Introduction
Numerous types of anticancer drugs are available to
treat colorectal cancer and combination therapies are often used
for the treatment of this type of cancer. Drugs that directly
damage DNA and drive cancer cells toward apoptosis have entered
mainstream treatment and are often used in combination with
molecular-targeted agents (1);
however, studies focusing on therapies that inhibit cancer cell
repair are limited. Our previous study focused on DNA damage
recognition (DDR) during the cell cycle and suggested that tumor
proliferation can be suppressed by preventing DNA-damaged cancer
cells from repairing themselves (2). In particular, ataxia telangiectasia
and Rad3-related (ATR) is an apical kinase that regulates the DNA
damage response and organizes cellular processes, such as repair of
arrested replication forks (replication stress), and associated DNA
double-strand and single-strand breaks (3,4).
Recently, several potent and selective ATR
inhibitors have been developed, including M6620 (Merck KGaA),
AZD6738 (AstraZeneca) and BAY1895344 (United States Biological),
which are in clinical development (5). Investigations using BAY1895344 and
M6620 have provided clinical evidence of the antitumor activity of
ATR inhibitors in patients with advanced cancer and ataxia
telangiectasia mutated (ATM) abnormalities (loss of ATM protein
expression and/or ATM adverse mutations) (6,7). In
p53-mutant or ATM-deficient cells, AZD6738 treatment has been
reported to cause replication fork arrest and result in the
accumulation of unrepaired DNA damage; subsequently, when
AZD6738-treated cells progress to mitosis, cell death by mitotic
catastrophe occurs (8). However,
the effects of AZD6738 in the absence of ATM abnormalities are
unknown. It is believed that in colorectal cancer, AZD6738 may
enhance tumor suppression in combination with anticancer agents
(9). The effects of AZD6738 have
been reported in various types of cancer, including in pancreatic,
gastric, hepatocellular and biliary tract cancer (10–13).
AZD6738 acts on different sites based on its
concentration. It inhibits Chk1 phosphorylation at the
G2/M checkpoint, thereby inhibiting cell repair. Cancer
cells that are not repaired may adapt to the cell cycle and
subsequently undergo apoptosis (14,15).
In the presence of ATR depletion, p53 mutations are lethal
(16); however, little is known
about the effects of DDR in p53 wild-type cells (17,18).
Notably, ~50% of cases of colorectal cancer have p53 mutations
(19), whereas the other 50% have
wild-type p53, indicating that AZD6738 may be ineffective. Cell
repair in p53-mutant colorectal cancer cells is dependent on the
G2/M checkpoint; therefore, a strategy to inhibit
phosphorylation of Chk1 at the G2/M checkpoint using
AZD6738 is logical. However, we cannot conclude that AZD6738 is
ineffective in p53 wild-type colorectal cancer cells; therefore, it
is necessary to verify whether it is effective or not.
Our previous study demonstrated the
treatment-enhancing effects of combination therapy with AZD6738 and
5-fluorouracil (5-FU) (20). 5-FU
acts outside the S phase, and DNA-damaged cancer cells can be
repaired at various cell cycle checkpoints. This fact complicates
the evaluation of the effects of AZD6738. In cancer, loss of
G1 checkpoint regulation and activation of
replication-promoting oncogenes increases the likelihood of cancer
cells entering the S phase with increased replication stress
(5). Trifluridine (FTD) replaces
thymidine in DNA during the S phase, resulting in DNA dysfunction.
However, the DNA is repaired soon thereafter by inducing the
phosphorylation of Chk1 at the 345th residue at the G2/M
checkpoint (21). FTD also has a
notably higher rate of incorporation into DNA than other drugs
(22). The presence of the S phase
just before the G2/M checkpoint and the high rate of
incorporation of FTD into the DNA are two reasons that led us to
conclude that FTD may suppress tumors more efficiently than 5-FU.
In a previous study, combination therapy with FTD and a Chk1
inhibitor significantly inhibited the proliferation of xenograft
tumors derived from esophageal squamous cell carcinoma (23). Although FTD is ineffective at
shrinking tumors (24), AZD6738 may
enhance the effects of FTD. The combination of FTD and AZD6738, if
effective, may be an alternative treatment for colorectal cancer,
which has poor outcomes with the use of conventional drugs. The
present study aimed to determine the in vitro and in
vivo effects of combined treatment with AZD6738 and FTD on
tumor proliferation and the expression of proteins involved in DNA
damage checkpoints.
Materials and methods
Cell lines
The human colorectal cancer cell lines HT29, HCT116,
DLD-1 and SW480 were purchased from the American Type Culture
Collection. These cancer cell lines were certified by short tandem
repeat profiling. HT29 and HCT116 cells were cultured in DMEM
(FUJIFILM Wako Pure Chemical Corporation) supplemented with 10%
fetal bovine serum [(FBS); Gibco; Thermo Fisher Scientific Inc.]
and 1% penicillin/streptomycin. DLD-1 cells were cultured in RPMI
(FUJIFILM Wako Pure Chemical Corp.) supplemented with 10% FBS and
1% penicillin/streptomycin. SW480 cells were cultured in
Leibovitz's L-15 Medium (Gibco; Thermo Fisher Scientific Inc.)
supplemented with 10% FBS and 1% penicillin/streptomycin. HT29,
HCT116 and DLD-1 cells were cultured at 37°C in 5% CO2,
whereas SW480 cells were cultured at 37°C in 100% atmospheric air
according to the datasheets for each cell line. Only the HT29 cell
line was a BRAF mutant and only the HCT116 cell line was p53
wild-type.
