Introduction
Liver cancer is the fifth most common type of
cancer, and the third most common cause of cancer-related death
worldwide (1). Liver cancer usually
occurs in patients with chronic hepatitis and cirrhosis, which
limits the feasibility of curative therapies such as surgical
resection and locoregional ablation therapy. Systemic chemotherapy,
such as sorafenib and Lenvatinib, is used to treat patients with
advanced liver cancer, which is associated with vascular invasion
and metastasis. Recent advances in diagnostic imaging and
supportive care for liver cancer have increased the duration of
treatment periods and the quality of life of patients. However, the
long-term survival in liver cancer remains unsatisfactory, with a
median survival time of 12.3 months with sorafenib and 13.6 months
with Lenvatinib treatment (2).
Therefore, novel treatment strategies for liver cancer are required
to achieve higher rates of patient survival.
Acanthopanax senticosus (Rupr. et Maxim)
Harms (ASH), also known as Siberian ginseng or eleuthero, is a
small hardy shrub native to China, Korea, Russia and the northern
region of Japan (3). ASH is a
well-known traditional Chinese medicinal herb, that possesses
various pharmacological properties such as anti-fatigue,
antioxidant, anti-protective and antibacterial activities (4–7). ASH
is also known to exhibit therapeutic effects in several diseases,
such as heart disease, hypertension, allergies (8), chronic bronchitis, diabetes (9), gastric ulcers (10), rheumatoid arthritis (11) and neurodegenerative diseases
(12). Previous studies have also
shown that ASH exhibits a cytotoxic effect on several cancer cell
types. The stem bark of ASH inhibits tumor growth in stomach cancer
(13), breast cancer (14) and leukemia (15), as well as the growth of sarcoma
cells (16). However, the effect of
ASH on liver cancer cells remains unknown. In the present study,
the effects of ASH root extract on liver cancer cell lines was
examined.
Materials and methods
Preparation of ASH extract
The ASH root extract (ASHE) used in the present
study was prepared as described previously (17). Briefly, the roots of ASH were
collected from the native area of Heilongjiang, China. The
collected ASH roots (fresh weight 10 kg) were cut and immersed in
water for 3 h at 80°C to obtain extracts. The extract was then
evaporated in vacuo, yielding ~500 g powder using a
spray-drying method (10). ASHE
(lot no. 8142) used in this experiment was prepared and supplied by
Sun Chlorella Corp., and dissolved in distilled water to a
concentration of 100 mg/ml for use in subsequent experiments.
Chemicals reagents
Chloroquine (CQ), bafilomycin A1 and 3-methyladenine
(3-MA) were purchased from Sigma-Aldrich; Merck KGaA. The working
concentrations of CQ were 0.5 µM (HuH-7) or 2 µM (HepG2). The
working concentrations of bafilomycin A1 were 50 nM (HuH-7) or 125
nM (HepG2). The working concentrations of 3-MA were 0.1 mM (HuH-7)
or 0.3 mM (HepG2). Treatment duration with CQ and 3-MA was 72 h.
Treatment duration of bafilomycin A1 was 2 h.
Cell lines and culture conditions
The human liver cancer cell lines, HuH-7 (JCRB0403)
and HepG2 (JCRB1054), were obtained from the Japanese Collection of
Research Bioresources (JCRB), and cultured in DMEM supplemented
with 10% FBS, 10 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin (all from Sigma-Aldrich; Merck KGaA) at 37°C in a
humidified incubator with 5% CO2.
Cell viability assay
HuH-7 and HepG2 cells were treated for 24, 48 or 72
h with 62, 125, 250, 500 or 1,000 µg/ml ASHE in 96-well plates
(Thermo Fisher Scientific, Inc.). Subsequently, cell viability was
determined using the Premix WST-1 Cell Proliferation assay kit
(Takara Bio Inc.) according to the manufacturer's protocol.
Absorbance at 440 nm was measured using a microplate reader (Thermo
Scientific Multiskan FC; Thermo Fisher Scientific, Inc.).
Colony formation assay
HuH-7 (1×105) and HepG2
(5×104) cells were seeded in 6-well plates (Thermo
Fisher Scientific, Inc.), and treated with the indicated
concentrations of ASHE for 5 days. After treatment, the colonies
were fixed in 4% paraformaldehyde for 15 min at 4°C, stained with
0.25% crystal violet for 15 min at room temperature, observed using
a light microscope (magnification, ×400) and imaged. A colony was
defined to consist of ≥50 cells.
Cell cycle assay
HuH-7 and HepG2 cells were allowed to adhere to
6-well plates for 24 h prior to treatment with ASHE. After
incubation, cells were fixed with cold 70% ethanol for 2 h,
followed by staining with FxCycle propidium iodide (PI)/RNase
Staining Solution (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Stained cells were then analyzed using a
BD FACSCanto II flow cytometer (BD Biosciences) with FACSDiva
version 8.0.1 (BD Biosciences). Cell cycle analysis was performed
using FlowJo version (FlowJo LLC).
Apoptosis assay
HuH-7 and HepG2 cells were allowed to adhere to
6-well plates, and were treated with ASHE for 72 h. Treated cells
were then harvested and washed, followed by staining with a
phycoerythrin-conjugated Annexin V antibody (1:20) and 7-AAD (1:20)
for 15 min at room temperature in the dark (BD Pharmingen).
Apoptotic cells were analyzed using a BD FACSCanto II flow
cytometer and the FACSDiva software. The percentage of apoptotic
cells was calculated by dividing the percentage of either Annexin
V-positive or 7-AAD-positive cells by the total number of
cells.
