Introduction
Despite recent improvements in long-term survival
rates for acute lymphoblastic leukaemia (ALL) (1) and acute myeloid leukaemia (AML)
(2), subgroups of patients
continue to experience poor long-term outcomes. TP53
alterations are a high-risk genomic feature present in 16% of newly
diagnosed ALL (3,4) and 13% of newly diagnosed AML cases
(4). The presence of a TP53
alteration is associated with poor long-term overall survival
compared with TP53 wild-type (TP53WT)
cases in both ALL (24 months vs. not reached, P=0.001) and AML (6
vs. 26 months, P<0.001) (3). To
date, no targeted therapies are currently available to treat
TP53-mutated (TP53MUT) acute leukaemia
outside of clinical trials, and more efficacious therapeutic
options are required to improve long-term outcomes.
CBL0137 is a small molecule curaxin that
epigenetically modulates multiple cancer-related signalling
pathways (5,6), including casein kinase 2
(CK2)-mediated activation of the TP53-encoded p53 protein
through the Facilitates Chromatin Transcription (FACT) complex
(7). CBL0137 is effective in
pre-clinical ALL patient-derived xenograft (PDX) models, most
notably in infant KMT2A-rearranged (KMT2Ar) ALL
(8,9). KMT2Ar is present in 5–10% of
newly-diagnosed acute leukaemia cases overall (10), including 70–80% of infant ALL and
35–50% of infant AML diagnoses (11), and TP53 mutations occur in
~16% of KMT2Ar ALL cases (4). Outcomes for KMT2Ar leukaemia
are exceptionally poor across all age groups, with five-year
event-free survival (EFS) rates of 30–50% (12). Evidently, advances in treatment
options are required to improve patient outcomes.
In a study by Lock et al (8), CBL0137 induced complete remission in
a KMT2Ar infant ALL PDX model, and partial responses were
observed in several B-cell ALL (B-ALL) and T-cell ALL (T-ALL)
models of unknown genomic subtype. Somers et al (9) similarly reported CBL0137 efficacy in
infant KMT2Ar B-ALL PDX models, with responses ranging from
delayed cancer progression to maintained complete remission. These
early data suggested that the role of CBL0137 deserves further
exploration. For instance, the presence of loss-of-function
TP53 alterations, a reasonably common event in KMT2Ar
B-ALL, may reasonably be expected to confer resistance.
Furthermore, given its in vitro efficacy against
KMT2Ar B-ALL, CBL0137 is also likely to be active against
other subtypes of B-ALL. In the present study, it was confirmed
that CBL0137 induces leukaemic cell apoptosis in a number of cell
lines with various driver alterations, including those with
KMT2A rearrangements, with LC50 concentrations in
the range of 166 to 676 nM. Notably, it was also demonstrated that
the potency of CBL0137 is attenuated in the presence of loss of
function TP53 alterations.
Materials and methods
Cell line maintenance
U-937 (CRL-1593.2), MV4;11 (CRL-9591), Jurkat
(TIB-152), THP-1 (TIB-202) and RS4;11 (CRL-1873) cell lines were
purchased from the American Type Culture Collection (ATCC). RCH-ACV
(ACC 548), REH (ACC 22) and NALM-19 (ACC 522) cell lines were
purchased from DSMZ. All cell lines were maintained in culture at
37°C in RPMI-1640 media (cat. no. R0883; Sigma-Aldrich; Merck)
containing foetal calf serum (FCS) (AU-FBS/SF; CellSera), 5 mM
L-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin. THP-1
cells were cultured in 20% FCS, and all other cell lines were
cultured in 10% FCS. Cells up to 20 passages from the original
stock were used for experiments.
Inhibitor storage
CBL0137 (cat. no. S0507; Selleck Chemicals) was
stored long-term at 10 mM in DMSO at −80°C and diluted in DMSO
immediately prior to use.
Generation of CRISPR/Cas9 TP53
knock-out RS4;11 cell lines
Vectors FUCas9mCherry and FgH1tUTG were a gift from
Professor Marco Herald (Walter and Eliza Hall Institute of Medical
Research, Melbourne, Australia) (13). Two independent single guide RNA
(sgRNA) targets were designed, to account for off-target effects.
sgRNA target sequences were designed to generate random indel and
frameshift mutations in exon 4 of TP53, to disrupt the p53
protein prior to the critical DNA-binding domain, where deleterious
TP53 mutations identified in human leukaemia cell lines are
located (Fig. 1A). sgRNA
oligonucleotides were designed with online Benchling®
Software (Benchling) and purchased from Sigma-Aldrich; Merck KGaA:
TP53 Oligo 1 forward, 5′-TCCCACCAGCAGCTCCTACACCGG-3′ and reverse,
5′-AAACCCGGTGTAGGAGCTGCTGGT-3′; and TP53 Oligo 2 forward,
5′-TCCCCCATTGTTCAATATCGTCCG-3′ and reverse,
5′AAACCGGACGATATTGAACAATGG-3′.
