar
|
|
23
|
Yuzawa S, Kano MR, Einama T and Nishihara
H: PDGFRβ expression in tumor stroma of pancreatic adenocarcinoma
as a reliable prognostic marker. Med Oncol. 29:2824–2830. 2012.
View Article : Google Scholar
|
|
24
|
Abe M, Kortylewicz ZP, Enke CA, Mack E and
Baranowska-Kortylewicz J: Activation of PDGFr-β signaling pathway
after imatinib and radioimmunotherapy treatment in experimental
pancreatic cancer. Cancers (Basel). 3:2501–2515. 2011. View Article : Google Scholar
|
|
25
|
Weissmueller S, Manchado E, Saborowski M,
Morris JP IV, Wagenblast E, Davis CA, Moon SH, Pfister NT,
Tschaharganeh DF, Kitzing T, et al: Mutant p53 drives pancreatic
cancer metastasis through cell-autonomous PDGF receptor β
signaling. Cell. 157:382–394. 2014. View Article : Google Scholar
|
|
26
|
Kuo TL, Cheng KH, Shan YS, Chen LT and
Hung WC: β-catenin-activated autocrine PDGF/Src signaling is a
therapeutic target in pancreatic cancer. Theranostics. 9:324–336.
2019. View Article : Google Scholar
|
|
27
|
Su A, He S, Tian B, Hu W and Zhang Z:
MicroRNA-221 mediates the effects of PDGF-BB on migration,
proliferation, and the epithelial-mesenchymal transition in
pancreatic cancer cells. PLoS One. 8:e713092013. View Article : Google Scholar
|
|
28
|
Hiram-Bab S, Katz LS, Shapira H, Sandbank
J, Gershengorn MC and Oron Y: Platelet-derived growth factor BB
mimics serum-induced dispersal of pancreatic epithelial cell
clusters. J Cell Physiol. 229:743–751. 2014. View Article : Google Scholar
|
|
29
|
Lee J, Lee J, Yun JH, Choi C, Cho S, Kim
SJ and Kim JH: Autocrine DUSP28 signaling mediates pancreatic
cancer malignancy via regulation of PDGF-A. Sci Rep. 7:127602017.
View Article : Google Scholar
|
|
30
|
Shen J, Vil MD, Zhang H, Tonra JR, Rong
LL, Damoci C, Prewett M, Deevi DS, Kearney J, Surguladze D, et al:
An antibody directed against PDGF receptor beta enhances the
antitumor and the anti-angiogenic activities of an anti-VEGF
receptor 2 antibody. Biochem Biophys Res Commun. 357:1142–1147.
2007. View Article : Google Scholar
|
|
31
|
Moench R, Grimmig T, Kannen V, Tripathi S,
Faber M, Moll EM, Chandraker A, Lissner R, Germer CT, Waaga-Gasser
AM and Gasser M: Exclusive inhibition of PI3K/Akt/mTOR signaling is
not sufficient to prevent PDGF-mediated effects on glycolysis and
proliferation in colorectal cancer. Oncotarget. 7:68749–68767.
2016. View Article : Google Scholar
|
|
32
|
Haemmerle M, Taylor ML, Gutschner T,
Pradeep S, Cho MS, Sheng J, Lyons YM, Nagaraja AS, Dood RL, Wen Y,
et al: Platelets reduce anoikis and promote metastasis by
activating YAP1 signaling. Nat Commun. 8:3102017. View Article : Google Scholar
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Jenning S, Pham T, Ireland SK, Ruoslahti E
and Biliran H: Bit1 in anoikis resistance and tumor metastasis.
Cancer Lett. 333:147–151. 2013. View Article : Google Scholar
|
|
35
|
Ou C, Sun Z, Li S, Li G, Li X and Ma J:
Dual roles of yes-associated protein (YAP) in colorectal cancer.
