Introduction
Multidrug resistance (MDR) is an innate or acquired
ability of cancer cells to evade the effects of multiple
chemotherapeutic drugs with various mechanisms (1). MDR is a major issue in cancer
chemotherapy, but the detailed mechanisms of MDR are not completely
understood (2). Generally, several
mechanisms such as overexpression of various members of the ATP
binding cassette (ABC) transport proteins, autophagy induction,
apoptosis inhibition, enhanced DNA repair, cancer stem cell
regulation, miRNA regulation, hypoxia induction and epigenetic
regulation are considered to be the factors of MDR occurrence
(2).
Autophagy is a complex cell behavior characterized
by a lysosomal degradation pathway that degrades damaged or
superfluous cell components into basic molecules (3,4). During
autophagy, the cytosolic form of LC3 (LC3I) is conjugated to
phosphatidylethanolamine to form LC3-phosphatidylethanolamine
conjugate (LC3II), which is recruited to autophagosomal membranes
(5). The conversion of LC3I to LC3II
via phosphatidylethanolamine conjugation has been accepted as the
gold standard for autophagosome formation (6), and since the amount of LC3II is
associated with the number of autophagosomes, the ratio of
LC3II/LC3I is considered to be a measurement of autophagy activity;
a high LC3II/LC3I ratio indicates the activation of autophagy
(7–10)
Accumulating evidence suggests that autophagy activation serves a
crucial role in MDR by preventing death from apoptosis, hypoxia and
stress responses (11,12). For example, autophagy activation can
protect breast cancer cells from apoptosis induced by Epirubicin
and Adriamycin (13,14). However, autophagy inhibition can also
desensitize cancer cells to anticancer drug treatment (15,16). To
date, no previous studies have clearly explained the role of
autophagy in MDR occurrence; a deeper understanding of autophagy in
MDR cells is needed for cancer therapeutic purposes.
MicroRNAs (miRNAs) are a class of small noncoding
RNAs comprising 19–22 nucleotides that have been reported to serve
important roles in autophagy regulation in MDR bladder and gastric
cancer cells (17,18). Notably, miR-199a-5p is involved in
autophagy and drug resistance in osteosarcoma cells (19). Previous studies have demonstrated the
miR-199a-5p reverses drug resistance by targeting DNA
damage-regulated autophagy modulator 1 to inhibit protective
autophagy or by repressing the Wnt2-mediated autophagy signaling
pathway in leukemia cells (20,21).
However, the direct target and detailed mechanism of miR-199a-5p in
autophagy regulation in multidrug-resistant lung cancer cells have
not been clarified to date.
The present study aimed to investigate the
association between autophagy and MDR development in
multidrug-resistant A549/T cells, and to determine the role and
mechanism of miR-199a-5p in autophagy regulation and reversal of
MDR in lung cancer cells.
Materials and methods
Cell lines and cell culture
Human kidney cell line 293T were cultured in DMEM
(Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(FBS; Biological Industries) and antibiotics (100 U/ml penicillin
and 100 µg/ml streptomycin; Biological Industries) at 37°C with 5%
CO2. Human lung carcinoma cell lines A549, H1299 and
H661, and human lung adenocarcinoma cell lines H522 and H1944, and
Paclitaxel (PTX)-selected ABCB1-overexperssing lung carcinoma cell
line A549/T were cultured in RPMI-1640 medium (Thermo Fisher
Scientific, Inc.) containing 10% FBS and antibiotics at 37°C with
5% CO2. The A549/T cell line was generously provided by
Professor Liguang Lou (Shanghai Institute of Materia Medica,
Chinese Academy of Sciences, Shanghai, China), and all other cell
lines were obtained from the American Type Culture Collection.
Induction of a PTX-resistant H1299
cell line
The PTX-resistant H1299 cell line was established
using increasing concentrations of PTX as previously described
(22). Briefly, the IC50
value of PTX in H1299 cells was determined by the Cell Counting
Kit-8 (CCK-8) assay (GlpBio Technology, Inc.) as described below to
be ~30 nM. H1299 cells were incubated with 15 nM PTX (half of the
corresponding IC50) for 72 h, following which PTX was
withdrawn and cells were cultured without PTX until cells recovered
and tolerated for PTX at this concentration. The same process was
repeated until the cells were resistant to the initial
concentration; subsequently, the concentration of PTX doubled and
gradually increased until a final concentration of 240 nM, and the
protocol with each concentration was repeated twice. When the
induced cells could survive in 240 nM, the cells were considered to
be PTX-resistant and termed H1299/T cells.
Cell Counting Kit-8 (CCK-8) assay
To assess the sensitivity of cells to
chemotherapeutic drugs, H1299, H1299/T, A549 and A549/T cells
(untransfected and transfected) were plated in 96-well plates
(3.5×103 cells/well) and allowed to attach overnight,
and various concentrations of chemotherapeutic drugs PTX (0.3 to 3
µM for H1299 and H1299/T cells; 1 to 10 µM for A549/T cells; 0.01
to 100 nM for A549 cells), docetaxel (DOC; 1 nM to 3 µM for A549/T
cells; 0.03 to 300 nM for A549 cells), topotecan (1 nM to 3 µM for
A549/T cells; 0.0001 to 0.3 nM for A549 cells), SN38 (3 nM to 1 µM
for A549/T cells; 0.01 to 300 nM for A549 cells), oxaliplatin (OXA;
3 nM to 10 µM for A549/T and A549 cells) and vinorelbine (NVB; 0.3
nM to 3 µM for A549/T cells; 0.01 to 3 nM for A549 cells) were
added to the wells. Following a 72-h incubation, 10 µl/well CCK-8
reagent was added and incubated for an additional 1 h in a
humidified atmosphere at 37°C with 5% CO2. The
absorbance was measured at 450 nm using a SpectraMax M5 microplate
reader (Molecular Devices, LLC). The inhibition rate was calculated
using the following formula: Inhibition rate=[(ODcontrol
cells-ODtreated cells)/ODcontrol cells]
×100%. The IC50 values were calculated using the
survival curves (Fig. S4) by
GraphPad Prism 5.0 software (GraphPad Software, Inc.) using at
least eight concentrations for each cell line. The fold-resistance
was calculated by dividing the IC50 of drug-resistant
cell lines by that obtained in the parent cell lines.
Cell proliferation was determined using CCK-8 assay
as previously described (23).
Briefly, A549 and A549/T cells (untransfected and transfected) were
plated in 96-well plates (1.5×103 cells/well). Following
incubation in a humidified atmosphere at 37°C with 5%
CO2 for 24, 48 and 72 h, 10 µl CCK-8 reagent was added
to each well and incubated for an additional 1 h in a humidified
atmosphere at 37°C with 5% CO2, and absorbance at 450 nm
was determined by SpectraMax M5 microplate reader (Molecular
Devices, LLC.).
Reverse transcription-quantitative
(RT-q)PCR
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from all cell lines
according to the manufacturer's instructions. To obtain cDNA, 2 µg
total RNA was reverse-transcribed using M-MLV RTase cDNA Synthesis
Kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer's instructions at 42°C for 60 min and 70°C for 10 min,
followed by 4°C. qPCR was performed using the SYBR®
Premix Ex Taq (Takara Biotechnology Co., Ltd.) on a CFX96 Real-Time
PCR detection system (Bio-Rad Laboratories, Inc.). The
thermocycling conditions were as follows: Pretreatment at 95°C for
20 min, followed by 49 cycles at 95°C for 10 sec, 59°C for 20 sec
and 72°C for 10 sec, a melt curve at 65–95°C with increments of
0.5°C every 5 sec, and finally 4°C for preservation. Specific PCR
primers used in the present study were synthesized by Sangon
Biotech Co., Ltd. and are listed in Table SI. For the detection of miRNA levels,
RNU6-1 level was used as an internal reference for normalization,
whereas β-actin levels were used as an internal reference for
mRNAs. The relative expression was calculated by the
2−∆∆Cq method (24).