Western blotting
Western blotting was performed as previously
described (18). Briefly, the
separated proteins were transferred to PVDF membranes and blotted
with specific primary antibodies overnight at 4°C. The primary
antibodies used were AMPKα (1:1,000; Cell Signaling Technology,
Inc.; cat. no. 2532), phospho-AMPKα (Thr172) (1:1,000; Cell
Signaling Technology, Inc.; cat. no. 2535), LC3B (1:1,000; Cell
Signaling Technology, Inc.; cat. no. 3868), Run domain
Beclin-1-interacting and cysteine-rich domain-containing (Rubicon;
1:1,000; Cell Signaling Technology, Inc.; cat. no. 8465), c-Jun
(1:1,000; Cell Signaling Technology, Inc.; cat. no. 9165),
phospho-c-Jun (Ser63) (1:1,000; Cell Signaling Technology, Inc.;
cat. no. 2361), SAPK/JNK (1:1,000; Cell Signaling Technology, Inc.;
cat. no. 9252), phospho-SAPK/JNK (Thr183/Tyr185) (1:1,000; Cell
Signaling Technology, Inc.; cat. no. 9251) or actin (1:3,000; Santa
Cruz Biotechnology, Inc.; cat. no. sc1615). After incubation for 1
h at room temperature with the secondary antibody, a horseradish
peroxidase-conjugated anti-rabbit IgG (1:2,000; Cell Signaling
Technology, Inc.; cat. no. 7074S), signals were visualized using
ECL Select Western Blotting Detection Reagent (GE Healthcare).
Finally, the bands were imaged using either ChemiDoc XRS Plus
(Bio-Rad Laboratories, Inc.) or WSE-6100H LuminoGraphI (ATTO).
Fluorescence microscopy
HuH-7 and HepG2 cells were plated at a density of
1.2×105 cells/well in 12-well plates and allowed to
adhere overnight. After ASHE treatment for 72 h, the cells were
incubated with 75 nM DAPGreen (Dojindo Molecular Technologies,
Inc.) and NucBlue Live ReadyProbes Reagent (Thermo Fisher
Scientific, Inc.) at 37°C for 30 min. After washing with PBS,
fluorescence imaging was performed using a Leica DMi8 fluorescence
microscope (magnification, ×400) and LAS X version 3.3 (Leica
Microsystems, Inc.).
Transmission electron microscopy
ASHE-treated and untreated HuH-7 cells were washed
with PBS and fixed in 2% glutaraldehyde (in 0.1 M PBS, pH 7.4) for
10 min at room temperature, and subsequently post-fixed with 2%
osmium tetra-oxide for 2 h in an ice bath. Next, the specimens were
dehydrated in graded concentrations of ethanol (30, 50, 70, 90 and
100%) and embedded in epoxy resin. Ultrathin sections were prepared
using an ultramicrotome. These sections were stained with uranyl
acetate for 15 min at room temperature and lead staining solution
for 5 min at room temperature, and were observed using a HITACHI
H-7600 transmission electron microscope (magnification,
×5,000)(Hitachi, Ltd.) at a high voltage of 100 kV.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA from HuH-7 and HepG2 cells was isolated