WST-1 assays
HT29, HCT116, DLD-1 and SW480 cells were treated
with FTD (Tokyo Kasei Kogyo Co. Ltd.) at concentrations of 0, 0.1,
1, 10, 100 and 1,000 µM with or without 0.5 µM AZD6738. Cell
viability was assessed by WST-1 assay using the Premix WST-1 Cell
Viability Assay System (Takara Bio, Inc.). The combination index
scores were calculated to show the synergistic effects (25). The AZD6738 concentration of 0.5 µM
was previously determined via cytotoxicity experiments in our
laboratory (20). Cells
(1.0×104 cells/well in 100 µl) were seeded in 96-well
plates and allowed to adhere for 24 h at 37°C. Thereafter, the
cells were treated with different concentrations of FTD with or
without AZD6738 for 72 h at 37°C. WST-1 was diluted 10-fold and
then added to the cells. After 1 h of incubation at 37°C, the
absorbance of cells in each well was measured at a wavelength of
450 nm using a microplate reader (SpectraMax ABC; Molecular
Devices, LLC). In addition, the HT29 and HCT116 cells were divided
into the following groups: Control (no drug added), AZD6738
(AZD6738 alone), FTD (FTD alone) and FTD + AZD6738 (FTD and AZD6738
combined). The concentrations of FTD used to treat HT29 and HCT116
cells were the IC50 values of 70 and 5 µM, respectively;
for the concentration of AZD, 0.5 µM was adopted. Cell viability
was assessed at 24, 48 and 72 h at 37°C.
After oral administration of FTD, FTD is rapidly
hydrolyzed by thymidine phosphorylase (TP) in the liver to an
inactive form (26). The addition
of tipiracil to FTD at a molar ratio of 1:0.5 or a weight ratio of
1:0.471 inhibits the degradation of FTD, and sufficient drug levels
can be achieved in the blood (27).
The mixture of FTD and tipiracil is called TAS-102. The present
study investigated tipiracil (MedChemExpress) toxicity in HT29
cells. Cell viability was evaluated using the WST-1 assay at each
of the following concentrations of FTD in TAS-102: 0, 0.1, 1, 10,
100 and 1,000 µM. The results were compared with the group treated
with FTD alone. Cell viability was assessed after treatment for 72
h at 37°C.
Flow cytometry
HT29 and HCT116 cells were divided into control,
AZD6738, FTD and FTD + AZD6738 groups. After culturing the cells
under their respective culturing conditions for 0, 24, 48 and 72 h,
they were collected and stored in 70% ethanol at −20°C. After 1
week, cell cycle progression was examined using flow cytometry. A
cell suspension was prepared using the BD Cycletest Plus DNA Kit
(BD Biosciences); cell concentration was adjusted to
1.0×106 cells/ml using Buffer Solution. The cell
suspension was centrifuged at 400 × g for 5 min at room temperature
(20–25°C) and the supernatant was carefully decanted. For enzymatic
degradation of solid tissue fragments, and digestion of the cell
membrane and cytoskeleton, cells were placed in a liquid in
tetrahydrochlorospermine detergent buffer containing trypsin and
mixed by hand with light tapping. The cells were incubated at room
temperature (20–25°C) for 10 min.
To inhibit the activity of trypsin and digest RNA, a
solution containing trypsin inhibitor and ribonuclease A was added
there and mixed with light tapping. The mixture was incubated at
room temperature (20–25°C) for 10 min. To bind PI to DNA, a
solution containing PI and spermine tetrahydrochloride was further
added at 2–8°C and mixed with light tapping. The solution was
incubated on ice 2–8°C in the dark for 10 min. Cell cycle
localization at each stage was measured using the FACSCanto II flow
cytometer (BD Biosciences) at a wavelength of 488 nm and the data
were analyzed using the BD FACSDiva Software version 8.0.2 (BD
Biosciences).
Western blotting
HT29 and HCT116 cells were divided into the control,
AZD6738, FTD and FTD + AZD6738 groups. After culturing for 48 and
72 h, the cells were collected and SDS sample buffer [2.75% SDS (pH
6.8), 9% glycerol, 87.5 mmol/l Tris-HCl, 150 mmol/l dithiothreitol,
and 0.003% bromophenol blue] was added. Subsequently,
1×106 cells were suspended in 150 µl SDS sample buffer.
After heating at 98°C for 5 min, 15 µl cell sample was loaded onto
the gel (2,20,28).
Proteins in the lysate were separated on 10% or 4–20% Mini-PROTEAN
TGX Precast Gels (Bio-Rad Laboratories, Inc.) according to the
molecular weight of the target protein. Separated proteins were
transferred onto nitrocellulose membranes at 100 V for 1 h at 4°C.
The membranes were then blocked with 5% skim milk (BD Biosciences)
in Tris-buffered saline-Tween 20 (10%) for 30 min at 20–25°C and
then incubated with the primary antibody diluted in 1% skim milk at
4°C overnight while being rotated (Rotator RT-50; TAITEC
Corporation). Subsequently, the membranes were incubated with the
secondary antibodies diluted in 1% skim milk at 4°C for 1 h while
being rotated. When phosphorylated (p)-Chk1 was being detected,
Western BLoT Immuno Booster Solution 1 and Western BLoT Immuno
Booster Solution 2 (cat. no. T7111A; Takara Bio, Inc.) were used to
dilute the primary and secondary antibodies, instead of skim milk,
respectively. The following primary antibodies were used: p-Chk1
Ser345 (1:1,000; cat. no. 2348; Cell Signaling Technology, Inc.),
Chk1 (1:1,000; cat. no. C9358; MilliporeSigma), β-actin (1:1,000;
cat. no 3700; Cell Signaling Technology, Inc.), Apoptosis Western
Blot Cocktail (1:250; cat. no. ab136812; Abcam) for cleaved
caspase-3, procaspase-3 and PARP, H2A.X variant histone (H2A.X;
1:1,000; cat. no ab11175; Abcam) and p-H2A.X (γH2A.X; 1:2,000; cat.
no. 05-636; Merck KGaA). The secondary antibodies used included
horseradish peroxidase HRP-conjugated goat anti-mouse
immunoglobulin (1:1,000; cat. no. P0447; Agilent Technologies,
Inc.) for Chk1, γH2A.X and β-actin, and HRP-conjugated goat
anti-rabbit immunoglobulin (1:1,000; cat. no. P0448; Agilent
Technologies, Inc.) for p-Chk1 and H2A.X. For the primary antibody
Apoptosis Western Blot Cocktail, the HRP-conjugated secondary
antibody cocktail from this kit was used (1:100; cat. no. ab136812;
Abcam) for cleaved caspase-3, procaspase-3, PARP. The following
reagents were used as HRP chemiluminescent reagents: SuperSignal
West Pico Chemiluminescent Substrate and SuperSignal West Femto
Chemiluminescent Substrate were used to visualize p-Chk1, and
Pierce ECL Western Blotting Substrate was used to visualize Chk1,
γH2A.X, H2A.X, cleaved caspase-3, procaspase-3, PARP and β-actin
(all reagents obtained from Thermo Fisher Scientific, Inc.).