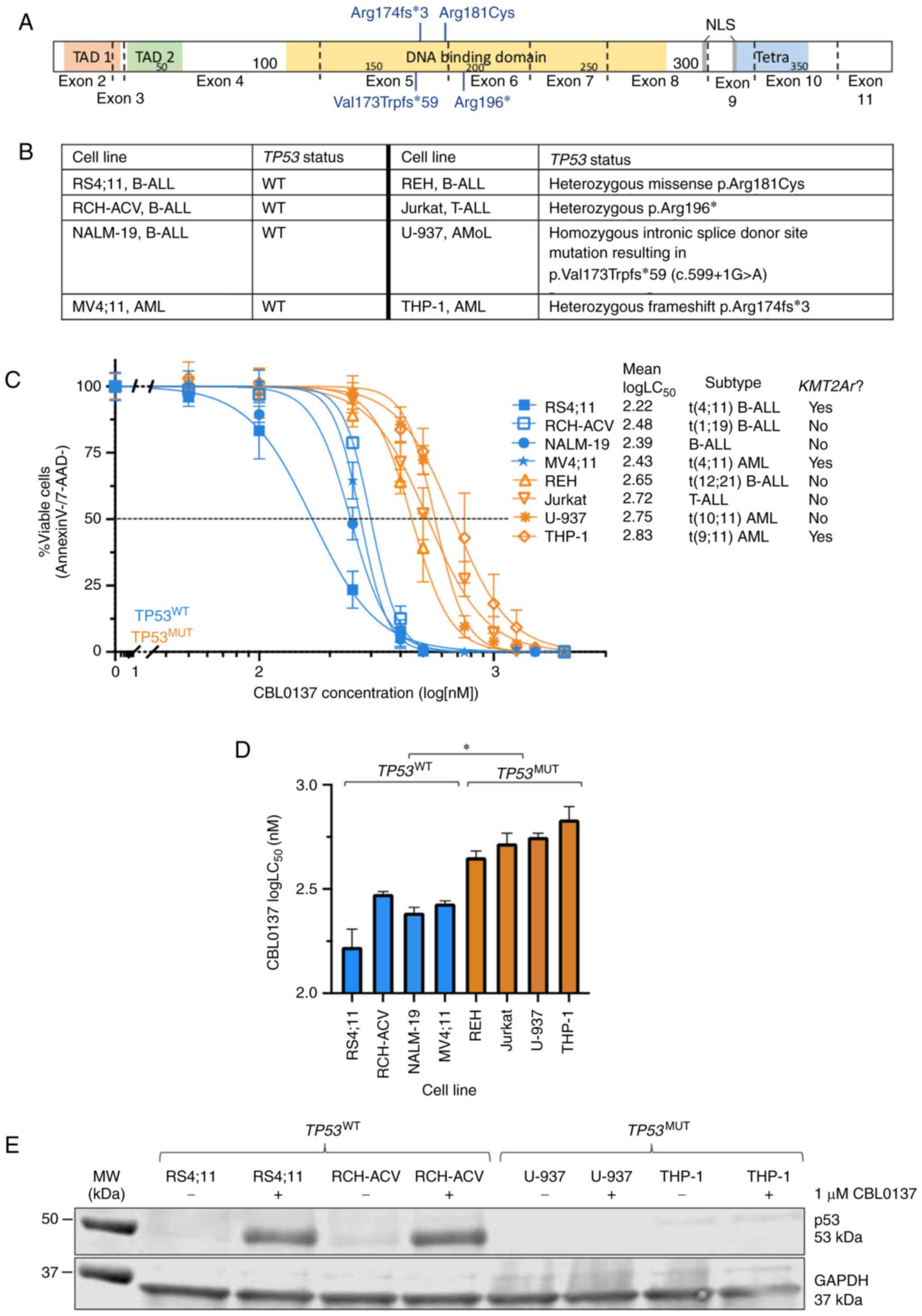 | Figure 1.CBL0137 induces apoptosis in acute
leukaemia cell lines, and LC50 are higher in
TP53-mutated cell lines. (A) TP53 mutations identified in
leukaemia cell lines. Data was obtained from the Broad Institute
Cancer Cell Line Encyclopedia portal. (B) Protein domain schematic
of p53, with mutations identified in cell lines annotated. Exons
are denoted by dotted lines. (C) Human acute leukaemia cell lines
were exposed to increasing doses of CBL0137 in duplicate and
apoptosis was measured by Annexin-V/7-AAD staining after 72 h.