using TRIzol® reagent (Thermo Fisher Scientific, Inc.).
cDNA was synthesized from 2 µg total RNA using a SuperScript VILO
cDNA Synthesis kit according to the manufacturer's protocol (Thermo
Fisher Scientific, Inc.). The primer sequences for human Rubicon
and β-actin genes were as follows: RUBCN forward,
5′-GATTACTGGCAGTTCGTGAAAGA-3′ and reverse,
5′-CTGCTCTGGTCGTTCTCGTG-3′; ACTB (β-actin) forward,
5′-GGCATCCTCACCCTGAAGTA-3′ and reverse, 5′-GAAGGTGTGGTGCCAGATTT-3′.
qPCR was performed in triplicate using Power SYBR Green PCR mix
(Thermo Fisher Scientific, Inc.). The thermocycling conditions were
3 min at 95°C, followed by 40 cycles of 95°C for 3 sec, and 60°C
for 20 sec. Changes in relative gene expression between cDNA
samples were determined using the 2−ΔΔCq method
(19).
Statistical analysis
All data are presented as the mean ± standard
deviation of three independent experiments. SPSS version 21 (IBM
Corp.) was used to compare data. A two-tailed unpaired Student's
t-test was used compare differences between two groups. Comparisons
between control (non-treated) and ASHE-treated cells were performed
using a one-way ANOVA followed by a post-hoc Tukey's test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ASHE inhibits the proliferation of
HuH-7 and HepG2 cells
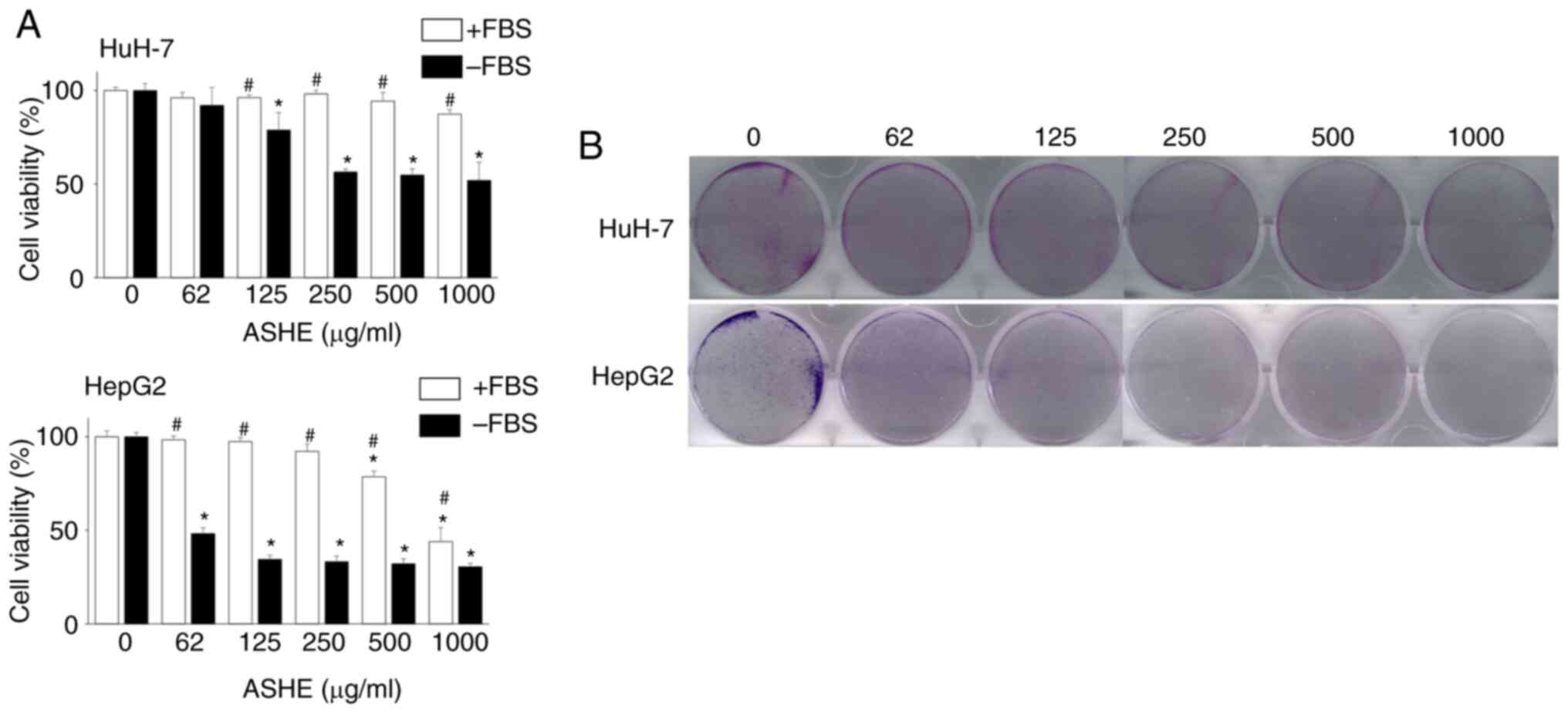
HuH-7 and HepG2 cells were treated with different
concentrations of ASHE (62-1,000 µg/ml) for 72 h to investigate the
effects of ASHE on cell viability. ASHE had minimal effects on cell
viability in these cell lines in the presence of FBS. However, in
the absence of FBS, cell viability was significantly reduced in a
dose-dependent manner (Fig. 1A).
Colony formation assay also revealed the inhibitory effect of ASHE
on the colony forming capacity of HuH-7 and HepG2 cells (Fig. 1B).
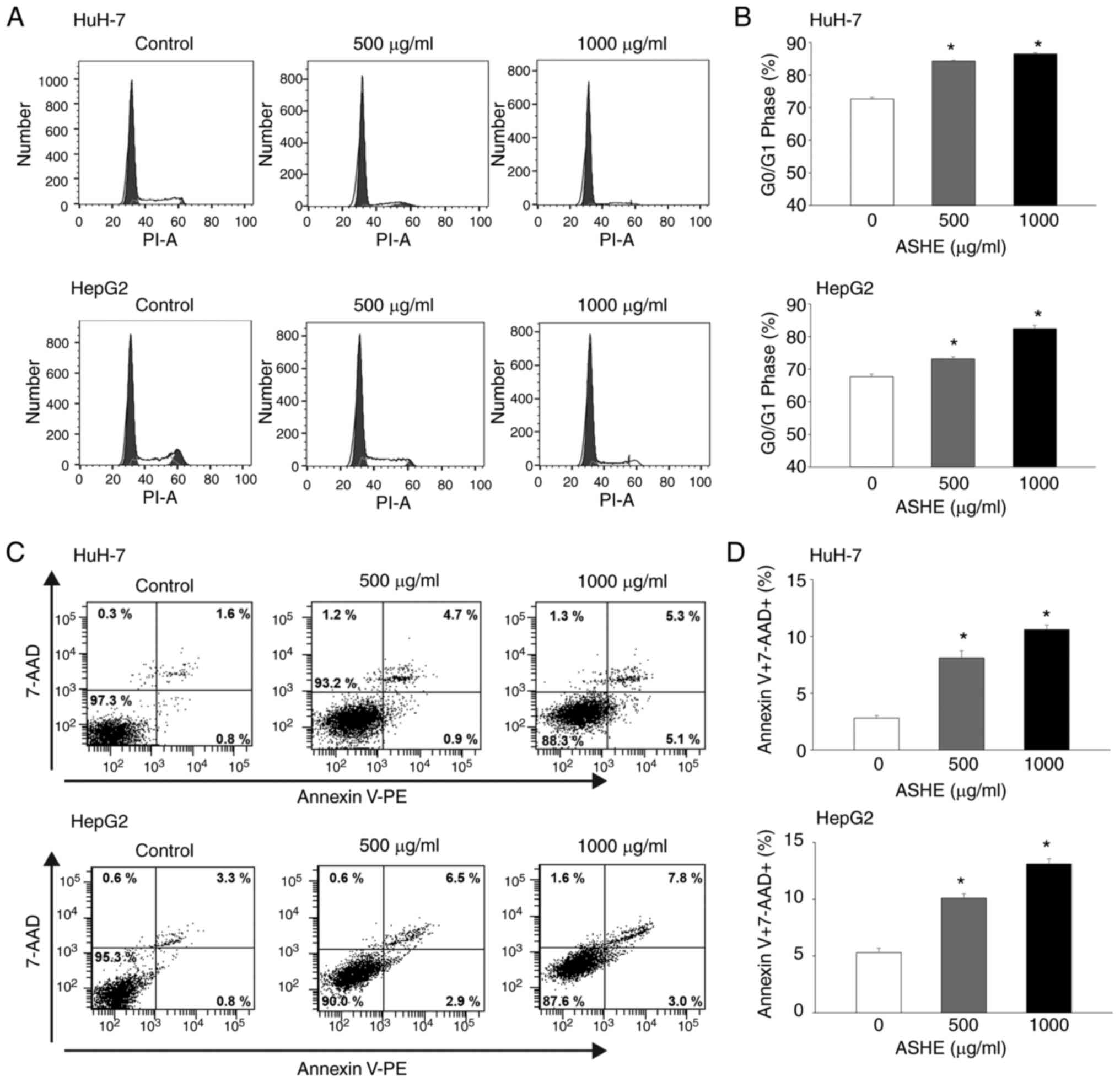
ASHE induces cell cycle arrest at the
G0/G1 phase and inhibits cell viability
Flow cytometry of PI stained cells showed
accumulation of cells in the G0/G1 phase, whereas the proportion of
cells in the S phase decreased in both HuH-7 and HepG2 cells
(Fig. 2A and B). Apoptosis analysis
was used to examine whether cell cycle arrest at the G0/G1 phase
could inhibit cell viability. Because the fluorescence intensity of
both Annexin V-negative and 7-AAD-negative cells was increased in
the cells treated with ASHE compared with the untreated cells, ASHE
was determined to affect the fluorescence intensity (Fig. 2C). The number of both Annexin
V-positive and 7-AAD-positive cells increased after treatment with
ASHE (Fig. 2D), suggesting that
apoptotic cell death occurred upon ASHE treatment.