Immunoreactive protein bands were detected using the ImageQuant
LAS-4000 mini control software (Cytiva). Each resulting band was
semi-quantified via densitometric analysis using ImageJ software
version 1.53 (National Institutes of Health).
Tumor generation and grouping
A total of 24 male BALB/c nu/nu mice (age, 4 weeks;
mean weight, ~22.45 g) were purchased from Japan SLC Inc., and drug
efficacy studies were performed using a xenotransplanted nude mouse
model. The mice were acclimated to the environment, and water was
orally administered using an oral sonde for 2 weeks. Mice were
housed in standard Plexiglas cages (n=3 mice/cage) at a constant
temperature (20–26°C) and humidity (40–60%), under a 12-h
light/dark cycle. Mice were given ad libitum access to
normal autoclaved feed and drinking water. An individual
identification number (cat. no. KN-295; Natsume Seisakusho Co.,
Ltd.) was attached to the right auricle using an ear punch method
to enable the identification of individual animals.
Seventh-generation HT29 cells (mycoplasma-free; mycoplasma
detection kit supplied by Lonza Group, Ltd.) were injected
subcutaneously to induce tumors. Subsequently, 2% isoflurane
(product. no. 099-06571; FUJIFILM Wako Pure Chemical Corp.) was
administered using a vaporizer (Rodent Circuit Controller;
Vetequip, Inc.) as general anesthesia. HT29 cells (5×106
in 200 µl phosphate-buffered saline) were injected into the right
lateral abdomen of each mouse. Subcutaneous swelling was observed
in all mice. Mice were randomly divided into the following groups
(n=6 mice/group): Control, AZD6738, TAS-102 and TAS-102 + AZD6738.
The Control group received only FTD and tipiracil, and the AZD6738
solvent. Similarly, the AZD6738 group received the FTD and
tipiracil solvent, and the FTD and tipiracil group received the
AZD6738 solvent.
Drug administration
FTD and tipiracil were dissolved in 0.5 w/v%
hydroxypropylmethyl cellulose (HPMC; Shin-Etsu Chemical Co., Ltd.;
TC-5®; grade, R; indicated viscosity is 6 mPa·S). FTD
and tipiracil were combined in a molar ratio of 1:0.5 and a weight
ratio of 1:0.471, and were dissolved in 0.5 w/v% HPMC. The final
FTD equivalent concentration was 7.5 mg/ml and the suspension was
vortexed to dissolve the drug. AZD6738 was dissolved in 5% DMSO,
40% propylene glycol and ddH2O according to the
datasheet. TAS-102 was administered at a dosage of 200 mg/kg/day
(100 mg/kg twice daily, 6-h interval), and body weights were
measured daily to determine the drug dosage (29). The duration of the study was 3 weeks
(5 days dosing, 2 days rest). AZD6738 was administered at a dose of
25 mg/kg based on previous studies (30,31).
Syringes (cat. no. IC-1-4908-01; Terumo Corporation) and an 18-G
oral sonde (cat. no. VS-493-18GS; Natsume Seisakakusho Co., Ltd.)
were used for the oral administration of TAS-102 and AZD6738.
Tumor assessment and humane
endpoints
The long and short diameters of the tumors were
measured using calipers (Shinwa Co., Ltd.) and tumor volume was
calculated using the following equation: (longest tumor diameter) ×
(shortest tumor diameter)2/2 (20). Drug administration was initiated
when an average tumor volume of 100 mm3 was achieved.
Tumors and body weights were evaluated daily for 3 weeks. A total
of 3 weeks after drug administration, all mice were euthanized by
cervical dislocation under general anesthesia using 2% isoflurane
and death was confirmed by cardiac arrest. Humane endpoints were
defined as maximum tumor weight >10% of body weight, maximum
tumor diameter >20 mm, tumor ulceration, necrosis, infection,
gait disturbance, impaired water and food intake, maximum weight
loss >20, and >25% at 7 days compared with the controls, and
cachexia. No mice met these criteria during the study. All animal
experiments were conducted with the approval of the Animal Welfare
and Use Committee of the Nagoya City University Graduate School of
Medicine (approval no. 22-006; Nagoya, Japan). The animal rooms and
laboratories in the Center for Laboratory Animal Research and
Education, Nagoya City University are equipped with P2A-level
diffusion prevention measures and certified (certification no.
FM3).