Neutral red staining
Neutral red staining assay was performed to assess
the quantity of the lysosome in cells. H1299, H1299/T, A549 and
A549/T cells (untransfected and transfected) were plated in 6-well
plates at the density of 5×104 cells per well and
allowed to attach overnight. Following treatment with chloroquine
(CQ) for 24 h (25–27) or cultured with serum-free medium
(starvation) for 6 h, respectively, the cells were stained with
neutral red stain solution (1:10 in 1X PBS) for 30 min in a
humidified atmosphere at 37°C with 5% CO2. Images were
captured by an Olympus IX3 fluorescence microscope (Olympus
Corporation) in three or four fields per sample.
miR-199a-5p knockdown (KD) and
overexpression (OE)
The transfections of sh-miR-199a-5p into A549/T
cells and miR-199a-5p into A549 cells were performed as previously
described (23). The plasmids were
designed with a GFP expression sequence to evaluate the success of
the transfection. The miR-199a-5p shRNA coding sequence was cloned
into pLV.I vector. The recombinant sh-miR-199a-5p plasmid was
transfected into 293T cells (5×105 cells/well) along
with pCMV–VSV-G, pMDLg/pRRE and pRSV-Rev using Lipofectamine™ 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) to generate lentiviral
supernatants containing the miR-199a-5p shRNA sequence. Different
concentrations of plasmids were used, but the quantity of all
plasmids was 3 µg, and the transfection duration was 48 h.
Subsequently, the lentivirus supernatants (2 ml/well) were
transduced into A549/T cells following ultrafiltration at 37°C for
24 h, and puromycin (10 µg/ml) was used to select monoclonal
miR-199a-5p-KD and vector control cells at 37°C for 2 weeks.
miR-199a-5p-OE cells were constructed using the same method; the
miR-199a-5p coding sequence was cloned into a pLVGFP vector, and
the recombinant miR-199a-5p plasmid was transfected into 293T cells
along with pCMV–VSV-G, pMDLg/pRRE and pRSV-Rev using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) to generate lentiviral supernatants containing
the miR-199a-5p coding sequence. The lentivirus supernatants (2
ml/well) were transduced into A549 cells following ultrafiltration
at 37°C for 24 h, and puromycin (10 µg/ml) was used to select
monoclonal miR-199a-5p-OE and vector control cells at 37°C for 2
weeks. The monoclonal miR-199a-5p-KD and vector control cells were
termed A549/T-miR-199a-5p-KD and A549/T-pLV.I-vector, and the
monoclonal miR-199a-5p-OE and vector control cells which were
termed A549-miR-199a-5p-OE and A549-pGFP-vector, respectively.
Western blotting
Following various treatments, cells were lysed with
RIPA buffer (Thermo Fisher Scientific, Inc.) and 100X phosphatase
inhibitor cocktail (Thermo Fisher Scientific, Inc.). The protein
concentration was determined using a BCA assay (Takara
Biotechnology Co., Ltd.) and equalized prior to loading; a total of
20 µg of proteins were separated by 8–10% SDS-PAGE and transferred
to a PVDF membrane. The membrane was blocked with 5% skimmed milk
in PBS containing 0.1% Tween-800 (TPBS) for 1 h at room temperature
and blotted with the relevant primary antibodies at 4°C for 10–12
h, followed by incubation with HRP-conjugated secondary antibodies
at room temperature for 1 h and visualization using the WB Femto
ECL Substrate (CWBIO). β-Actin was used as the loading control. The
primary antibodies used were as follows: LC3B (1:800; cat. no.
2775S), eukaryotic elongation factor 2 kinase (eEF2K; 1:1,000; cat.
no. 3692S), PI3K (1:1,000; cat. no. 4263S), Akt (1:1,000; cat. no.
4691S), phosphor (p)-Akt (S473, D9E; 1:1,000; cat. no. 4060S), mTOR
(7C10; cat. no. 2983S), p-mTOR (D9C2; 1:1,000; cat. no. 5536S)(all
Cell Signaling Technology, Inc.), p62/SQSTM1 (1:2,500; cat. no.
18420-1-AP; Protein Tech Group, Inc.), autophagy-related 5 (ATG5;
1:1,000; cat. no. GTX113309; GeneTex, Inc.), ABCB1 (1:1,000; cat.
no. P7965; MilliporeSigma) and β-Actin (1:5,000; cat. no. sc-1615;
Santa Cruz Biotechnology, Inc).
Dual-luciferase reporter assay
Luciferase activity was measured using the
Dual-Luciferase Reporter Assay system (Promega Corporation)
according to the manufacturer's instructions. Briefly,
A549-miR-199a-5p-OE and A549-pGFP-vector cells were plated in
96-well plates at the density of 3.5×103 cells/well and
allowed to attach overnight at 37°C. psiCHECK-2 reporter plasmids
(200 ng) containing the wild-type sequence which was
5′-GACACTTCGG-3′ or mutated coding sequence which was
5′-GGGTGTGTCG-3′ were transfected into the cells at 90% confluence.
The wild-type sequence was obtained from the National Center for
Biotechnology Information database (https://www.ncbi.nlm.nih.gov/gene/8878) and determined
using Snapgene Software (GSL Biotech, LLC). Following 24-h
transfection, the cells were lysed using the Passive Lysis Buffer
from the Dual-Luciferase Reporter Assay system kit, and the
luciferase activity was measured by a SpectraMax M5 Multi-Mode
microplate reader (Molecular Devices, LLC). The firefly luciferase
activity was normalized to that of Renilla luciferase, and
the relative luciferase activity was calculated by dividing the
luciferase activity value of the A549-miR-199a-5p-OE group by that
obtained in vector group. The sequences used for plasmid sequencing
were Rluc-Forward 5′-AGGACGCTCCAGATGAAATG-3′ and
psiCHECK-2-Reverse, 5′-ACTCATTTAGATCCTCACAC-3′.
MiRNA target gene prediction
The target genes of miR-199a-5p were predicted using
the online miRNA target prediction software TargetMiner (https://tools4mirs.org/software/target_prediction/targetminer/).
The database analysis predicted 905 target genes of miR-199a-5p.
Their binding to miR-199a-5p was compared, and the predicted genes
were screened for those associated with autophagy. The identified
targets were verified in A549-miR-199a-5p-OE and A549-pGFP-vector
cells.
Statistical analysis
Data are presented as the mean ± SEM of three
independent experiments. The statistical analysis was performed by
GraphPad Prism software. Unpaired Student's t-test and one-way
ANOVA with Dunnett's multiple comparison test were used to compare
the samples. Pearson's correlation analysis was used to determine
the relationships between continuous variables. P<0.05 was
considered to indicate a statistically significant difference.
Results
Autophagy inhibition in drug-resistant
A549/T and H1299/T lung cancer cells
Four lung cancer cell lines were used in the present
study, including the A549 and H1299 parental cell lines and
PTX-resistant A549/T and H1299/T cells. The results of the CCK-8
assay demonstrated that the IC50 values of A549/T and
H1299/T cells were significantly higher compared with those of the
parental cell lines (Table I).
 | Table I.Effects of paclitaxel on the
IC50 of A549, A549/T, H1299 and H1299/T cell lines. |
Table I.
Effects of paclitaxel on the
IC50 of A549, A549/T, H1299 and H1299/T cell lines.
| Cell line | IC50,
µM | Fold
resistance |
P-valuea |
|---|
| A549 | 0.012±0.007 | 1.000 |
| A549/T | 22.180±2.688 | 1,848.333 | <0.001 |
| H1299 | 0.030±0.013 | 1.000 |
| H1299/T | 0.294±0.022 | 9.800 | <0.001 |
To determine the association between autophagy and
MDR, the protein expression levels of LC3I and LC3II were detected
by western blotting. As presented in Fig.
1A, LC3I accumulated in A549/T cells, and the LC3II/LC3I ratio
was lower compared with that in A549 cells following CQ treatment.
Similar results were observed in the H1299/T cell line (Fig. 1B). Additionally, neutral red staining
was used to determine intracellular lysosome formation. Neutral red
accumulates in acidic vesicle organelles and is used for staining
lysosomes and as an indicator of autophagy (27). As demonstrated in Fig. 1C, compared with A549/T cells, A549
cells displayed a higher number of red dots in the cytoplasm.
Following CQ treatment and serum-free culture (6 h starvation),
A549 cells exhibited higher accumulation of lysosomes with red
staining compared with that in A549/T cells, suggesting that A549
cells had a higher basal autophagy level compared with A549/T
cells, and A549/T displayed a lower response of autophagy induction
by starvation. Similar results were observed in H1299 and H1299/T
cells, which confirmed that autophagy was inhibited in
drug-resistant lung cancer cells compared with that in the parental
cell lines. Next, the expression levels of autophagy-related
proteins were determined in A549 and A549/T cells. The results
demonstrated that the PI3K/Akt/mTOR signaling pathway-related
proteins and eEF2K were highly expressed in A549/T cells. By
contrast, the ATG5 and p62 expression levels was significantly
attenuated in A549/T cells compared with those in the parental A549
cells (Fig. 1D). These results
suggested that autophagy was suppressed in A549/T and H1299/T
cells, and that negative regulation of autophagy may be involved in
MDR in lung cancer cells.