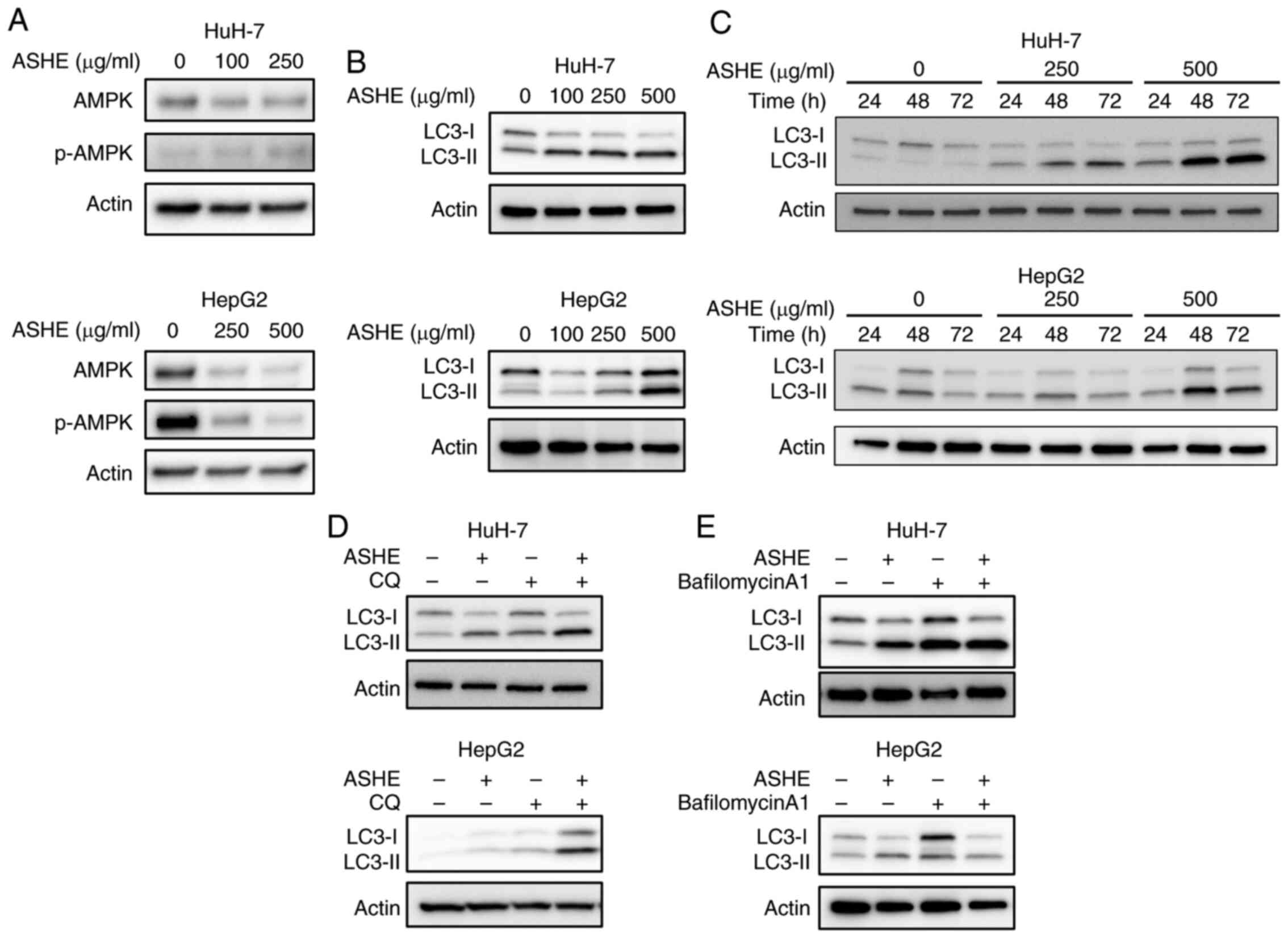
ASHE promotes autophagic cell death in
HuH-7 and HepG2 cells
Previous studies on mice models showed that the
fruit of the ASH tree modulates the autophagy regulator, AMPK
(20), and that autophagy may
trigger cell death in certain types of cells (21–23).
Therefore, the effect of ASHE on AMPK activity and autophagy was
assessed. No significant increase in the phosphorylation of AMPK
was observed in ASHE-treated HuH-7 and HepG2 cells (Fig. 3A). Furthermore, LC3-II protein
levels, a widely used marker for autophagic activity (24), were elevated in ASHE-treated HuH-7
and HepG2 cells compared with the untreated cells following
treatment for 48 and 72 h (Fig. 3B and
C). Thereafter, an LC3 turnover assay was performed to
determine whether the increase in LC3-II protein levels was due to
autophagy induction or inhibition (25). When ASHE-treated HuH-7 and HepG2
cells were co-treated with CQ, an inhibitor of degradation in
autolysosomal compartments (26),
LC3-II levels were further increased compared with ASHE treatment
alone (Fig. 3D). In contrast, in
the presence of bafilomycin A1, an autophagy inhibitor that blocks
the fusion of autophagosomes and lysosomes (26), LC3-II levels remained unchanged
irrespective of ASHE treatment (Fig.