Immunohistochemistry
Tumors were fixed in 4% paraformaldehyde for 6 h at
4°C and then embedded in paraffin. Paraffin-embedded tissues were
cut into 3-µm sections and mounted on slides. Sections were
deparaffinized twice with Hemo-De for 10 min, hydrated twice with
100, 90, 80 and 70% ethanol (5 min each time), and washed with
running water. For antigen retrieval, the slides were immersed in
10 mM citrate buffer (pH 6.0) and heated in a 600-Watt microwave
for 10 min. Endogenous peroxidase activity was blocked by immersion
in a mixed solution of 0.3% hydrogen peroxide and 100% methanol at
20–25°C for 30 min. Blocking was performed in a humidified box at
20–25°C for 10 min with 4% Block Ace powder (cat. no. UKB80; DS
Pharma Biomedical Co., Ltd.). Sections were stained overnight at
4°C with γH2A.X primary antibody (1:500; cat. no. 05-636; Merck
KGaA) and stained with anti-mouse EnVision+ HRP-conjugated polymer
as the secondary antibody (1:1,500; cat. no. K4001; Dako; Agilent
Technologies, Inc.) for 45 min at 20–25°C. To detect antibody
binding, 3,3-diaminobenzidine substrate (cat. no. K3467; Dako;
Agilent Technologies, Inc.) was used as a chromogenic agent. The
slides were incubated in the substrate solution for 10 min at
20–25°C. After rinsing in running water, hematoxylin was used for
contrast staining (30 sec at 20–25°C). A total of 10 fields of view
were examined for each tumor. The mean percentage of
γH2A.X-positive cells per high magnification field of view ± SD was
determined. Images of the slides were captured using a light
microscope (BZ-X710; Keyence Corporation) and were analyzed using
the BZ-X710 Analyzer software version 1.4.0.1 (Keyence
Corporation).
Statistical analysis
In vitro experiments were generally performed
at least three times; in vivo experiments were performed
only once. Statistical analysis was performed using the EZR
software (Easy R) version 1.41 (https://www.jichi.ac.jp/saitama-sct/SaitamaHP.files/statmed.html;
Saitama Medical Center, Jichi Medical University, Saitama, Japan).
All data are presented as the mean ± SD. Two groups were compared
using unpaired Student's t-test, whereas two or more groups were
compared using one-way ANOVA followed by Tukey's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effects of combining AZD6738 with
FTD
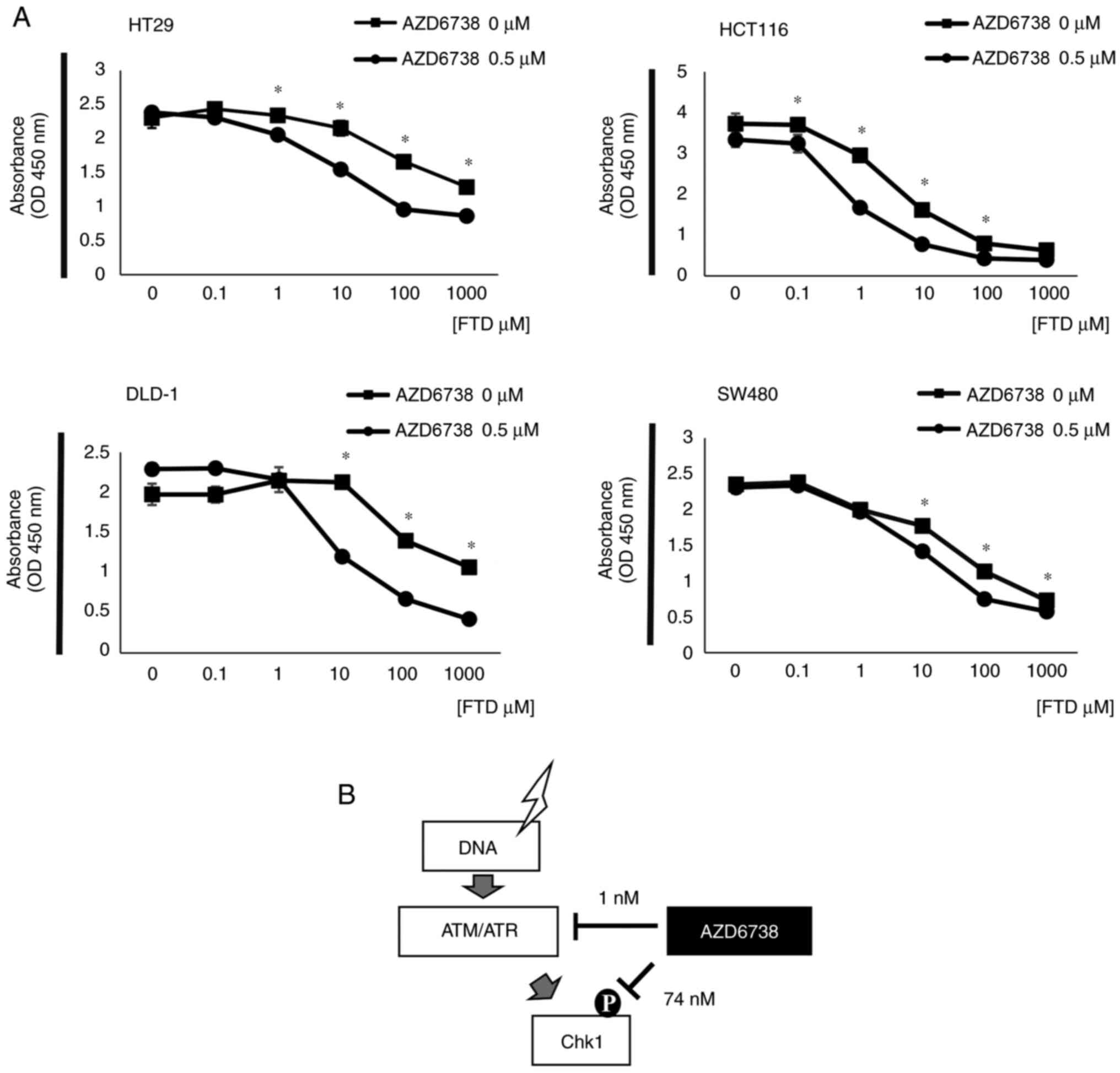
The viability of HT29, HCT116, DLD-1 and SW480 cells
with or without 0.5 µM AZD6738 treatment was determined (Fig. 1A). The combined use of AZD6738 and
FTD at multiple concentrations inhibited cell viability in all four
cell lines compared with FTD alone. The combination index scores of
HT29, HCT116, DLD-1 and SW480 were 0.775, 0.231, 0.134 and 0.718,
indicating a synergistic effect (25). The point of action of AZD6738 varies
with its concentration, as stated in the data sheet obtained from
MedChemExpress. AZD6738 is a potent inhibitor of ATR kinase
activity with an IC50 of 0.001 µM against the isolated
enzyme and 0.074 µM against ATR kinase-dependent CHK1
phosphorylation in cells (Fig. 1B).