Oncotarget. 8:75727–75741. 2017. View Article : Google Scholar
|
|
36
|
Hussein MR, Haemel AK and Wood GS:
Apoptosis and melanoma: Molecular mechanisms. J Pathol.
199:275–288. 2003. View Article : Google Scholar
|
|
37
|
Lu J, Yang Y, Guo G, Liu Y, Zhang Z, Dong
S, Nan Y, Zhao Z, Zhong Y and Huang Q: IKBKE regulates cell
proliferation and epithelial-mesenchymal transition of human
malignant glioma via the Hippo pathway. Oncotarget. 8:49502–49514.
2017. View Article : Google Scholar
|
|
38
|
Zygulska AL, Krzemieniecki K and
Pierzchalski P: Hippo pathway-brief overview of its relevance in
cancer. J Physiol Pharmacol. 68:311–335. 2017.
|
|
39
|
Chen Q, Zhang N, Gray RS, Li H, Ewald AJ,
Zahnow CA and Pan D: A temporal requirement for Hippo signaling in
mammary gland differentiation, growth, and tumorigenesis. Genes
Dev. 28:432–437. 2014. View Article : Google Scholar
|
|
40
|
Piccolo S, Dupont S and Cordenonsi M: The
biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev.
94:1287–1312. 2014. View Article : Google Scholar
|
|
41
|
Zanconato F, Cordenonsi M and Piccolo S:
YAP/TAZ at the roots of cancer. Cancer Cell. 29:783–803. 2016.
View Article : Google Scholar
|
|
42
|
Hansen CG, Moroishi T and Guan KL: YAP and
TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol.
25:499–513. 2015. View Article : Google Scholar
|
|
43
|
Azzolin L, Panciera T, Soligo S, Enzo E,
Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V,
et al: YAP/TAZ incorporation in the β-catenin destruction complex
orchestrates the Wnt response. Cell. 158:157–170. 2014. View Article : Google Scholar
|
|
44
|
Fitamant J, Kottakis F, Benhamouche S,
Tian HS, Chuvin N, Parachoniak CA, Nagle JM, Perera RM, Lapouge M,
Deshpande V, et al: YAP inhibition restores hepatocyte
differentiation in advanced HCC, leading to tumor regression. Cell
Rep. 10:1692–1707. 2015. View Article : Google Scholar
|
|
45
|
Yuan WC, Pepe-Mooney B, Galli GG, Dill MT,
Huang HT, Hao M, Wang Y, Liang H, Calogero RA and Camargo FD: NUAK2
is a critical YAP target in liver cancer. Nat Commun. 9:48342018.
View Article : Google Scholar
|
|
46
|
Park JA and Kwon YG: Hippo-YAP/TAZ
signaling in angiogenesis. BMB Rep. 51:157–162. 2018. View Article : Google Scholar
|
|
47
|
Zhao B, Li L, Tumaneng K, Wang CY and Guan
KL: A coordinated phosphorylation by Lats and CK1 regulates YAP
stability through SCF (beta-TRCP). Genes Dev. 24:72–85. 2010.
View Article : Google Scholar
|
|
48
|
Lee YA, Noon LA, Akat KM, Ybanez MD, Lee
TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, et
al: Autophagy is a gatekeeper of hepatic differentiation and
carcinogenesis by controlling the degradation of Yap. Nat Commun.
9:49622018. View Article : Google Scholar
|
|
49
|
Ansari D, Ohlsson H, Althini C, Bauden M,
Zhou Q, Hu D and Andersson R: The Hippo signaling pathway in
pancreatic cancer. Anticancer Res. 39:3317–3321. 2019. View Article : Google Scholar
|
|
50
|
Ben Mimoun S and Mauviel A: Molecular
mechanisms underlying TGF-β/Hippo signaling crosstalks - Role of
baso-apical epithelial cell polarity. Int J Biochem Cell Biol.