Concentration-response curves and logLC50 values were
extrapolated using non-linear regression analysis in GraphPad
Prism. Data are mean + SD for 3 biological replicates. (D) Mean
CBL0137 logLC50 values for each cell line are shown.
LC50 values are provided in Table SII. For statistical analysis,
*P<0.05 for comparison of TP53 wild-type (TP53WT) and mutated
(TP53MUT) mean logLC50, Kruskal-Wallis test with
multiple comparisons. (E) Representative immunoblot of total p53
protein in DMSO vehicle-treated (−) and 1 µM CBL0137-treated (+)
cell lines. AML, acute myeloid leukaemia; AMoL, acute monocytic
leukaemia; B-ALL, B-lineage acute lymphoblastic leukaemia; MUT,
mutated; WT, wild-type; T-ALL, T-lineage acute lymphoblastic
leukaemia; TAD, transactivation domain; NLS, nuclear localisation
signal; Tetra, Tetramerisation motif. |
TP53 FgH1tUTG sgRNA vectors were generated by
BsmBI digestion and T4 DNA ligation, incubated overnight at
4°C and transformed by 42°C heat shock for 2 min in
DH5a™-T1R chemically competent E. coli
(Thermo Fisher Scientific, Inc.), and selected based on ampicillin
resistance (50 µg/ml) conferred by transformation of the FgH1tUTG
plasmid. Successful ligation of inserts was confirmed by Sanger
sequencing with the BigDye™ Terminator v3.1 Cycle
Sequencing Kit (used according to manufacturer's protocol) and
SeqStudio™ Genetic Analyser System (both from Thermo
Fisher Scientific, Inc.). DNA chromatogram results were analysed
using online Benchling® Software.
293T cells (CRL-3216; purchased from ATCC) were
co-transfected with FUCas9mCherry (4.2 µg) or FgH1tUTG sgRNA (4.2
µg) vectors, and 2nd generation lentiviral packaging vectors pMD2.G
(1.6 µg), pMDLg/pRRE (2.4 µg) and pRSV-Rev (1.1 µg) (Addgene,
Inc.). Each transfection was prepared in 450 µl
Opti-MEM™ Reduced Serum Media and 30 µl
Lipofectamine™ 2000 transfection reagent (both from
Thermo Fisher Scientific, Inc.), and incubated for 1 h at room
temperature prior to application to 293T cells. Viral supernatant
was harvested from 293T cultures 48 h post-transfection (MOI not
quantified), and RS4;11 cells were transduced by spinfection in the
presence of 4 mg/ml Polybrene® (Santa Cruz
Biotechnology, Inc.) at 220 × g for 1 h at room temperature in
six-well tissue culture plates. One week post-transduction, GFP and
mCherry double-positive populations were sorted with a BD
FACSFusion flow cytometer (BD Biosciences). Sorted populations were
activated with doxycycline (1 µg/ml) for 3 days, and genomic DNA
was extracted by phenol-chloroform to confirm induction of
frameshift mutations by PCR amplification of TP53 exon 4 using
Q5® High-Fidelity DNA Polymerase (cat. no. M0491; New
England Biolabs, Inc.) and the following primers: TP53 intron 4
forward, 5′-TCCTCTGACTGCTCTTTTCACCCAT-3′ and reverse,
5′-AATATTCAACTTTGGGACAGGAGTCAGAGA-3′. Thermocycling conditions were
as follows: 98°C for 1 min, followed by 33 cycles at 98°C for 10
sec, 64°C for 15 sec and 72°C for 2 min, followed by 1 cycle at
72°C for 10 min. Samples were then stored at 4°C until they were
visualised in 2% agarose gel containing 1:10,000 GelRed (Biotium,
Inc.).
Sanger sequencing was utilised to identify the
profile of mutations present (Fig.
S1). Sanger sequencing was performed using the
BigDye™ Terminator v3.1 Cycle Sequencing Kit according
to manufacturer's instructions (Thermo Fisher Scientific, Inc.) and
SeqStudio™ Genetic Analyser System (Thermo Fisher
Scientific, Inc.). DNA chromatogram results were analysed using
online Benchling® Software. RNA was harvested after 7
and 14 days in culture, to quantify TP53 expression by
quantitative reverse transcription-quantitative PCR (RT-qPCR). As a
comparator, RS4;11 cells expressing Cas9 vector only were used as a
TP53WT control in all relevant experiments.
RT-qPCR thermocycling conditions were as follows: 10 min at 95°C
for one cycle, followed by 40 cycles at 95°C for 15 sec and 60°C
for 60 sec. RT-qPCR cycling reactions were performed on a
QuantStudio 7 Real-Time PCR system (Thermo Fisher Scientific,
Inc.).