Therefore, cell viability assays were performed after adding 1 nM
AZD6738 to cells treated with each concentration of FTD to confirm
that cell viability was not inhibited (Fig. S1). The IC50 values of
AZD6738, calculated from 72-h MTT dose-response curves, are ≥1 µM
for HCT116 and HT29 cells (32).
The minimum concentration at which the drug acts adequately is
unknown. Therefore, to determine the minimum concentration at which
AZD6738 acts, the effects of each concentration of FTD and AZD6738
with an IC50 of 70 µM on HT29 cells were evaluated by
flow cytometry (data not shown). The HT29 cell line was treated
with 70 µM FTD combined with 0, 0.5, 5, 50, 100, 250, 500, 1,000
and 5,000 nM AZD6738. Flow cytometry was used to evaluate the
localization of each cell cycle. The cell cycle localization did
not change in response to 0.5, 5, 50, 100 and 250 nM AZD6738 at 48
h, but the number of cells in the G2/M phase began to
decrease at 500 nM (0.5 µM). Therefore, it was concluded that the
lowest concentration at which AZD6738 begins to act on the HT29
cell line is 0.5 µM. Further toxicity experiments revealed that the
optimal cell concentration at which AZD6738 acts is 0.5 µM. The
IC50 for FTD was 70 µM in HT29 cells and 5 µM in HCT116
cells (33,34).
Inhibitory effects of combined FTD and
AZD6738 treatment on cell cycle progression and cell viability
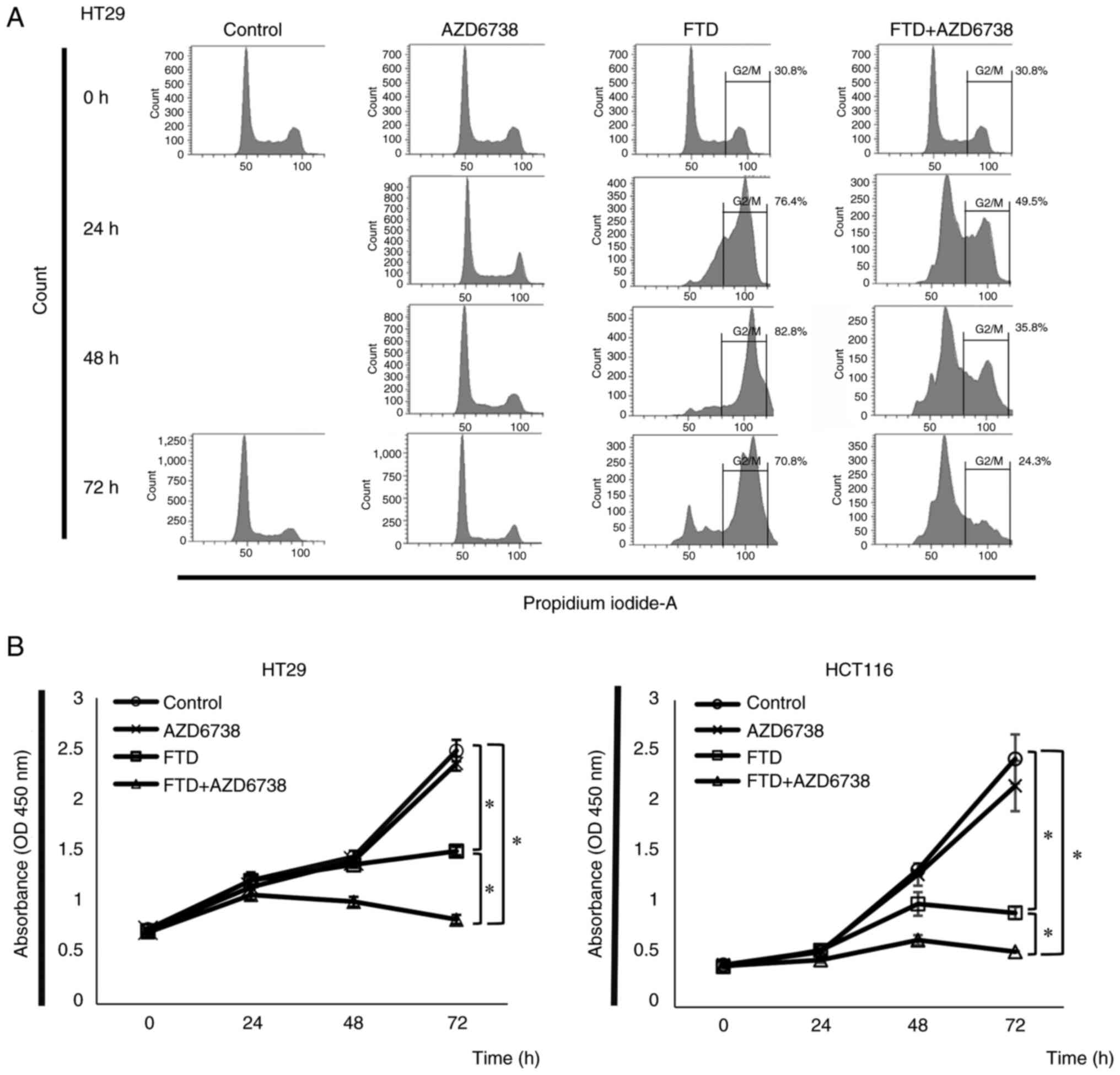
HT29 and HCT116 cells were divided into the control,
AZD6738, FTD, and FTD + AZD6738 groups, and were cultured for 0,
24, 48 and 72 h. The cell cycle progression of each treatment group
is shown in Fig. 2A. Cell cycle
arrest was detected at the G2/M checkpoint in the FTD
group. By contrast, the FTD + AZD6738 group showed a decrease in
the number of cells localized to the G2/M checkpoint. Cell
viability was measured at each elapsed time, and a significant
difference in cell viability was observed between the FTD + AZD6738
and FTD groups at 72 h for HT29 and HCT116 cells (Fig. 2B).