3E). Thus, the LC3 turnover assay results suggested that ASHE
enhanced autophagy by promoting autophagosome-lysosome fusion
process, and not the degradative activity.
Next, DAPGreen was used to visualize the autophagic
vacuoles in the ASHE-treated cells under a fluorescence microscope.
As shown in Fig. 4A, the number of
fluorescence-stained autophagic vacuoles increased in HuH-7 and
HepG2 cells in an ASHE dose-dependent manner. The ultrastructure of
ASHE-treated HuH-7 cells was assessed using transmission electron
microscopy to further confirm whether the vacuoles were the result
of autophagy (Fig. 4B). Both the
number and the percentage area of autophagic structures was
increased in ASHE-treated cells compared with those in the
untreated cells in a dose-dependent manner (Fig. S1A, B). Furthermore, to examine the
effect of autophagy on cell viability, the cells were treated with
3-MA, which inhibits autophagy at an early stage, instead of CQ and
bafilomycin A1. Growth inhibition was partially attenuated in HuH-7
cells co-treated with 3-MA and ASHE compared with cells treated
with ASHE alone (Fig. 4C). A
similar result was also observed in HepG2 cells at low ASHE
concentrations.
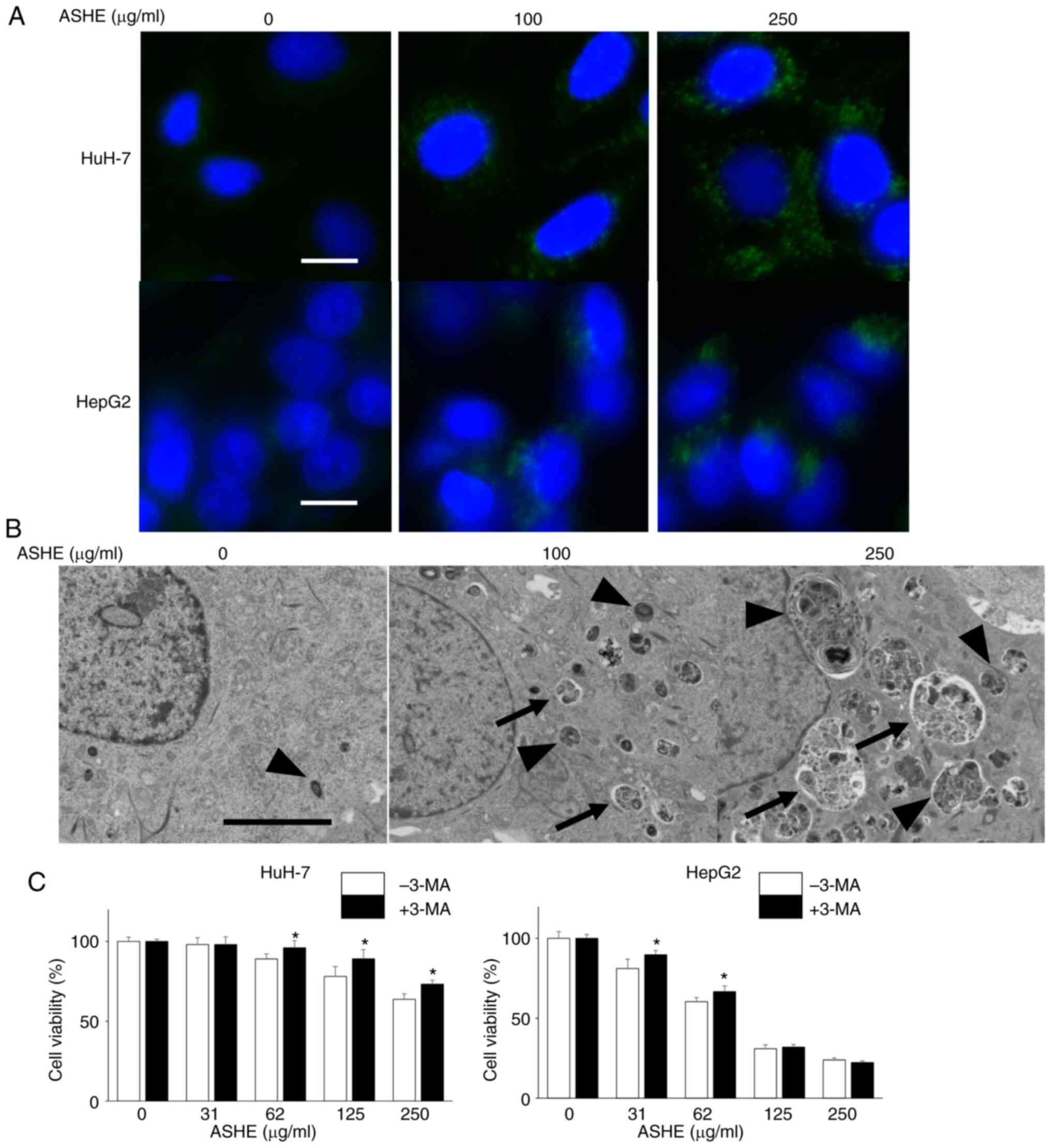 | Figure 4.Autophagic vesicle quantity is
increased in ASHE-treated liver cancer cells. (A) Fluorescence
micrographs of HuH-7 and HepG2 cells treated with ASHE in the
absence of FBS for 72 h. Following treatment, cells were incubated
with DAPGreen (50 nM) for 30 min. Magnification, ×400. Scale bar,
10 µm. (B) Representative transmission electron micrographs of
HuH-7 cells treated with ASHE in the absence of FBS. Arrows
indicate autophagosomes, and arrowheads indicate autolysosomes.
Magnification, ×5,000. Scale bar, 5 µm. (C) ASHE-treated HuH-7 and
HepG2 cells were co-treated with 3-MA (0.1 or 0.3 mM) for 72 h,
followed by measurement of cell viability. Data are presented as
the mean ± standard deviation of three independent experiments.