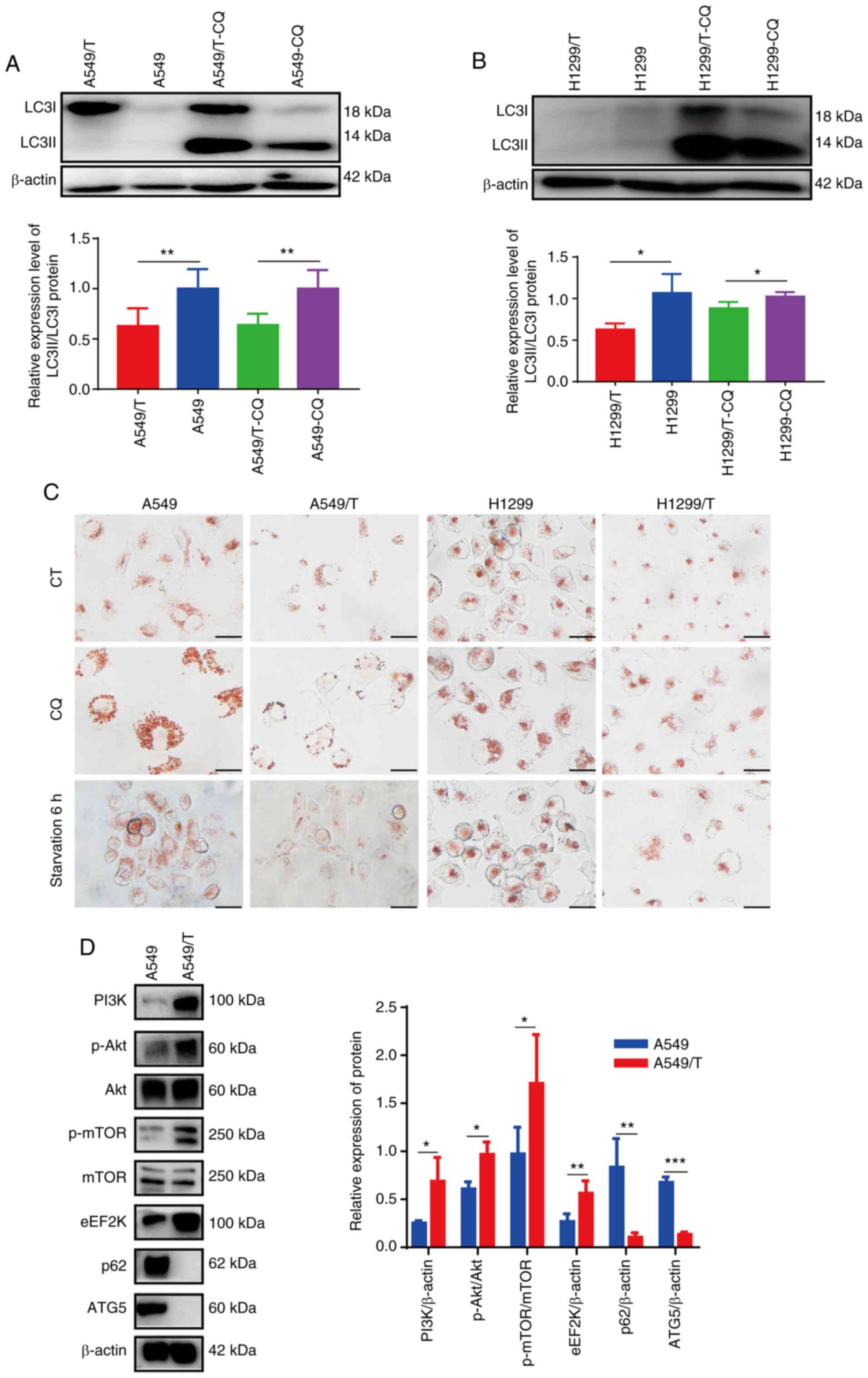 | Figure 1.Autophagy inhibition in MDR A549/T
and H1299/T cells. (A and B) Western blot analysis of LC3I and
LC3II protein expression in cells treated with or without 10 µM CQ
and the LC3II/LC3I ratio. β-actin was used as the loading control.
(C) Neutral red staining for lysosomes analysis. Scale bar, 10 µm.
(D) Western blot analysis of the expression levels of PI3K/Akt/mTOR
pathway, eEF2K, p62 and ATG5 proteins, and the quantitative
analysis results. The experiments were repeated three times, and
representative bands are presented. *P<0.05, **P<0.01 and
***P<0.001. eEF2K, eukaryotic elongation factor 2 kinase; ATG5,
autophagy-related 5; CQ, chloroquine; CT, control; p-,
phosphorylated. |
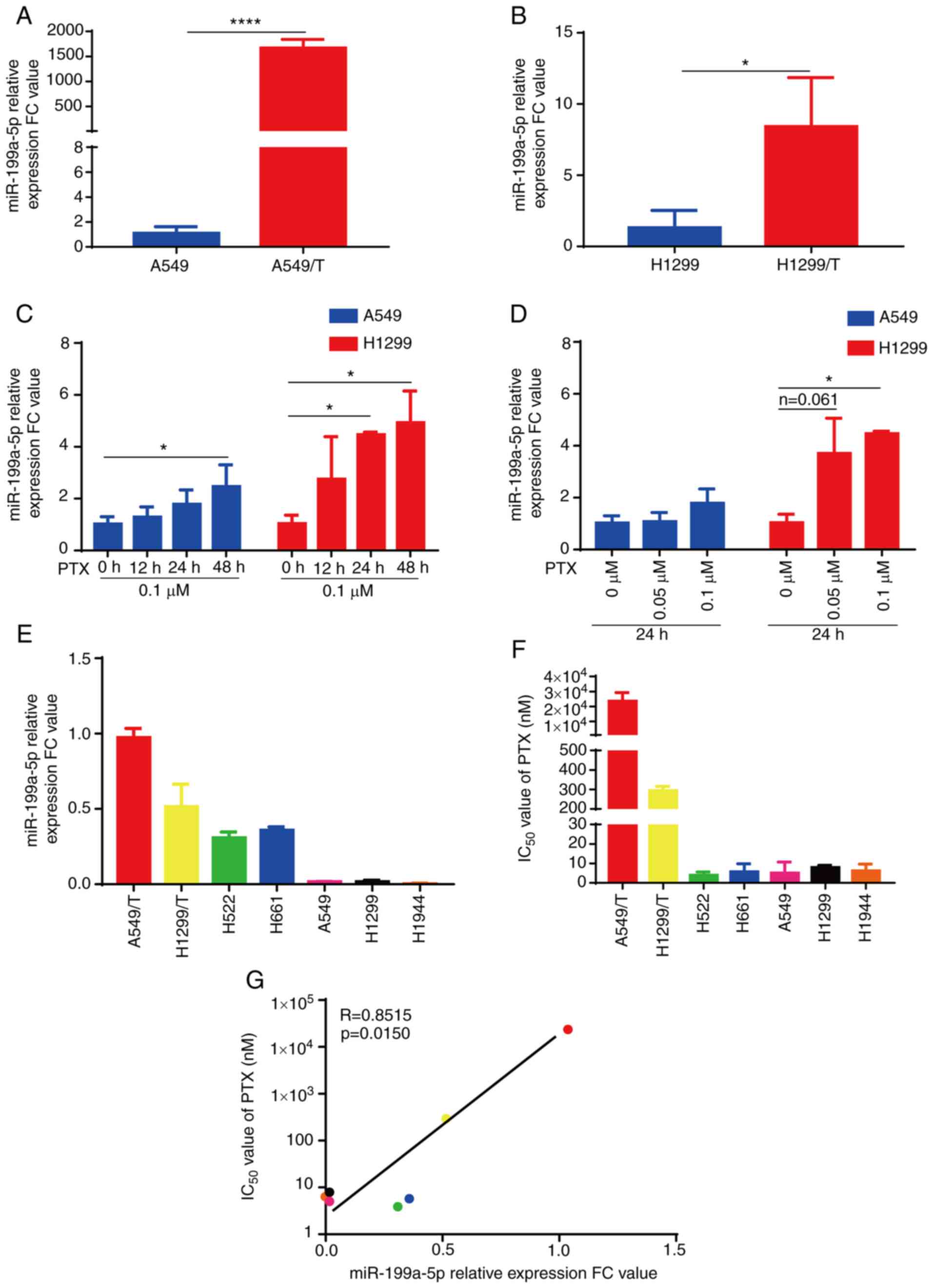
miR-199a-5p is upregulated in
drug-resistant A549/T and H1299/T cells
As aforementioned, miR-199a-5p is associated with
MDR (19). To determine the effects
of miR-199a-5p on MDR, the present study evaluated the expression
levels of miR-199a-5p in four lung cancer cell lines by RT-qPCR.