98:75–81. 2018. View Article : Google Scholar
|
|
51
|
Dobrokhotov O, Samsonov M, Sokabe M and
Hirata H: Mechanoregulation and pathology of YAP/TAZ via Hippo and
non-Hippo mechanisms. Clin Transl Med. 7:232018. View Article : Google Scholar
|
|
52
|
Totaro A, Panciera T and Piccolo S:
YAP/TAZ upstream signals and downstream responses. Nat Cell Biol.
20:888–899. 2018. View Article : Google Scholar
|
|
53
|
Pei T, Li Y, Wang J, Wang H, Liang Y, Shi
H, Sun B, Yin D, Sun J, Song R, et al: YAP is a critical oncogene
in human cholangiocarcinoma. Oncotarget. 6:17206–17220. 2015.
View Article : Google Scholar
|
|
54
|
Gibault F, Corvaisier M, Bailly F, Huet G,
Melnyk P and Cotelle P: Non-photoinduced biological properties of
verteporfin. Curr Med Chem. 23:1171–1184. 2016. View Article : Google Scholar
|
|
55
|
Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong
J, Sun X and Li J: Verteporfin suppresses cell survival,
angiogenesis and vasculogenic mimicry of pancreatic ductal
adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci.
108:478–487. 2017. View Article : Google Scholar
|
|
56
|
Yang S, Zhang L, Purohit V, Shukla SK,
Chen X, Yu F, Fu K, Chen Y, Solheim J, Singh PK, et al: Active YAP
promotes pancreatic cancer cell motility, invasion and
tumorigenesis in a mitotic phosphorylation-dependent manner through
LPAR3. Oncotarget. 6:36019–36031. 2015. View Article : Google Scholar
|
|
57
|
Yamaguchi R, Lartigue L and Perkins G:
Targeting Mcl-1 and other Bcl-2 family member proteins in cancer
therapy. Pharmacol Ther. 195:13–20. 2019. View Article : Google Scholar
|
|
58
|
Boisvert-Adamo K, Longmate W, Abel EV and
Aplin AE: Mcl-1 is required for melanoma cell resistance to
anoikis. Mol Cancer Res. 7:549–556. 2009. View Article : Google Scholar
|
|
59
|
Su H, Si XY, Tang WR and Luo Y: The
regulation of anoikis in tumor invasion and metastasis. Yi Chuan.
35:10–16. 2013.(In Chinese). View Article : Google Scholar
|
|
60
|
Jang JW, Kim MK and Bae SC: Reciprocal
regulation of YAP/TAZ by the Hippo pathway and the Small GTPase
pathway. Small GTPases. 11:280–288. 2018. View Article : Google Scholar
|
|
61
|
Zhang J, Lauf PK and Adragna NC: PDGF
activates K-Cl cotransport through phosphoinositide 3-kinase and
protein phosphatase-1 in primary cultures of vascular smooth muscle
cells. Life Sci. 77:953–965. 2005. View Article : Google Scholar
|
|
62
|
Li L, Li J, Wang JY, Yang CQ, Jia ML and
Jiang W: Role of RhoA in platelet-derived growth factor-BB-induced
migration of rat hepatic stellate cells. Chin Med J (Engl).
123:2502–2509. 2010.
|
|
63
|
Smoot RL, Werneburg NW, Sugihara T,
Hernandez MC, Yang L, Mehner C, Graham RP, Bronk SF, Truty MJ and
Gores GJ: Platelet-derived growth factor regulates YAP
transcriptional activity via Src family kinase dependent tyrosine
phosphorylation. J Cell Biochem. 119:824–836. 2018. View Article : Google Scholar
|
|
64
|
Wu LMN, Deng Y, Wang J, Zhao C, Wang J,
Rao R, Xu L, Zhou W, Choi K, Rizvi TA, et al: Programming of
schwann cells by Lats1/2-TAZ/YAP signaling drives malignant
peripheral nerve sheath tumorigenesis. Cancer Cell. 33:292–308.e7.
2018. View Article : Google Scholar
|