*P<0.05 vs. ASHE-treated cells in the absence of 3-MA. ASHE,
Acanthopanax senticosus Harms root extract; 3-MA,
3-methyladenine. |
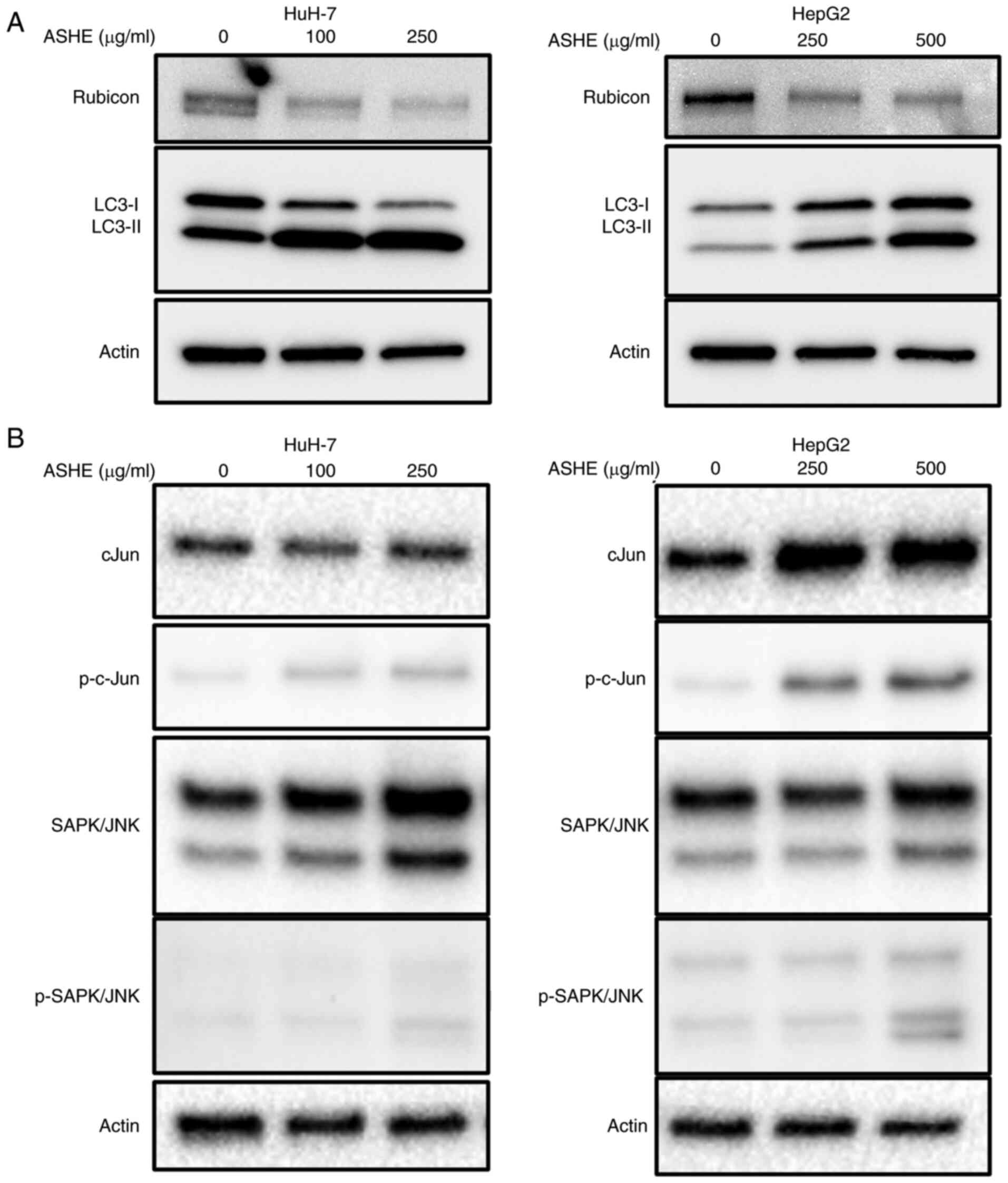
ASHE decreases Rubicon expression in
HuH-7 and HepG2 cells
As co-treatment with bafilomycin A1 did not enhance
LC3-II expression in ASHE-treated cells (Fig. 3E), the protein expression of Rubicon
was examined, which inhibits autophagosome and lysosome fusion by
binding to the UVRAG complex. UVRAG serves as an
autophagy-associated protein by binding to Beclin-1 and functions
in the autophagy maturation process (27). The protein expression of Rubicon in
ASHE-treated HepG2 and HuH-7 cells was lower than that in untreated
cells (Fig. 5A). Regulation of
Rubicon protein expression is mediated by the JNK signaling pathway
in hepatocytes (28). However,
western blotting results showed no decrease in the phosphorylation
of either c-Jun or SAPK/JNK in both HuH-7 and HepG2 cells following
treatment with ASHE (Fig. 5B).
Notably, no significant change in Rubicon expression was observed
in HuH-7 and HepG2 cells treated with ASHE in the presence of
either CQ or bafilomycin A1 (Fig.
S2). Moreover, no decrease in RUBCN mRNA expression was
observed in ASHE-treated cells compared with the untreated cells
(Fig. S3).
Discussion
In the present study, it was demonstrated that ASHE
exhibits cytostatic effects mediated by apoptosis and autophagy in
liver cancer cells by inhibiting Rubicon protein expression. This
effect was partially attenuated by co-treatment with the autophagy
inhibitor, 3-MA, suggesting that the mechanism underlying the
reduction in cell viability of liver cancer cells by ASHE may
involve autophagy.
ASHE is a widely used component of Chinese
traditional medicine prescribed for the treatment of several
diseases, such as heart disease, hypertension, allergies (8), chronic bronchitis, diabetes (9), gastric ulcers (10), rheumatoid arthritis (11) and neurodegenerative disease
(12). Previous studies have shown
that ASHE triggers apoptosis in stomach (13) and breast cancer cells (14); however, this effect is not evident
in leukemia (15) and sarcoma cells
(16). Thus, ASHE exhibits diverse
cytotoxic effects in cells of different cancer types. Moreover, a
previous study showed that the polysaccharides extracted from ASH
root caused pronounced apoptosis in HepG2 cells (29). As crude ASHE was used in the present
study, the apoptotic effect of ASHE was not observed.