The expression levels of miR-199a-5p were significantly higher in
A549/T and H1299/T cells compared with those in their parental
cells (Fig. 2A and B). Notably, PTX
treatment increased the expression levels of miR-199a-5p in A549
and H1299 cells with a tendency of dose- and time-dependence
(Fig. 2C and D). In addition, the
expression levels of miR-199a-5p and the IC50 values of
cells to PTX were determined in six lung cancer cell lines. As
demonstrated in Fig. 2E-G, the
expression levels of miR-199a-5p were positively correlated with
the IC50 value of PTX in these cell lines (R=0.8515;
P=0.015). These results suggested that miR-199a-5p was upregulated
in drug-resistant A549/T and H1299/T cells, miR-199a-5p
upregulation was PTX-dependent in lung cancer cells, and the
expression levels of miR-199a-5p were associated with the
resistance of lung cancer cells to PTX treatment.
Knockdown of miR-199a-5p reverses the
resistance of A549/T cells to chemotherapeutic drugs, and
overexpression of miR-199a-5p induces drug resistance in A549
cells
To further investigate the involvement of
miR-199a-5p in MDR, miR-199a-5p-KD A549/T and miR-199a-5p-OE A549
cell lines were established. The GFP fluorescence observed in the
four transfected cell lines demonstrated that the plasmid
transfection was successful (Fig.
S1). miR-199a-5p expression levels were downregulated in
A549/T-miR-199a-5p-KD cells upregulated in A549-miR-199a-5p-OE
cells compared with those in the corresponding vector groups
(Fig. 3A and B). The CCK-8 assay
results demonstrated that A549/T-miR-199a-5p-KD cells were
sensitive to PTX, DOC, SN38 and OXA, as indicated by the reduced
cell viability following treatment compared with that in the
vector-transfected cells (Fig. 3C;
Table II), whereas
A549-miR-199a-5p-OE cells exhibited lower sensitivity to PTX, DOC,
OXA and NVB compared with that in the corresponding vector control
group (Fig. 3D; Table II).
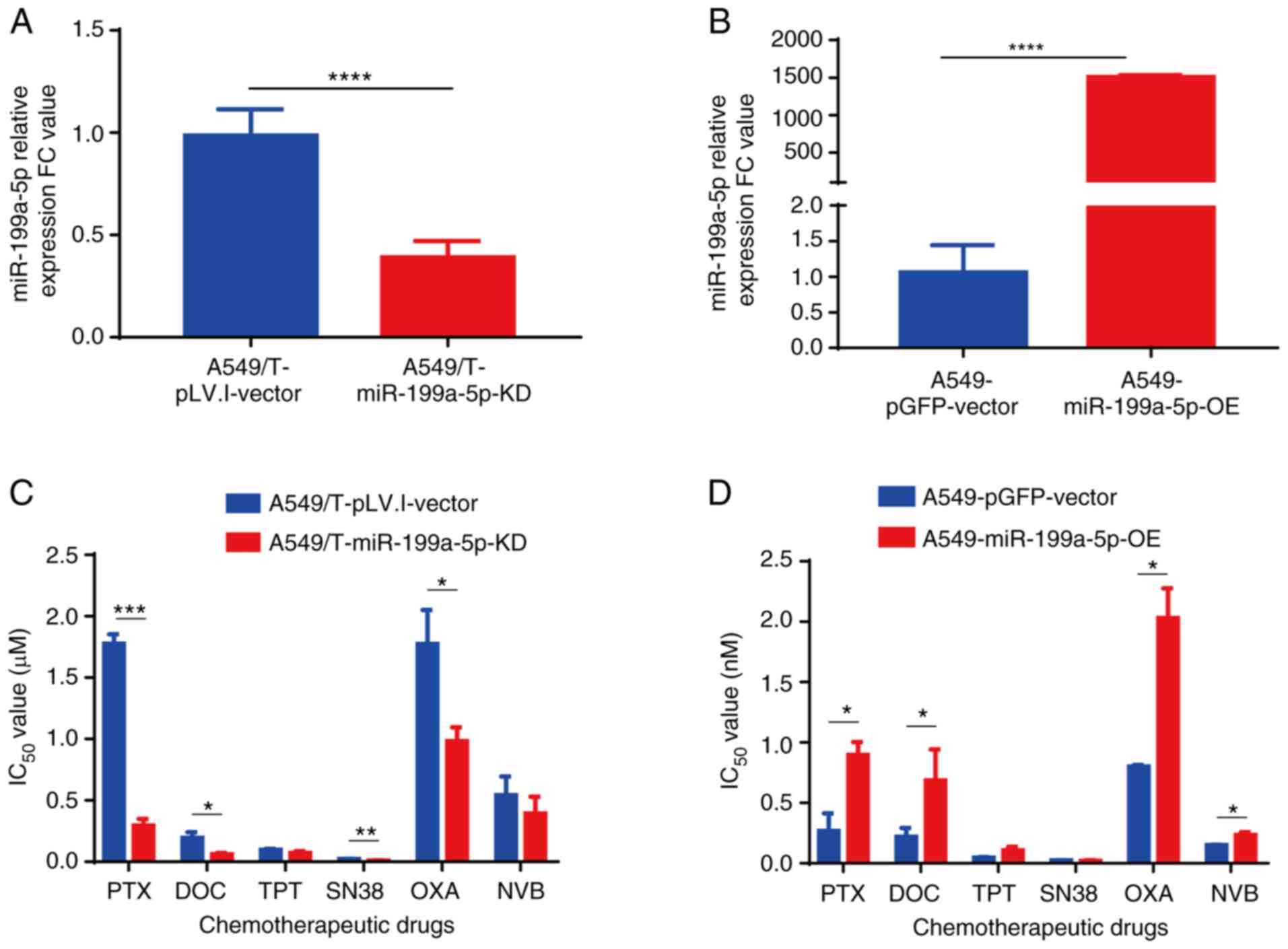 | Figure 3.Knockdown and overexpression of
miR-199a-5p affects cell sensitivity to chemotherapeutic drugs. (A
and B) Reverse transcription-quantitative PCR analysis of
miR-199a-5p expression levels in (A) A549/T-miR-199a-5p-KD and (B)
A549-miR-199a-5p-OE cells. (C and D) The effects of various
chemotherapeutic drugs on the viability of A549/T-miR-199a-5p-KD
and A549-miR-199a-5p-OE cells determined by the Cell Counting Kit-8
assay. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001.
miR, microRNA; KD, knockdown; OE, overexpression; PTX, paclitaxel;
DOC, docetaxel; TPT, topotecan; OXA, oxaliplatin; NVB,
vinorelbine. |
| Table II.Effects of miR-199a-5p on the
sensitivity of cells to chemotherapeutic drugs. |
Table II.
Effects of miR-199a-5p on the
sensitivity of cells to chemotherapeutic drugs.
|
| IC50,
µM |
|
| IC50,
nM |
|
|
|---|
|
|
|
|
|
|
|
|
|---|
| Drug |
A549/T-pLV.I-vector |
A549/T-miR-199a-5p-KD | Fold-reversal | P-value |
A549-pGFP-vector |
A549-miR-199a-5p-OE |
Fold-resistance | P-value |
|---|
| PTX | 1.776±0.078 | 0.295±0.053 | 6.020 | <0.001 | 0.267±0.147 | 0.897±0.107 | 3.340 | 0.011 |
| DOC | 0.194±0.047 | 0.062±0.010 | 3.129 | 0.015 | 0.220±0.072 | 0.686±0.256 | 3.118 | 0.039 |
| TPT | 0.096±0.010 | 0.071±0.016 | 1.352 | 0.083 | 0.047±0.005 | 0.107±0.030 | 2.277 | 0.076 |
| SN38 | 0.022±0.001 | 0.014±0.001 | 1.571 | 0.007 | 24.370±2.263 | 19.020±7.345 | 0.780 | 0.300 |
| OXA | 1.773±0.279 | 0.958±0.189 | 1.851 | 0.010 |
0.800±0.014a | 2.030±0.247 | 2.538 | 0.020 |
| NVB | 0.543±0.151 | 0.392±0.138 | 1.385 | 0.270 | 0.150±0.003 | 0.234±0.024 | 1.560 | 0.019 |
miR-199a-5p contributes to MDR by
suppressing autophagy
To further determine the underlying mechanism of MDR
regulation by miR-199a-5p, autophagy levels were assessed in
A549/T-miR-199a-5p-KD and A549-miR-199a-5p-OE cells. Western blot
analysis demonstrated that the LC3II/LC3I ratio in
A549/T-miR-199a-5p-KD cells was increased compared with that in the
corresponding vector group, whereas the LC3II/LC3I ratio in
A549-miR-199a-5p-OE cells was markedly decreased compared with the
vector group following CQ treatment (Fig.