Combined FTD and AZD6738 treatment
alters protein expression, leading to the accumulation of DNA
damage and cell death
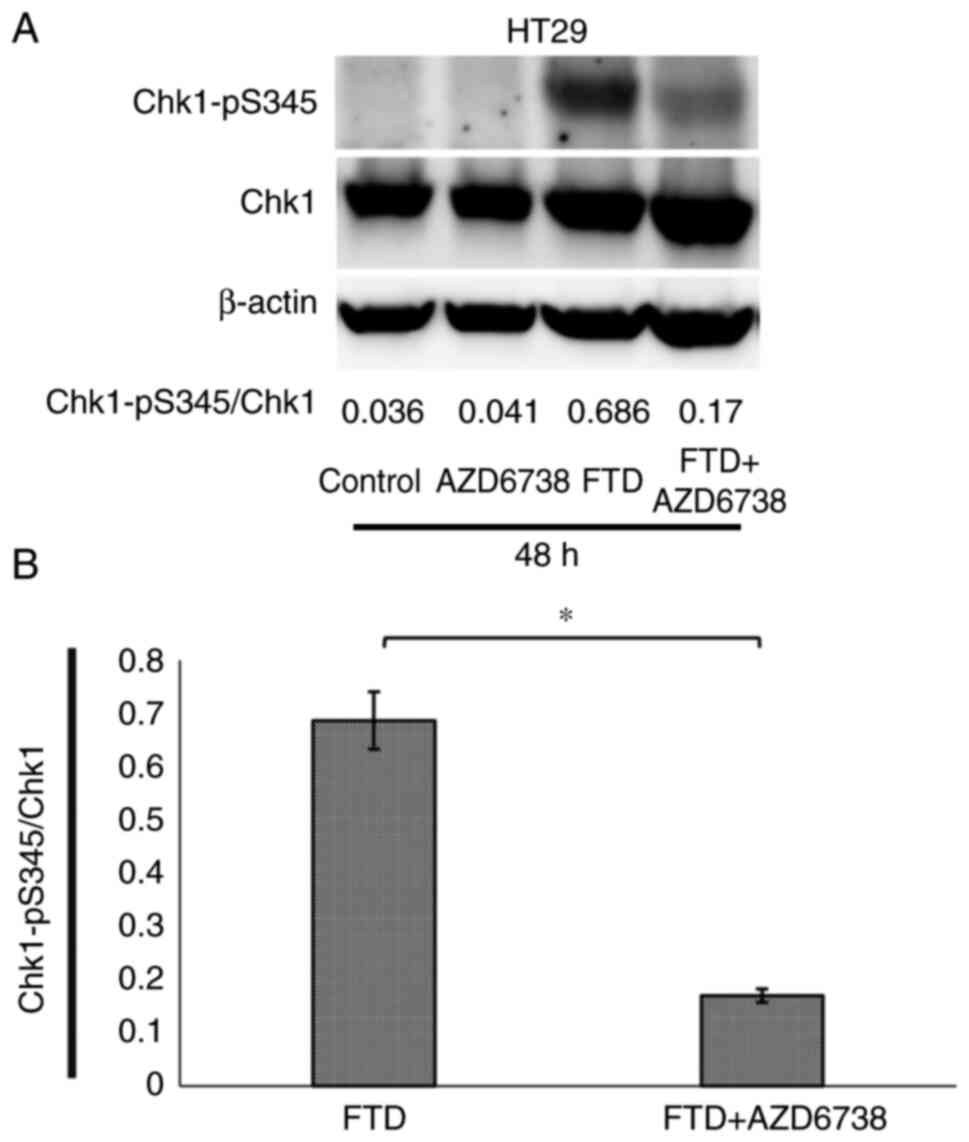
Chk1 phosphorylation in HT29 cells was suppressed in
the FTD + AZD6738 group compared with that in the FTD group at 48 h
(Fig. 3). Thus, AZD6738 was
confirmed to suppress Chk1 phosphorylation. In HT29 and HCT116
cells, DNA was more damaged in the FTD + AZD6738 group than that in
the FTD group, as confirmed by the increased expression levels of
γH2A.X (Fig. 4A and B) (35). Furthermore, the expression of
apoptotic proteins in HT29 and HCT116 cells was higher in the FTD +
AZD6738 group than that in the FTD group, as confirmed by the
expression levels of cleaved caspase-3 and cleaved poly(ADP-ribose)
polymerase (Fig. 4C and D). These
results indicated that DNA may be damaged in response to FTD +
AZD6738 treatment, leading to apoptosis.