Apoptosis detection via Annexin
V/7-Aminoactinomycin D staining
Cells were seeded at 2×105 cells/ml and
treated for 72 h with a range of CBL0137 doses in 0.3% DMSO, and
incubated in 96-well tissue culture plates at 37°C/5%
CO2 for 72 h. Treated cells were harvested at room
temperature by centrifugation at 220 × g for 5 min, washed twice in
flow cytometry binding buffer (Hank's Balanced Salt Solution (cat.
no. H9394; Sigma-Aldrich; Merck KGaA), 10 mM HEPES, 5 mM
CaCl2), and cells in each well stained with Annexin V
(cat. no. 556421; BD Biosciences) and 7-Aminoactinomycin (7-AAD)
(cat. no. A1310; Thermo Fisher Scientific, Inc.) according to the
supplier's protocol (BD Biosciences), and incubated on ice for 30
min. Assays were analysed on a BD FACSCanto flow cytometer (BD
Biosciences) and data were analysed using FlowJo version 10
software (FlowJo LLC) to determine apoptotic (Annexin V and/or
7-AAD positive) and non-apoptotic (Annexin V and 7-AAD negative)
populations.
The in vitro efficacy of CBL0137 to induce
apoptosis was assessed on four TP53WT (RS4;11,
RCH-ACV, NALM-19, MV4;11) and four TP53MUT (REH,
Jurkat, U-937 and THP-1) human acute leukaemia cell lines using
Annexin-V/7-AAD. The U-937 cell line was initially classified as
histiocytic lymphoma (14) but is
now classified as acute monocytic leukaemia (AMoL) and is therefore
referred to as such throughout the manuscript (15). TP53 mutations present in
each cell line investigated are provided in Fig. 1A, obtained from the Broad Institute
Cancer Cell Line Encyclopedia portal (16) (https://sites.broadinstitute.org/ccle/). All
identified TP53 mutations within the cell lines utilised are
predicted to be deleterious according to COSMIC (17) (https://cancer.sanger.ac.uk) or ClinVar (18) (https://www.ncbi.nlm.nih.gov/clinvar/) databases.
Later, the in vitro efficacy of CBL0137 to induce apoptosis
was assessed on CRISPR/Cas9 TP53 knock-out RS4;11 cell
lines.
RNA analysis
RNA was extracted from
5×106−1×107 cells using TRIzol®
reagent (Thermo Fisher Scientific, Inc.). RNA concentration and
purity were quantified with a NanoDrop 2000 Spectrophotometer
(Thermo Fisher Scientific, Inc.). The QuantiTect reverse
transcription kit (Qiagen GmbH) was used to synthesise cDNA from 1
µg of RNA as per manufacturer's instructions. RT-qPCR reactions
were prepared in duplicate as follows: 12.5 µl RT2
SYBR® Green ROX™ qPCR Mastermix (Qiagen
GmbH), 400 nM each of forward and reverse primers, 1 µl cDNA,
nuclease-free H2O to 25 µl final volume. Cycling
reactions were performed on a QuantStudio 7 Real-Time PCR system
(Thermo Fisher Scientific, Inc.) with the following primers: TP53
qPCR forward, 5′-GAAGGAAATTTGCGTGTGG-3′ and reverse,
5′-TGTTACACATGTAGTTGTAGTGG-3′. Values were normalised against
housekeeping gene ACTB (forward, 5′-GATCATTGCTCCTCCTGAGC-3′ and
reverse, 5′-TCTGCGCAAGTTAGGTTTTGTC-3′). The thermocycling
conditions were as aforementioned. Relative gene expression was
calculated by the ∆∆Cq method and fold-change expression
(2−∆∆Cq) was calculated relative to Cas9 (TP53-WT)
control (19).
Protein analysis
Cells were treated for 6 h in either 0.3% DMSO or 1
µM CBL0137 (0.3% DMSO) final concentration in RPMI-1640 + 2% FCS +
50 U/ml penicillin + 50 µg/ml streptomycin. Cells were pelleted by
centrifugation at 10,000 × g for 10 min at 4°C and lysed in 60 µl
1% NP-40 (IGEPAL®) buffer (Sigma-Aldrich; Merck KGaA).