4A). In addition, compared with that in the vector cells,
lysosomal accumulation appeared to be increased in
A549/T-miR-199a-5p-KD and decreased in A549-miR-199a-5p-OE cells,
and this effect was more pronounced following CQ treatment and
serum-free culture (Fig. 4B). Since
the ABCB1 expression levels and the proliferation of
A549/T-miR-199a-5p-KD and A549-miR-199a-5p-OE cells remained
unchanged compared with those in the corresponding vector groups
(Fig. S3A and B), these results
suggested that miR-199a-5p-mediated drug resistance in lung cancer
cells was independent of ABCB1 expression and cell proliferation,
but mainly associated with autophagy inhibition.
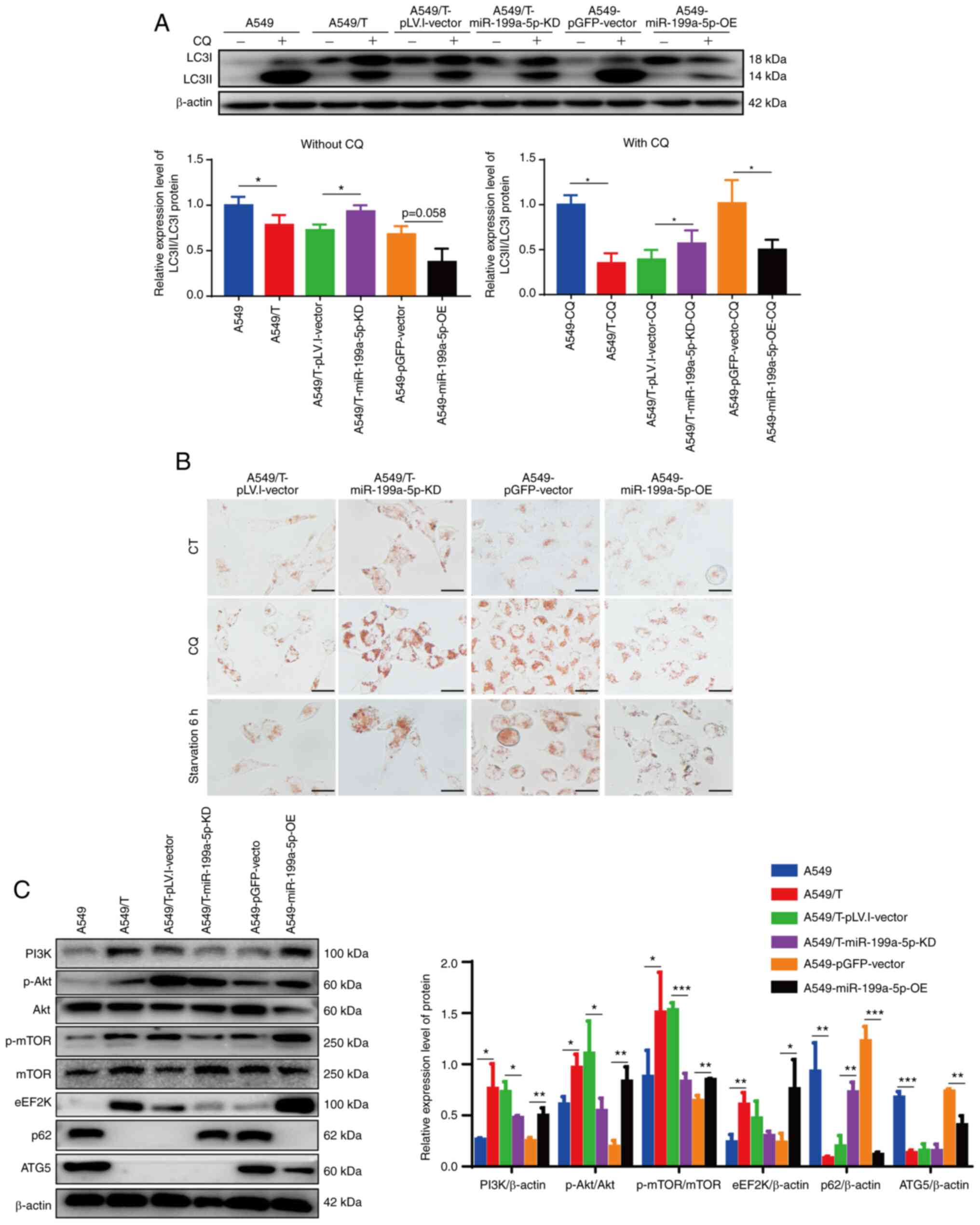 | Figure 4.Knockdown and overexpression of
miR-199a-5p affects cellular autophagy levels. (A) Western blot
analysis of LC3I and LC3II protein expression levels in various
cell lines and the LC3II/LC3I ratio with or without CQ treatment.
β-actin was used as the loading control. (B) Neutral red staining
for lysosome analysis. Scale bar, 10 µm. (C) Western blot analysis
of the protein levels of PI3K/Akt/mTOR pathway, eEF2K, p62 and ATG5
proteins and the quantitative analysis results. The experiments
were repeated three times and a representative data are presented.
*P<0.05, **P<0.01 and ***P<0.001. miR, microRNA; KD,
knockdown; OE, overexpression; eEF2K, eukaryotic elongation factor
2 kinase; ATG5, autophagy-related 5; CQ, chloroquine; CT, control;
p-, phosphorylated. |
miR-199a-5p regulates autophagy via
the PI3K/Akt/mTOR signaling pathway and autophagy-related
proteins
Western blotting experiment results demonstrated
that overexpression of miR-199a-5p increased the expression levels
of the PI3K/Akt/mTOR signaling pathway-related proteins and eEF2K,
whereas knockdown of miR-199a-5p decreased the expression levels of
these proteins compared with those in the corresponding vector
groups (Fig. 4C). TargetMiner
database analysis revealed that miR-199a-5p bound to the ATG5
(NM_004849) 3′-untranslated region, which suggested that ATG5 may
be a target of miR-199a-5p (Fig.
S2A). Further results demonstrated that transfection with
miR-199a-5p-OE attenuated ATG5 protein expression levels, whereas
miR-199a-5p-KD did not affect ATG5 expression. RT-qPCR analysis
revealed similar results: The mRNA expression levels of ATG5 were
significantly downregulated in A549-miR-199a-5p-OE cells compared
with those in the A549-pGFP-vector cells, but were not
significantly increased in A549/T-miR-199a-5p-KD cells compared
with those in the A549/T-pLV.I-vector cells (Fig. S2B). These results suggested that
knockdown of miR-199a-5p induced autophagy, whereas overexpression
of miR-199a-5p suppressed autophagy by activating PI3K/Akt/mTOR
pathway and repressing ATG5 expression.