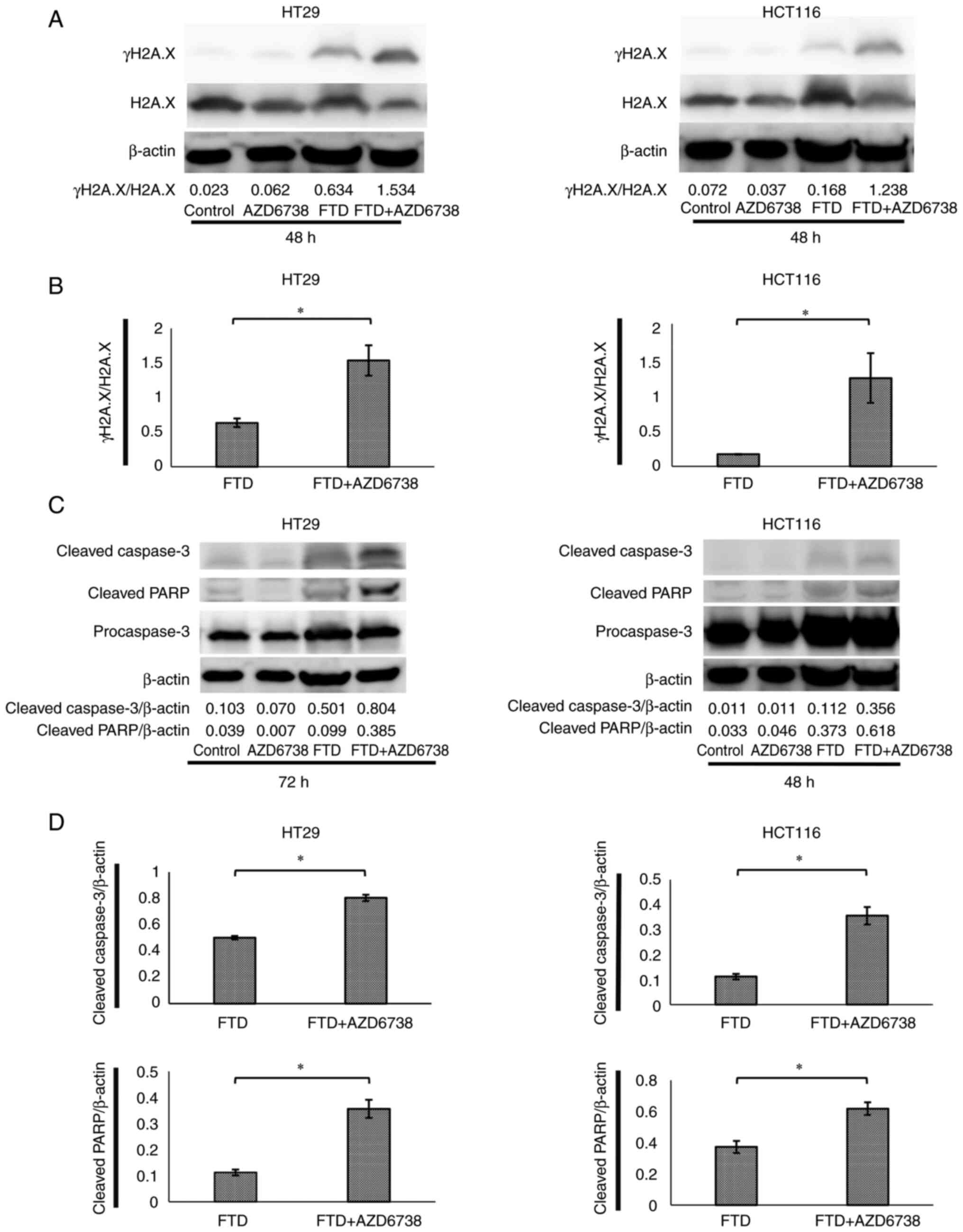 | Figure 4.AZD6738 in combination with FTD
results in the accumulation of more DNA damage and increased γH2A.X
protein levels compared with FTD alone. AZD6738 + FTD also
increased the expression of apoptotic proteins compared with FTD
alone. HT29 and HCT116 cells were divided into control, AZD6738
(0.5 µM), FTD (70 µM or 5 µM) and FTD (70 µM or 5 µM) + AZD6738
(0.5 µM) groups and treated until the indicated time. Whole-cell
extracts were subjected to western blotting with γH2A.X, H2A.X,
cleaved caspase-3, cleaved PARP and β-actin antibodies. For FTD
concentrations, the respective IC50 values were used: 70 µM for
HT29 and 5 µM for HCT116. (A) γH2A.X expression in HT29 and HCT116
cells after 48 h. (B) γH2A.X/H2A.X in HT29 and HCT116 cells after
48 h. (C) Cleaved caspase-3 and cleaved PARP levels in HT29 and
HCT116 cells at 48 and 72 h. (D) Cleaved caspase-3/β-actin and
cleaved PARP/β-actin in HT29 and HCT116 cells at 48 and 72 h. Data
are presented as the mean ± SD (n=3). Statistical significance was
determined using Student's t-test. *P<0.05. γH2A.X,
phosphorylated form of H2A.X; FTD, trifluridine; H2A.X, H2A.X
variant histone; PARP, poly(ADP-ribose) polymerase. |
Synergistic effects of AZD6738 and FTD
in vivo
The effects of FTD and TAS-102 on cell viability
were comparable, thus indicating that cell viability was not
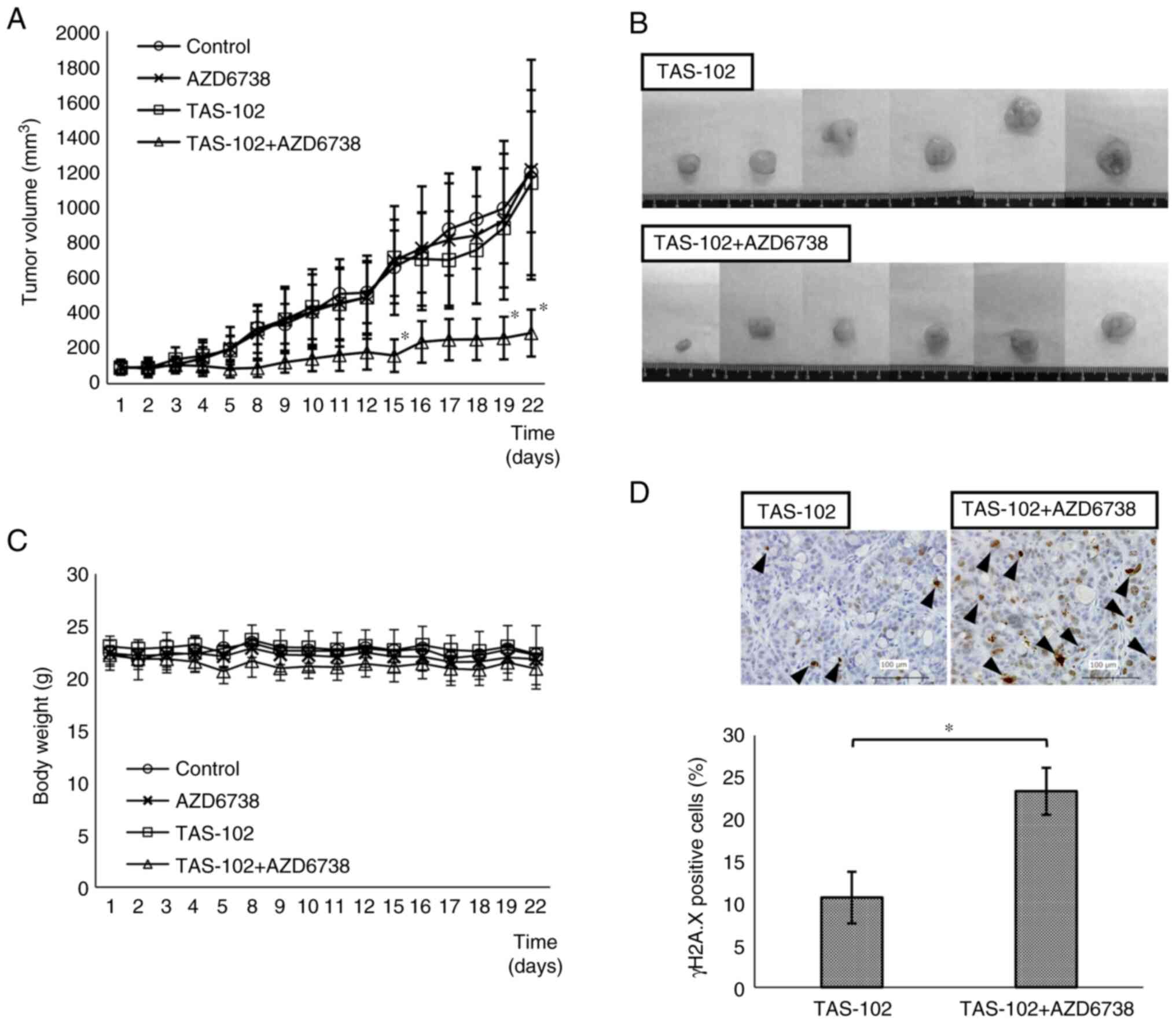
affected by the addition of tipiracil to FTD (Fig. S2). In the TAS-102 + AZD6738 group,
tumor volume was significantly smaller than that in the TAS-102
alone group on days 15, 19 and 22 (Fig.
5A). Images of the excised tumors in the TAS-102 and TAS-102 +
AZD6738 groups are shown in Fig.
5B. Measurements of the excised tumors were consistent with
those measured from the body surface. The TAS-102 + AZD6738 group
exhibited slight weight loss compared with the other groups, but
this was not significant (Fig.
5C).
No humane endpoints were reached in the mice.
Furthermore, no significant differences in the weights of the
liver, kidney and testes were detected between the groups (Fig. S3). As demonstrated by tumor
immunostaining, γH2A.X protein levels were significantly higher in
the TAS-102 + AZD6738 group than those in the TAS-102 group
(Fig. 5D). The higher expression of
γH2A.X in the TAS-102 + AZD6738 group compared with that in the
TAS-102 group indicated that DNA damage is accumulated without
repair.
Discussion
AZD6738 has an IC50 of 0.074 µM for
inhibiting Chk1 Ser345 phosphorylation (ATR substrate) in cells
(30). AZD6738 exerts different
effects depending on its concentration, including inhibition of
Chk1 phosphorylation and, at the same concentration, inhibition of