Protein concentration was determined by generating a standard curve
using a bicinchoninic acid (BCA) assay. Lysates were denatured and
60 µg protein was electrophoresed on 4–15% pre-cast gels (Bio-Rad
Laboratories, Inc.). Proteins were transferred to polyvinylidene
difluoride membranes (Bio-Rad Laboratories, Inc.) and blocked with
1X Intercept blocking buffer (LI-COR Biosciences) for 1 h at room
temperature before incubation with primary antibodies (mouse
anti-p53, product no. 2524; rabbit anti-p21, product no. 2947; and
rabbit anti-GAPDH, product no. 2118) at 4°C for 17–48 h. All
primary antibodies were purchased from Cell Signaling Technology,
Inc. and diluted at 1:1,000 in 1X Intercept blocking buffer. After
washing with TBS-T (containing 0.1% Tween-20) and TBS, membranes
were incubated with secondary antibodies [IRDye® 800CW
anti-mouse (1:10,000) or IRDye® 800CW anti-rabbit
(1:10,000)] (cat nos. 926-32212 and 925-32213, respectively; LI-COR
Biosciences) for 2 h in the dark at room temperature and visualised
on the LI-COR Odyssey® fluorescent scanner. For
calculation of p21 expression fold change, Empiria
Studio® Software version 2.0 (https://www.licor.com/bio/empiria-studio/) was used to
normalise protein expression to housekeeping GAPDH protein. Fold
expression change was then calculated for CBL0137-treated samples,
relative to untreated samples for each respective cell line.
Statistical analysis
All calculations and statistical analysis of data
were performed using GraphPad Prism Version 9.2.0 for Mac OS
(GraphPad Software, Inc.). Kruskal-Wallis test with Dunn's multiple
comparison post hoc test or Mann-Whitney U tests were performed to
compare mean logLC50 values between each cell line
tested. LogLC50 and LC50 values were
extrapolated using non-linear regression analysis. All data was
generated from 3 independent biological replicates. P<0.05 was
considered to indicate a statistically significant difference.
Results
All identified TP53 mutations within the cell
lines REH, Jurkat, U-937 and THP-1 occur within a critical region
of the p53 DNA binding domain (Fig. 1A
and B). The presence of TP53 loss-of-function genomic
alterations significantly reduced the sensitivity of cells to
CBL0137, independent of other genomic lesions present in each cell
line (Fig. 1C and D). The
aggregate mean logLC50 was greater for
TP53MUT cell lines compared with
TP53WT cell lines (mean logLC50=2.38
nM vs. 2.75 nM, P=0.046; Fig. 1D),
with a more than two-fold increase in LC50 (Table SI). Notably, the KMT2A WT
B-ALL cell lines NALM-19 and RCH-ACV exhibited low
logLC50 values (logLC50=2.39 and 2.48 nM
respectively; Fig. 1C). The
KMT2Ar cell line THP-1 was the most resistant cell line
investigated (logLC50=2.83 nM; Fig. 1C and D). A total of four cell
lines, including two TP53WT and two
TP53MUT were probed for the presence of total p53
protein, demonstrating increased levels of p53 following CBL0137
treatment in TP53WT cell lines only (Fig. 1E).
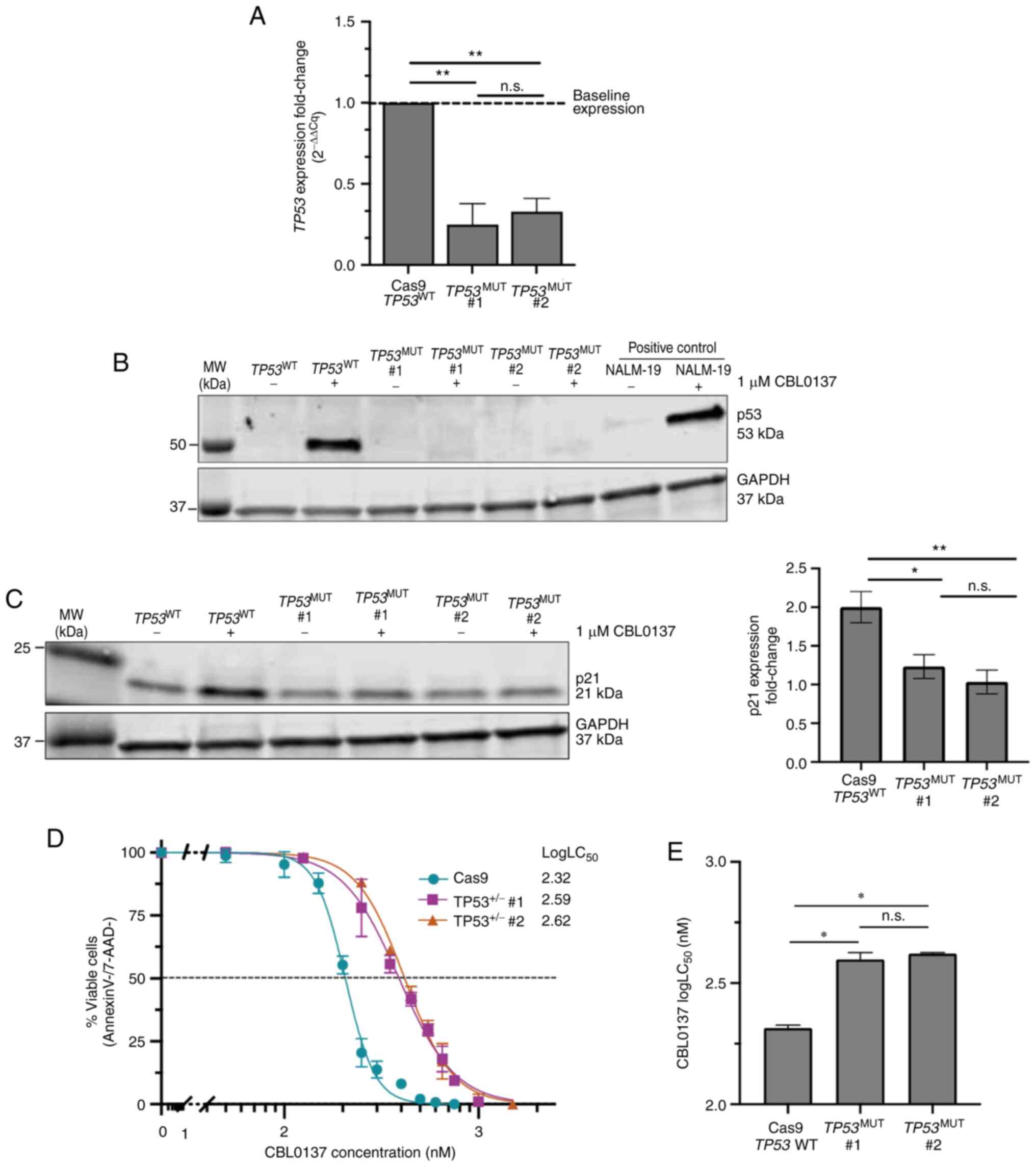
To further investigate the role of p53 in CBL0137
efficacy, heterozygous TP53 loss-of-function cell lines
(TP53+/−) were generated using CRISPR/Cas9 in the