Autophagy is a catabolic process that delivers
endogenous cytoplasmic content to the lysosome, followed by
degradation and recycling of proteins and organelles. Dysregulation
of autophagy is known to contribute towards protection of organisms
against pathologies, such as infections, heart disease,
neurodegeneration, aging and cancer (30). Autophagy functions as either an
inducer or inhibitor of cell death in cancer cells. In the present
study, ASHE-mediated autophagy promoted cell death in cooperation
with apoptosis in liver cancer cells. As the cytotoxicity of ASHE
was most evident in the absence of FBS, it is proposed that FBS is
an inhibitor of ASHE-induced autophagy.
During autophagy induction, LC3 protein is
synthesized and processed by Atg4 into LC3-I, followed by
conversion to LC3-II, which is conjugated with
phosphatidylethanolamine. Therefore, LC3 is commonly used for
autophagy assays (24). The results
of the present study showed that LC3-II protein expression levels
were increased in ASHE-treated cells compared with the untreated
cells. The difference in LC3-I levels between HuH-7 and HepG2 cells
suggests different LC3-I to LC3-II conversion rates in the two cell
types.
Autophagy serves dual roles in cancer development
depending on the cancer type, stage or genetic context (21,30).
Rubicon protein inhibits autophagy by suppressing the fusion of
autophagosomes and lysosomes (27).
This protein is upregulated by the JNK pathway in palmitate-treated
hepatocytes in fatty liver (28).
In the present study, the total amount of both c-Jun or SAPK/JNK
proteins in HuH-7 and HepG2 cells increased after ASHE treatment,
suggesting the presence of a factor that promotes the transcription
of these proteins in ASHE. Nevertheless, neither the JNK pathway
nor the transcription of RUBCN was inhibited in ASHE-treated
liver cancer cells. Notably, the expression of Rubicon protein in
ASHE-treated HepG2 and HuH-7 cells was decreased. Thus, inhibiting
Rubicon protein may be a promising approach for the treatment of
fatty liver (28), and therefore,
validation of this effect using other liver cancer cell lines is
required. Additionally, as ASHE induces autophagy as well as
inhibiting Rubicon expression in cell lines other than liver cancer
cell lines, autophagy-related diseases of other organs may also be
treated using ASHE.
In the present study, the increase in autophagic
flux after ASHE treatment in HuH-7 and HepG2 cells was independent
of the inhibition of Rubicon protein expression. Thus, unlike
metformin, which suppresses the mammalian target of the rapamycin
(mTOR) pathway (31), ASHE showed
no positive regulatory effect on AMPK activation. ASHE has four
major bioactive components, isofraxidin, eleutheroside B,
eleutheroside E and chlorogenic acid (3). Isofraxidin, eleutheroside B, and
chlorogenic acid offers protection against damage from radiation
and oxidation. Eleutheroside E is reported to exhibit
anti-inflammatory effects. As these components are not known to
induce autophagy, to the best of our knowledge, further studies are
required to determine the mechanism underlying ASHE-mediated
autophagy promotion.
In conclusion, the mechanism of ASHE action in liver
cancer cells was shown to involve autophagy. Decreased expression
of Rubicon protein upon ASHE treatment promotes the fusion of
autophagosomes and lysosomes, and leads to autophagy. As crude ASHE
affected liver cancer cell autophagy in the absence of FBS,
treatment with ASHE may not be practical in clinical settings,
where there are sufficient nutrients. Isolating the ASHE components
that inhibit Rubicon expression and promote autophagic flux, even
in the presence of FBS, may facilitate its application for the
treatment of diseases associated with autophagy dysregulation.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Miss Rieko Naganuma (Hanaichi
UltraStructure Research Institute Co. Ltd.), Miss Yukie Nakamura
(Health Sciences University of Hokkaido), and Miss Mayumi
Shitamichi (Health Sciences University of Hokkaido) for their
assistance with this research project.
Funding
This study was funded by Sun Chlorella Co., Ltd.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YK, MT and OU performed the experiments. MF, EO and
HTakek provided and prepared the ASH extracts. YK, KT and HTaked
participated in the design of the study. YK, OU and YA participated
in the writing of the manuscript and data analysis. YK and MT
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
MF, EO and HT are employees of Sun Chlorella Co.,
Ltd. They all had no role in the interpretation, drafting or
decision to publish the manuscript.
References
|
1
|
Marrero JA, Kulik LM, Sirlin CB, Zhu AX,
Finn RS, Abecassis MM, Roberts LR and Heimbach JK: Diagnosis,
staging, and management of hepatocellular carcinoma: 2018 practice
guidance by the American association for the study of liver
diseases. Hepatology. 68:723–750. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kudo M, Finn RS, Qin S, Han KH, Ikeda K,
Pscaglia F, Bari A, Park JW, Han G, Jassem J, et al: Lenvatinib
versus sorafenib in first-line treatment of patients with
unresectable hepatocellular carcinoma: A randomised phase 3
non-inferiority trial. Lancet. 391:1163–1173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davydov M and Krikorian AD:
Eleutherococcus senticosus (Rupr. & Maxim.) Maxim.
(Araliaceae) as an adaptogen: A closer look. J Ethnopharmacol.
72:345–393. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishibe S, Kinoshita H, Takeda H and Okano
G: Phenolic compounds from stem bark of Acanthopanax
senticosus and their pharmacological effect in chronic swimming
stressed rats. Chem Pharm Bull (Tokyo). 38:1763–1765. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee S, Son D, Ryu J, Lee YS, Jung SH, Kang
J, Lee SY, Kim HS and Shin KH: Anti-oxidant activities of
Acanthopanax senticosus stems and their lignan components.