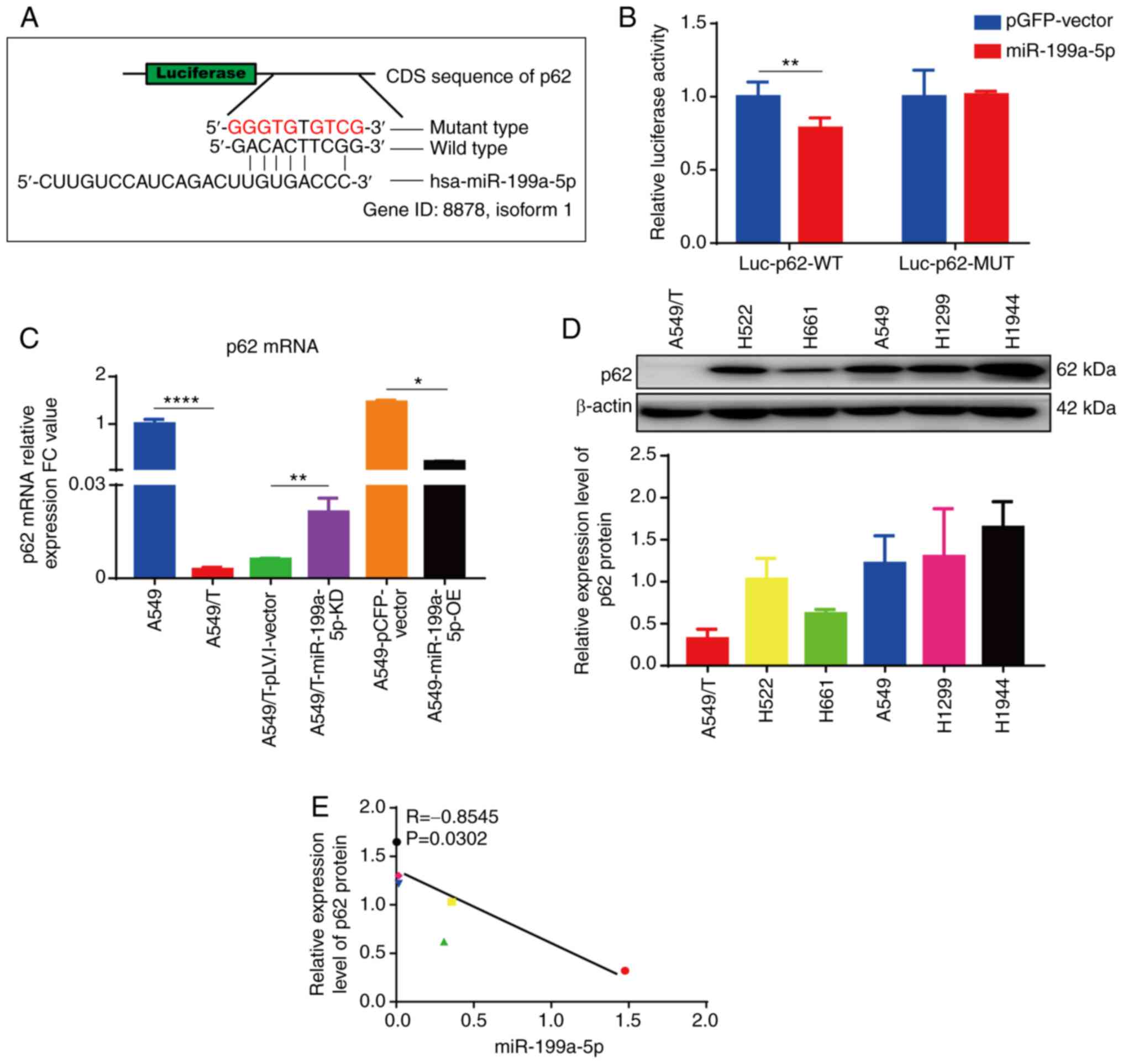
miR-199a-5p targets p62
The expression levels of p62 were significantly
decreased in A549/T compared with those in the parental A549 cells
(Fig. 1D) and in A549-miR-199a-5p-OE
compared with those in the vector-transfected cells (Fig. 4C). In addition, knockdown of
miR-199a-5p rescued the expression of p62 in A549/T cells (Fig. 4C). Therefore, we hypothesized that
miR-199a-5p may directly target p62. Consistent with this
hypothesis, analysis of the p62 coding sequence revealed six
complementary bases paired with miR-199a-5p (Fig. 5A). Additionally, dual-luciferase
reporter assay revealed that overexpression of miR-199a-5p reduced
the luciferase activity of the p62 wild-type reporter compared with
that in the mutated group, which confirmed that p62 was one of the
direct targets of miR-199a-5p (Fig.
5B). In addition, overexpression of miR-199a-5p significantly
downregulated, whereas knockdown of miR-199a-5p significantly
upregulated the mRNA expression levels of p62 compared with those
in the corresponding vector groups (Fig.
5C). Subsequently, the present study investigated the
relationship between p62 protein (Fig.
5D) and miR-199a-5p (Fig. 2E)
expression levels in six lung cancer cell lines. The results
demonstrated that the protein expression levels of p62 were
negatively correlated with miR-199a-5p expression levels in these
lung cancer cell lines (R=−0.8545; P=0.0302) (Fig. 5E). These results suggested that p62
mRNA was a downstream target of miR-199a-5p, and that miR-199a-5p
negatively regulated the levels of p62 protein.
Discussion
Autophagy malfunction has been proposed to serve a
crucial role in cancer development and resistance to chemotherapy
(3). Autophagy activation may
contribute to drug resistance (11,13,14) and
may prevent cell death by apoptosis, necrosis or pyroptosis
(28). Autophagy is crucial for
reducing cellular stress (12), ROS
production (11) and genomic
instability (29), and direct
evidence has demonstrated that autophagy activation regulates DNA
damage response to capsaicin, which induces drug resistance
(30). In addition, as a prototypical
damage-associated molecular pattern molecule, high-mobility group
B1 is released following autophagy induction and promotes drug
resistance in ovarian (31),
colorectal (32) and lung (33) cancer. However, autophagy inhibition
also affects MDR. For instance, Kanzawa et al (15) have reported that the autophagy
inhibitor 3-MA suppresses the sequestration of cytoplasmic material
during autophagy to rescue the tumor cells and promote resistance
to chemotherapy. Lv et al (16) have demonstrated that curcumin
decreases the levels of colon cancer-associated transcript 1 and
sensitizes multidrug-resistant breast cancer cells to cisplatin via
autophagy activation. Depending on the cell type, environment and
stimulus, autophagy and cell death can have opposing, additive or
even synergistic effects, which also leads to the development of
drug resistance (34). In the present
study, autophagy was inhibited in MDR lung cancer cells by
miR-199a-5p overexpression, and the potential underlying mechanism
included the activation of the PI3K/Akt/mTOR signaling pathway,
upregulation of the protein expression levels of eEF2K and the
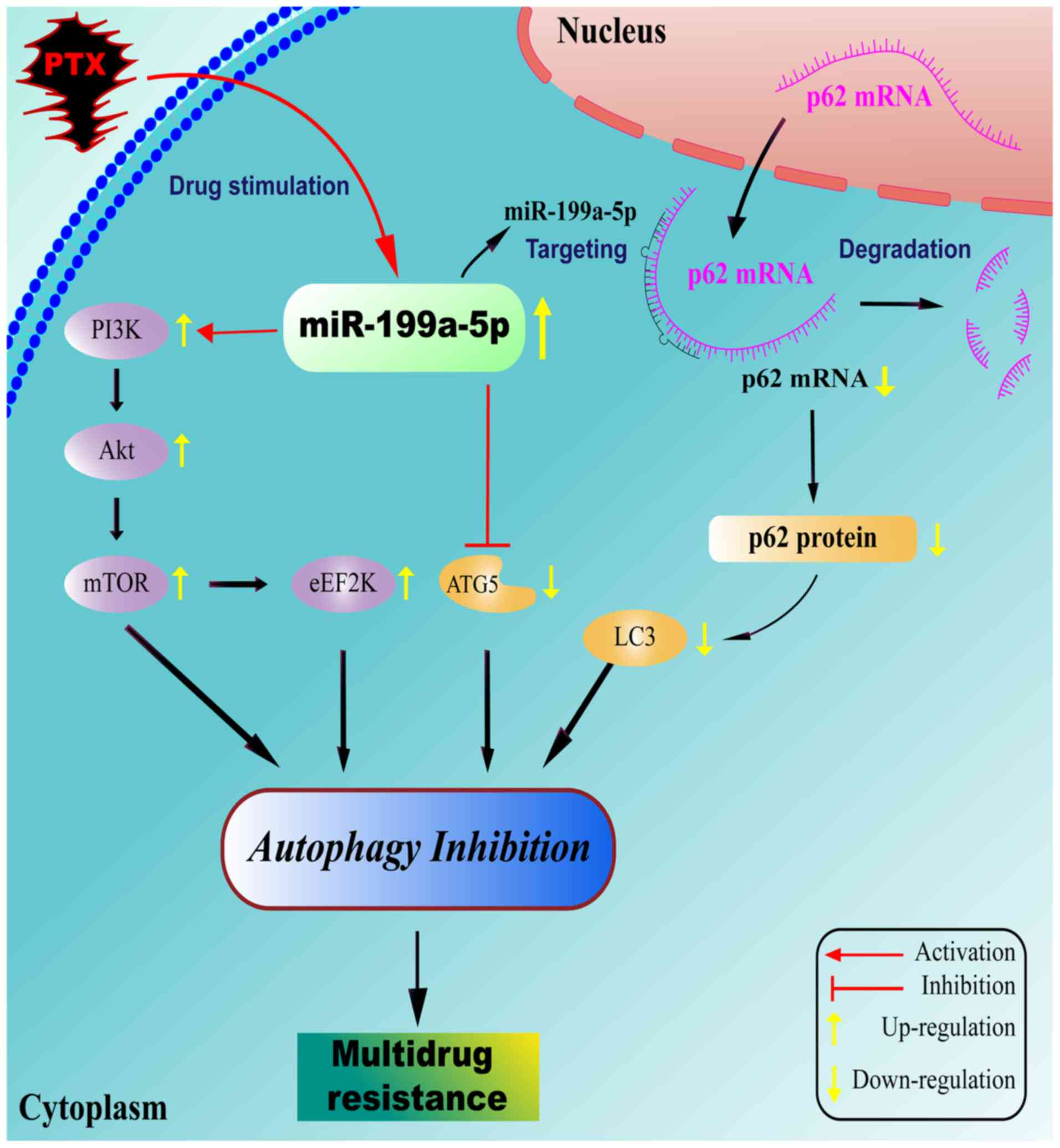
attenuation of ATG5.