human KMT2A-AFF1 ALL cell line RS4;11. The RS4;11 cell line
was selected as a representative of KMT2Ar cell lines as it
is highly sensitive to CBL0137 and expresses KMT2A-AFF1, the
most common KMT2Ar identified in B-ALL (10). Heterozygous TP53
loss-of-function was confirmed in cell lines by RT-qPCR and
immunoblot analysis (Fig. 2A and
B). Both TP53+/− cell lines exhibited a
significant reduction in TP53 expression by RT-qPCR,
compared with control TP53WT RS4;11 cells
(Fig. 2A). Immunoblot analysis
demonstrated that treatment with 1 µM CBL0137 stimulated p53
expression in WT RS4;11 and positive control NALM-19 cells, but
this effect was abrogated in both TP53+/− RS4;11
cell lines (Fig. 2B). The p53
effector protein p21 was upregulated ~two-fold in CBL0137-treated
TP53WT RS4;11 cells but not
TP53MUT cells, demonstrating that heterozygous
TP53 loss-of-function is sufficient to abrogate activation
of downstream p53 pathways (Fig.
2C). The cell senescence effector protein p21 is a
well-characterised p53 effector protein, where p21 is rapidly
activated following p53 activation, to induce cellular senescence
and apoptosis (20,21). Annexin-V/7-AAD staining of
TP53+/− cell lines revealed a ~two-fold increase
in CBL0137 logLC50 (mean logLC50 WT=2.32 nM
vs. TP53+/− #1=2.59 nM, P=0.043; WT vs.
TP53+/− #2=2.62 nM, P=0.021; Fig. 2D and E), indicating that
heterozygous TP53 loss-of-function is sufficient to cause a
significant reduction in the sensitivity of cell lines to
CBL0137.
Discussion
In the present study, it was demonstrated that
CBL0137 has similar potency in all tested TP53WT
acute leukaemia cell lines, regardless of the presence of any
additional genomic lesions, including KMT2Ar. However, the
presence of a TP53 loss-of-function mutation significantly
increased CBL0137 LC50, with a ~2-fold increase (range
1.7-4.0-fold) compared with TP53WT cell lines,
regardless of genotype. It was also demonstrated, for the first
time to the best of our knowledge, that heterozygous TP53
loss-of-function is alone sufficient to cause a significant
increase in the LC50 of CBL0137 in the KMT2Ar
B-ALL cell line RS4;11.
These results suggested that CBL0137 is indeed a
promising therapy which may be broadly applicable in acute
leukaemia. These data also indicated that cytotoxicity of CBL0137
is not specific to KMT2Ar, but rather the target depends on
the presence or absence of functional TP53. Reduced
sensitivity is expected in the context of TP53 mutated
malignancies, and further testing is warranted to understand the
clinical significance of TP53 mutation status in CBL0137
efficacy. It is important to note that thus far, all ALL xenograft
models tested by the Pediatric Preclinical Testing Program were
TP53WT (8). An
additional 6/31 paediatric solid tumour models exhibited tumour
growth delay, and 5/6 of these were TP53WT
(8). This should also be
investigated through further pre-clinical testing, and anticipated
in its clinical development program. For instance, two recently
completed Phase I studies of CBL0137 reported acceptable
tolerability and some clinical activity against cancers of the
liver, prostate, uterus, breast and ovary (22,23).