Arch Pharm Res. 27:106–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jang MH, Shin MC, Kim YJ, Kim CJ and Chung
JH: Protective effect of Acanthopanax senticosus against
ethanol-induced apoptosis of human neuroblastoma cell line SK-N-MC.
Am J Chin Med. 31:379–388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee S, Shin DS, Oh KB and Shin KH:
Antibacterial compounds from the leaves of Acanthopanax
senticosus. Arch Pharm Res. 26:40–42. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yi JM, Hong SH, Kim JH, Kim HK, Song HJ
and Kim HM: Effect of Acanthopanax senticosus stem on mast
cell-dependent anaphylaxis. J Ethnopharmacol. 79:347–352. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu KY, Wu YC, Liu IM, Yu WC and Cheng JT:
Release of acetylcholine by syringin, an active principle of
Eleutherococcus senticosus, to raise insulin secretion in
Wistar rats. Neurosci Lett. 434:195–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujikawa T, Yamaguchi A, Morita I, Takeda
H and Nishibe S: Protective effects of Acanthopanax
senticosus Harms from Hokkaido and its components on gastric
ulcer in restrained cold water stressed rats. Biol Pharm Bull.
19:1227–1230. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takahashi Y, Tanaka M, Murai R,
Kuribayashi K, Kobayashi D, Yanagihara N and Watanabe N:
Prophylactic and therapeutic effects of Acanthopanax
senticosus Harms extract on murine collagen-induced arthritis.
Phytother Res. 28:1513–1519. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu SM, Li XZ, Zhang SN, Yang ZM, Wang KX,
Lu F, Wang CZ and Yuan CS: Acanthopanax senticosus protects
structure and function of mesencephalic mitochondria in a mouse
model of Parkinson's disease. Chin J Integr Med. 24:835–843. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hibasami H, Fujikawa T, Takeda H, Nihibe
S, Satoh T, Fujisawa T and Nakashima K: Induction of apoptosis by
Acanthopanax senticosus HARMS and its component, sesamin in
human stomach cancer KATO III cells. Oncol Rep. 7:1213–1216.
2000.PubMed/NCBI
|
|
14
|
Siao AC, Hou CW, Kao YH and Jeng KC:
Effect of sesamin on apoptosis and cell cycle arrest in human
breast cancer mcf-7 cells. Asian Pac J Cancer Prev. 16:3779–3783.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang QY, Zhong H, Chen FY, Zhang MY, Cai
JY and Xhong JH: A preliminary study on epigenetic regulation of
Acanthopanax senticosus in leukemia cell lines. Exp Hematol.
44:466–473. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamazaki T, Shimosaka S, Sasaki H,
Matsumura T, Tukiyama T and Tokiwa T:
(+)-Syringaresinol-di-O-β-D-glucoside, a phenolic compound from
Acanthopanax senticosus Harms, suppresses proinflammatory
mediators in SW982 human synovial sarcoma cells by inhibiting
activating protein-1 and/or nuclear factor-κB activities. Toxicol
in Vitro. 21:1530–1537. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miyazaki S, Oikawa H, Takekoshi H,
Hoshizaki M, Ogata M and Fujikawa T: Anxiolytic effects of
Acanthopanax senticosus HARMS occur via regulation of
autonomic function and activate hippocampal BDNF-TrkB signaling.
Molecules. 24:1322018. View Article : Google Scholar
|
|
18
|
Ishikawa K, Kawano Y, Arihara Y, Kubo T,
Takada K, Murase K, Miyanishi K, Kobune M and Kato J: BH3 profiling
discriminates the antiapoptotic status of 5-fluoro-uracil-resistant
colon cancer cells. Oncol Rep. 42:2416–2425. 2019.PubMed/NCBI
|
|
19
|
Kamihara Y, Takada K, Sato T, Kawano Y,
Murase K, Arihara Y, Kikuchi S, Hayasaka N, Usami M, Iyama S, et
al: The iron chelator deferasirox induces apoptosis by targeting
oncogenic Pyk2/β-catenin signaling in human multiple myeloma.
Oncotarget. 7:64330–64341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saito T, Nishida M, Saito M, Tanabe A,
Eitsuka T, Yuan SH, Ikekawa N and Nishida H: The fruit of
Acanthopanax senticosus (Rupr. et Maxim.) Harms improves
insulin resistance and hepatic lipid accumulation by modulation of
liver adenosine monophosphate-activated protein kinase activity and
lipogenic gene expression in high-fat diet-fed obese mice. Nutr
Res. 36:1090–1097. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Debnath J, Baehrecke EH and Kroemer G:
Does autophagy contribute to cell death? Autophagy. 1:66–74. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Espert L, Denizot M, Grimaldi M,
Robert-Hebmann V, Gay B, Varbano M, Codogno P and Biard-Piechaczyk
M: Autophagy is involved in T cell death after binding of HIV-1
envelope proteins to CXCR4. J Clin Invest. 116:2161–2172. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iwashita H, Sakurai HT, Nagahora N,
Ishiyama M, Shioji K, Sasamoto K, Okuma K, Shimizu S and Ueno Y:
Small fluorescent molecules for monitoring autophagic flux. FEBS
Lett. 592:559–567. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two Beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suzuki A, Kakisaka K, Suzuki Y, Wang T and
Takikawa Y: C-Jun N-terminal kinase-mediated Rubicon expression
enhances hepatocyte lipoapoptosis and promotes hepatocyte
ballooning. World J Gastroenterol. 22:6509–6519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang H, Sun B, Zhang Z, Chen J, Hao Q, Sun
Y, Yang Y, Wang Z and Pei J: Effects of Acanthopanax
senticosus polysaccharide on the proliferation, apoptosis and
cell cycle in human HepG2 cells. Pharmazie. 71:201–204.
2016.PubMed/NCBI
|
|
30
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Xu W, Yan Z, Zhao W, Mi J, Li J
and Yan H: Metformin induces autophagy and G0/G1 phase cell cycle
arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways.
J Exp Clin Cancer Res. 37:632018. View Article : Google Scholar : PubMed/NCBI
|