The activation of the PI3K/Akt/mTOR signaling
pathway has been reported to induce the phosphorylation of ATG13
and block the initiation of autophagy (35). eEF2K is located downstream of the mTOR
signaling pathway and is activated by phosphorylation of p70S6K to
repress autophagy (35,36). By contrast, the suppression of eEF2K
promotes autophagy (37,38) and enhances the cytotoxicity of
fluoxetine in triple negative breast cancer cells (39). Therefore, high levels of eEF2K may
inhibit autophagy and promote MDR in A549/T cells. In addition,
ATG5 has been reported to serve an important role in the elongation
and expansion of phagophore membrane; low levels of ATG5 lead to
the failure of autophagosome maturation or inhibit the autophagy
process, confirming its involvement in autophagy inhibition
(4).
Previous studies have reported the downregulation of
miR-199a-5p levels in drug-resistant cell lines compared with those
in the corresponding parental cell lines (19–21).
However, the present study was the first to reveal that the levels
of miR-199a-5p were upregulated in MDR lung cancer cells compared
with those in the parental cell lines. In the present study,
RT-qPCR results demonstrated that the levels of miR-199a-5p were
upregulated in PTX-resistant A549/T cells compared with those in
the parental cells, which was different from imatinib-resistant
K562, cisplatin-treated MG63 and Adriamycin-resistant K562 cells in
previous studies (19–21). These results suggested that the
effects of miR-199a-5p on MDR induction were cell line-specific.
The upregulation of miR-199a-5p was negatively correlated with the
sensitivity of lung cancer cells to PTX, indicating that the levels
of miR-199a-5p may be used as a biomarker for the occurrence of MDR
in lung cancer.
The knockdown of miR-199a-5p has been reported to
result in the inhibition of pro-survival pathways, as observed by
the downregulation of p-Akt, p-ERK and β-catenin protein levels in
Huh7.5.1 cells infected with hepatitis C virus (40). Similarly, in the present study,
miR-199a-5p-OE activated the PI3K/Akt/mTOR signaling pathway. In
addition, the overexpression of miR-199a-5p increased the eEF2K
expression levels compared with those in the control cells. Online
miRNA target analysis software is normally used for target
prediction (41,42). In the present study, TargetMiner
database analysis revealed that ATG5 was a direct target of
miR-199a-5p. The results of the validation assay demonstrated that
the expression levels of ATG5 mRNA and protein were downregulated
in A549-miR-199a-5p-OE cells compared with those in the
vector-transfected cells, whereas A549/T-miR-199a-5p-KD cells
exhibited no changes of ATG5 mRNA and protein levels, which
suggested that miR-199a-5p may not be the only regulator of ATG5
(9). The activation of the
PI3K/Akt/mTOR signaling pathway, enhancement of eEF2K and
inhibition of ATG5 expression levels may be considered as the
mechanism of autophagy suppression by miR-199a-5p in MDR A549/T
cells (Fig. 6).
A notable result of the present study was that
miR-199a-5p directly targeted p62, which led to the downregulation
of p62 expression levels in autophagy-suppressed A549/T cells,
which has not been investigated to date. Although miR-199a-5p may
target other genes to induce MDR, p62 appears to be the major novel
target of miR-199a-5p (43). p62 is
considered to be a marker of autophagy; it accumulates in
autophagy-suppressed cells, but does not participate in autophagy
initiation (44). However, previous
studies have suggested that p62 also serves a role in the autophagy
process (43–45). Bjorkoy et al (45) have reported that the depletion of p62
inhibits the recruitment of LC3 to autophagosomes under starvation
conditions, which indicates the accessibility for p62 in autophagy
occurrence. In addition, the deficiency of p62 leads to impaired
formation of LC3II and affect autophagy (46). Gao et al (47) have demonstrated that the 20S
proteasome cleaves LC3 and disrupts the conjugation function of LC3
protein to further inhibit the autophagy process, whereas
proteolysis of LC3 by the 20S proteasome is inhibited by p62. These
findings suggest that p62 serves a positive role in autophagy
activation, whereas the deficiency of p62 negatively affects the
autophagy process (Fig. 6), which was
different from the normal autophagy inhibition process (48,49). To
the best of our knowledge, no previous studies have reported the
function of p62 in MDR development, and further research is
needed.
In conclusion, the present study for the first time
reported autophagy inhibition and miR-199a-5p upregulation in
drug-resistant A549/T and H1299/T cells compared with those in
their parental cells. The upregulation of miR-199a-5p expression
levels was not only stimulated by PTX, but also negatively
associated to the sensitivity of lung cancer cells to PTX.
Knockdown of miR-199a-5p induced autophagy and resensitized cells
to multiple chemotherapeutic drugs, whereas overexpression of
miR-199a-5p suppressed autophagy and desensitized cells to multiple
chemotherapeutic drugs. Activation of the PI3K/Akt/mTOR signaling
pathway, increase in the levels of eEF2K and decrease in the ATG5
expression levels may be the potential mechanism to suppress
autophagy and induce MDR. In addition, miR-199a-5p directly
targeted p62 mRNA and reduced p62 expression levels. Deficiency of
p62 might inhibit autophagy and induce MDR. Taken together, the
results of the present study revealed a novel function of
miR-199a-5p as an autophagy inhibitor and a promoter of MDR in lung
cancer cells. The regulatory effects of miR-199a-5p on autophagy
may provide a novel therapeutic strategy for future
multidrug-resistant lung cancer therapy and drug development.
Supplementary Material
Supporting Data
Acknowledgements
Paclitaxel-selected ATP-binding cassette
B1-overexperssing lung carcinoma cell lines A549/T were generously
provided by Professor Liguang Lou (Shanghai Institute of Materia
Medica, Chinese Academy of Sciences, Shanghai, China).
Funding
This work was funded by The National Nature Science
Foundation of China (grant nos. 81872496 and 81873056) and The
Science and Technology Commission of Shanghai Municipality (grant
nos. 20S11902200 and 16DZ2280100).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XL, XZ and TZ conceived and designed the study. XZ
and FZ analyzed the data. TZ and MX performed the experiments and
wrote the manuscript. WZ, XG and FZ performed certain experiments.
TZ, XL and XZ visualized the data and supervised the study. TZ, MX,
WZ, XG, FZ, XL and XZ confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alfarouk KO, Stock CM, Taylor S, Walsh M,
Muddathir AK, Verduzco D, Bashir AH, Mohammed OY, Elhassan GO,
Harguindey S, et al: Resistance to cancer chemotherapy: Failure in
drug response from ADME to P-gp. Cancer Cell Int. 15:712015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu Q, Yang Z, Nie Y, Shi Y and Fan D:
Multi-drug resistance in cancer chemotherapeutics: Mechanisms and
lab approaches. Cancer Lett. 347:159–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Runwal G, Stamatakou E, Siddiqi FH, Puri
C, Zhu Y and Rubinsztein DC: LC3-positive structures are prominent
in autophagy-deficient cells. Sci Rep. 9:101472019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oh S, Xiaofei E, Ni D, Pirooz SD, Lee JY,
Lee D, Zhao Z, Lee S, Lee H, Ku B, et al: Downregulation of
autophagy by Bcl-2 promotes MCF7 breast cancer cell growth
independent of its inhibition of apoptosis. Cell Death Differ.
18:452–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang J, Pi C and Wang G: Inhibition of
PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy
in hepatocellular carcinoma cells. Biomed Pharmacother.