The TP53 mutational status would optimise patient selection
and target those most likely to benefit from this drug for further
clinical testing. It is possible that mutations in p53 downstream
mediators may also influence CBL0137 potency. However, alterations
within p53 downstream mediators such as CDKN1A (p21) and
BCL2 (BCL-2) are rarely reported in acute leukaemia. It is
also possible that alterations within CK2 or the FACT complex may
influence CBL0137 potency, as CBL0137-mediated activation of p53
occurs via these pathways (7), but
data on the occurrence of mutations in these pathways in acute
leukaemia is currently lacking (24).
The present findings highlighted the importance of
p53 activation in CBL0137 efficacy in acute leukaemia, indicating
that TP53 mutation status is an important factor in the
clinical application of CBL0137. There is an ongoing need to
identify therapeutic strategies for patients with high-risk acute
leukaemia, such as those with TP53 mutations, to improve
outcomes for these patients. The data of the present study
indicated that CBL0137 is a promising anticancer therapy that
depends on WT p53 activity, which thus far has not been
therapeutically targetable. The clinical feasibility of CBL0137
across both TP53MUT and TP53WT
malignancies will become evident as further clinical trials are
performed. In the present study, it was clearly demonstrated that
the potency profile of CBL0137 is tightly linked to TP53
mutation status, and it was revealed for the first time that
heterozygous TP53 loss-of-function alone significantly
affects response to CBL0137 in vitro. These results support
the need for accurate determination of TP53 mutation status
for patients enrolling in future CBL0137 clinical trials.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Dr Randall Grose
(SAHMRI, Adelaide, Australia) for his technical assistance with
flow cytometric experiments.
Funding
The present study was supported in part by the National Health
and Medical Research Council (NHMRC), the Bristol-Meyers Squibb
company, the Tour de Cure Australia, the Leukaemia Foundation
Australia and the University of Adelaide.
Availability of data and materials
The datasets used and/or analysed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MOF conceptualised the presented idea. MOF performed
experiments and constructed the manuscript in consultation with
BJM, ECP, DTY, LNE and DLW. MOF and BJM confirm the authenticity of
all the raw data. All authors provided critical feedback and helped
shape the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was performed under the
International Bioethics Committee (IBC) certification number
BC02/2018, and all work contained within the present study was
approved by the Royal Adelaide Hospital HREC committee (approval
no. HREC/15/RAH/54).
Patient consent for publication
Not applicable.
Competing interests
DLW receives research support from Bristol-Meyers
Squibb, and Honoraria from Bristol-Meyers Squibb and Amgen. DTY
receives research support from Bristol-Meyers Squibb and Novartis,
and Honoraria from Bristol-Meyers Squibb, Novartis, Pfizer and
Amgen. None of these agencies have had a role in the preparation of
this manuscript. All other authors declare that they have no
competing interests.
Glossary
Abbreviations
Abbreviations:
|
ALL
|
acute lymphoblastic leukaemia
|
|
AML
|
acute myeloid leukaemia
|
|
KMT2Ar
|
KMT2A-rearranged
|
|
EFS
|
event-free survival
|
|
PDX
|
patient-derived xenograft
|
|
B-ALL
|
B-cell ALL
|
|
T-ALL
|
T-cell ALL
|
|
TP53WT
|
TP53-wild type
|
|
TP53MUT
|
TP53-mutated
|
References
|
1
|
Inaba H and Mullighan CG: Pediatric acute
lymphoblastic leukemia. Haematologica. 105:2524–2539. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shallis RM, Wang R, Davidoff A, Ma X and
Zeidan AM: Epidemiology of acute myeloid leukemia: Recent progress
and enduring challenges. Blood Rev. 36:70–87. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stengel A, Kern W, Haferlach T,
Meggendorfer M, Fasan A and Haferlach C: The impact of TP53
mutations and TP53 deletions on survival varies between AML, ALL,
MDS and CLL: An analysis of 3307 cases. Leukemia. 31:705–711. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stengel A, Schnittger S, Weissmann S,
Kuznia S, Kern W, Kohlmann A, Haferlach T and Haferlach C: TP53
mutations occur in 15.7% of ALL and are associated with
MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood.
124:251–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kantidze OL, Luzhin AV, Nizovtseva EV,
Safina A, Valieva ME, Golov AK, Velichko AK, Lyubitelev AV,
Feofanov AV, Gurova KV, et al: The anti-cancer drugs curaxins
target spatial genome organization. Nat Commun. 10:14412019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dallavalle S, Mattio LM, Artali R, Musso
L, Aviñó A, Fàbrega C, Eritja R, Gargallo R and Mazzini S:
Exploring the interaction of curaxin CBL0137 with G-quadruplex DNA
oligomers. Int J Mol Sci. 22:64762021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gasparian AV, Burkhart CA, Purmal AA,
Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA,
Pal S, et al: Curaxins: Anticancer compounds that simultaneously
suppress NF-κB and activate p53 by targeting FACT. Sci Transl Med.
3:95ra742011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lock R, Carol H, Maris JM, Kolb EA,
Gorlick R, Reynolds CP, Kang MH, Keir ST, Wu J, Purmal A, et al:
Initial testing (stage 1) of the curaxin CBL0137 by the pediatric
preclinical testing program. Pediatr Blood Cancer. 64:e262632017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Somers K, Kosciolek A, Bongers A,
El-Ayoubi A, Karsa M, Mayoh C, Wadham C, Middlemiss S, Neznanov N,
Kees UR, et al: Potent antileukemic activity of curaxin CBL0137
against MLL-rearranged leukemia. Int J Cancer. 146:1902–1916. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forgione MO, McClure BJ, Eadie LN, Yeung
DT and White DL: KMT2A rearranged acute lymphoblastic leukaemia:
Unravelling the genomic complexity and heterogeneity of this
high-risk disease. Cancer Lett. 469:410–418. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muntean AG and Hess JL: The pathogenesis
of mixed-lineage leukemia. Annu Rev Pathol. 7:283–301. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marks DI, Moorman AV, Chilton L, Paietta
E, Enshaie A, DeWald G, Harrison CJ, Fielding AK, Foroni L,
Goldstone AH, et al: The clinical characteristics, therapy and
outcome of 85 adults with acute lymphoblastic leukemia and
t(4;11)(q21;q23)/MLL-AFF1 prospectively treated in the
UKALLXII/ECOG2993 trial. Haematologica. 98:945–952. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS,
O'Connor L, Milla L, Wilcox S, Tai L, Strasser A and Herold MJ: An
inducible lentiviral guide RNA platform enables the identification
of tumor-essential genes and tumor-promoting mutations in vivo.
Cell Rep. 10:1422–1432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ralph P, Moore M and Nilsson K: Lysozyme
synthesis by established human and murine histiocytic lymphoma cell
lines. J Exp Med. 143:1528–1533. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chanput W, Peters V and Wichers H: THP-1
and U937 cells. The Impact of Food Bioactives on Health. Verhoeckx
K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A,
Requena T, Swiatecka D and Wichers H: Springer; Cham: pp. 147–159.
2015
|
|
16
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tate JG, Bamford S, Jubb HC, Sondka Z,
Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E,
et al: COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 47(D1): D941–D947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46(D1): D1062–D1067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang JD, Ruschhaupt M and Biczok R: ddCt
method for qRT-PCR data analysis. Bioconductor. 2013.http://www.bioconductor.org/packages/release/bioc/vignettes/ddCt/inst/doc/rtPCR.pdfOctober
26–2021
|
|
20
|
He G, Siddik ZH, Huang Z, Wang R, Koomen
J, Kobayashi R, Khokhar AR and Kuang J: Induction of p21 by p53
following DNA damage inhibits both Cdk4 and Cdk2 activities.
Oncogene. 24:2929–2943. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia M, Knezevic D and Vassilev LT: p21
does not protect cancer cells from apoptosis induced by
nongenotoxic p53 activation. Oncogene. 30:346–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sarantopoulos J, Mahalingam D, Sharma N,
Iyer RV, Ma WW, Ahluwalia MS, Johnson S, Purmal A, Shpigotskaya P,
Hards A, et al: Results of a completed phase I trial of CBL0137
administered intravenously (IV) to patients (Pts) with advanced
solid tumors. J Clin Oncol. 38 (Suppl 15):S35832020. View Article : Google Scholar
|
|
23
|
Fedyanin M, Tryakin A, Lisyanskaya AS,
Solovyeva E, Fadeeva N, Gladkov O, Moiseyenko V, Cheporov SV,
Shpigotskaya P, Purmal A, et al: Results of a completed
first-in-human phase Ib dose-escalation study of oral CBL0137 in
patients with advanced solid tumors. J Clin Oncol. 38 (Suppl
15):S36072020. View Article : Google Scholar
|
|
24
|
Chua MMJ, Lee M and Dominguez I:
Cancer-type dependent expression of CK2 transcripts. PLoS One.
12:e01888542017. View Article : Google Scholar : PubMed/NCBI
|