103:699–707. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tekirdag KA, Korkmaz G, Ozturk DG, Agami R
and Gozuacik D: MIR181A regulates starvation- and rapamycin-induced
autophagy through targeting of ATG5. Autophagy. 9:374–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang LM, Li B, Guan JJ, Xu HD, Shen GH,
Gao QG and Qin ZH: Transcription factor EB is involved in
autophagy-mediated chemoresistance to doxorubicin in human cancer
cells. Acta Pharmacol Sin. 38:1305–1316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mishima Y, Terui Y, Mishima Y, Taniyama A,
Kuniyoshi R, Takizawa T, Kimura S, Ozawa K and Hatake K: Autophagy
and autophagic cell death are next targets for elimination of the
resistance to tyrosine kinase inhibitors. Cancer Sci. 99:2200–2208.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang P, Liu X, Li H, Chen Z, Yao X, Jin J
and Ma X: TRPC5-induced autophagy promotes drug resistance in
breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci Rep.
7:31582017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lv XA, Wang B, Xu XH, Lei P, Bin W, Dong
XX, Zeng CH and Du QW: Curcumin re-sensitizes multidrug resistant
(MDR) breast cancer to cisplatin through inducing autophagy by
decreasing CCAT1 expression. RSC Adv. 7:33572–33579. 2017.
View Article : Google Scholar
|
|
17
|
Wu J, Li W, Ning J, Yu W, Rao T and Cheng
F: Long noncoding RNA UCA1 targets miR-582-5p and contributes to
the progression and drug resistance of bladder cancer cells through
ATG7-mediated autophagy inhibition. Onco Targets Ther. 12:495–508.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen S, Wu J, Jiao K, Wu Q, Ma J, Chen D,
Kang J and Zhao G, Shi Y, Fan D and Zhao G: MicroRNA-495-3p
inhibits multidrug resistance by modulating autophagy through
GRP78/mTOR axis in gastric cancer. Cell Death Dis. 9:10702018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Y, Jiang W, Hu Y, Da Z, Zeng C, Tu M,
Deng Z and Xiao W: MicroRNA-199a-5p inhibits cisplatin-induced drug
resistance via inhibition of autophagy in osteosarcoma cells. Oncol
Lett. 12:4203–4208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Zhang G, Wu B, Yang W and Liu Z:
MiR-199a-5p represses protective autophagy and overcomes
chemoresistance by directly targeting DRAM1 in acute myeloid
leukemia. J Oncol. 2019:56134172019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen PH, Liu AJ, Ho KH, Chiu YT, Anne Lin
ZH, Lee YT, Shih CM and Chen KC: MicroRNA-199a/b-5p enhance
imatinib efficacy via repressing WNT2 signaling-mediated protective
autophagy in imatinib-resistant chronic myeloid leukemia cells.
Chem Biol Interact. 291:144–151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Li L, Guan Y, Liu X, Meng Q and Guo
Q: MiR-92b regulates the cell growth, cisplatin chemosensitivity of
A549 non small cell lung cancer cell line and target PTEN. Biochem
Biophys Res Commun. 440:604–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Chen L, Feng L, Zhu M, Shen Q, Fang
Y, Liu X and Zhang X: NEK2 promotes proliferation, migration and
tumor growth of gastric cancer cells via regulating KDM5B/H3K4me3.
Am J Cancer Res. 9:2364–2378. 2019.PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moore CE, Wang X, Xie J, Pickford J,
Barron J, Regufe da Mota S, Versele M and Proud CG: Elongation
factor 2 kinase promotes cell survival by inhibiting protein
synthesis without inducing autophagy. Cell Signal. 28:284–293.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen XL, Liu P, Zhu WL and Lou LG:
DCZ5248, a novel dual inhibitor of Hsp90 and autophagy, exerts
antitumor activity against colon cancer. Acta Pharmacol Sin.
42:132–141. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duprez L, Wirawan E, Vanden Berghe T and
Vandenabeele P: Major cell death pathways at a glance. Microbes
Infect. 11:1050–1062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang H and Zou Z: Targeting autophagy to
overcome drug resistance: Further developments. J Hematol Oncol.
13:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoon JH, Ahn SG, Lee BH, Jung SH and Oh
SH: Role of autophagy in chemoresistance: Regulation of the
ATM-mediated DNA-damage signaling pathway through activation of
DNA-PKcs and PARP-1. Biochem Pharmacol. 83:747–757. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S and Wei Y: Association of HMGB1,
BRCA1 and P62 expression in ovarian cancer and chemotherapy
sensitivity. Oncol Lett. 15:9572–9576. 2018.PubMed/NCBI
|
|
32
|
Huang CY, Chiang SF, Chen WT, Ke TW, Chen
TW, You YS, Lin CY, Chao KSC and Huang CY: HMGB1 promotes
ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance
through RAGE in colorectal cancer. Cell Death Dis. 9:10042018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng H, Chen JN, Yu X, Jiang P, Yuan L,
Shen HS, Zhao LH, Chen PF and Yang M: HMGB1 enhances drug
resistance and promotes in vivo tumor growth of lung cancer cells.
DNA Cell Biol. 35:622–627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: For better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie T, Li SJ, Guo MR, Wu Y, Wang HY, Zhang
K, Zhang X, Ouyang L and Liu J: Untangling knots between autophagic
targets and candidate drugs, in cancer therapy. Cell Prolif.
48:119–139. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu H, Yang JM, Jin S, Zhang H and Hait WN:
Elongation factor-2 kinase regulates autophagy in human
glioblastoma cells. Cancer Res. 66:3015–3023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pan Z, Chen Y, Liu J, Jiang Q, Yang S, Guo
L and He G: Design, synthesis, and biological evaluation of
polo-like kinase 1/eukaryotic elongation factor 2 kinase
(PLK1/EEF2K) dual inhibitors for regulating breast cancer cells
apoptosis and autophagy. Eur J Med Chem. 144:517–528. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xie CM, Liu XY, Sham KW, Lai JM and Cheng
CH: Silencing of EEF2K (eukaryotic elongation factor-2 kinase)
reveals AMPK-ULK1-dependent autophagy in colon cancer cells.
Autophagy. 10:1495–1508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun D, Zhu L, Zhao Y, Jiang Y, Chen L, Yu
Y and Ouyang L: Fluoxetine induces autophagic cell death via
eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer.
Cell Prolif. 51:e124022018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Gao H, Duan S and Song X:
Inhibition of microRNA-199a-5p reduces the replication of HCV via
regulating the pro-survival pathway. Virus Res. 208:7–12. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang G, Sun D, Li W and Xin Y: AKNA is a
potential prognostic biomarker in gastric cancer and function as a
tumor suppressor by modulating EMT-related pathways. Biomed Res
Int. 2020:67267592020.PubMed/NCBI
|
|
42
|
Murugesan M and Premkumar K: Integrative
miRNA-mRNA functional analysis identifies miR-182 as a potential
prognostic biomarker in breast cancer. Mol Omics. Apr 22–2021.(Epub
ahead of print). doi: 10.1039/d0mo00160k. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang K, Zhang X, Cai Z, Zhou J, Cao R,
Zhao Y, Chen Z, Wang D, Ruan W, Zhao Q, et al: A novel class of
microRNA-recognition elements that function only within open
reading frames. Nat Struct Mol Biol. 25:1019–1027. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moscat J, Karin M and Diaz-Meco MT: p62 in
cancer: Signaling adaptor beyond autophagy. Cell. 167:606–609.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bjorkoy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Peng H, Yang J, Li G, You Q, Han W, Li T,
Gao D, Xie X, Lee BH, Du J, et al: Ubiquitylation of
p62/sequestosome1 activates its autophagy receptor function and
controls selective autophagy upon ubiquitin stress. Cell Res.
27:657–674. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao Z, Gammoh N, Wong PM,
Erdjument-Bromage H, Tempst P and Jiang X: Processing of autophagic
protein LC3 by the 20S proteasome. Autophagy. 6:126–137. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bjørkøy G, Lamark T, Pankiv S, Øvervatn A,
Brech A and Johansen T: Monitoring autophagic degradation of
p62/SQSTM1. Methods Enzymol. 452:181–197. 2009. View Article : Google Scholar
|
|
49
|
Zhang CF, Gruber F, Ni C, Mildner M,
Koenig U, Karner S, Barresi C, Rossiter H, Narzt MS, Nagelreiter
IM, et al: Suppression of autophagy dysregulates the antioxidant
response and causes premature senescence of melanocytes. J Invest
Dermatol. 135:1348–1357. 2015. View Article : Google Scholar : PubMed/NCBI
|