DNA damage repair. In the present study, AZD6738 suppressed cell
viability when combined with FTD despite using a concentration of
AZD6738 (0.5 µM) that did not kill cells in toxicity experiments
(20). These findings indicated
that combination therapy with FTD and AZD6738 inhibited the
viability of colorectal cancer cells. In HT29 colorectal cancer
cells, FTD-induced DNA damage arrested cells at the G2/M
checkpoint for repair, but the suppression of Chk1 phosphorylation
by AZD6738 prevented the repair of cancer cells, resulting in the
accumulation of DNA damage and, consequently, apoptosis.
Furthermore, combination therapy with TAS-102 and AZD6738
suppressed tumor growth in a xenograft mouse model.
Based on the results of the present study and a
previous study on 5-FU, the following may be inferred (20). In a HT29 ×enograft mouse model, the
volume of tumors in mice treated with 5-FU + AZD6738 was ~50% the
volume of tumors in mice treated with 5-FU alone. In the present
study, the volume of tumors in mice treated with TAS-102 + AZD6738
was ~25% of the volume of tumors in mice treated with TAS-102
alone. These results suggested that FTD was taken up more
efficiently than 5-FU, resulting in a higher tumor suppressive
effect. Cancer cells with p53 mutations at the G1
checkpoint depend on the G2/M checkpoint for repair
(36). However, it could be
hypothesized that HCT116 cells without p53 mutations have DNA
damage in the S phase, which is often repaired at the
G2/M checkpoint, leading to cell death. Although ATR
inhibitors were expected to be effective only in cells with p53
mutations, the present study demonstrated that ATR inhibitors were
also effective in cells without p53 mutations.
Notably, the present results indicated that some
anticancer drugs, including those that were considered ineffective,
can achieve tumor suppression when combined with AZD6738. BRAF
mutations occur in colorectal cancer with a low response rate to
systemic chemotherapy. These mutations occur in ~10% of patients
with metastatic colorectal cancer, and novel therapeutic agents are
urgently needed for this type of cancer (37). The HT29 cells used in the present
study are BRAF mutants. Notably, in the present study, FTD alone
was unable to inhibit cell viability; however, the combination of
FTD and AZD6738 inhibited the viability of HT29 cells, suggesting
the acquisition of a chemotherapy response.
FTD exerts different effects on different cell
lines. HCT116 cells are BRAF wild-type, and the IC50 of
FTD in this cell line was 5 µM, which is sufficient for its
efficacy. Conversely, HT29 cells are BRAF mutant, and the
IC50 of FTD in this cell line is ~100 µM; therefore, the
effect of drug therapy is generally poor (33). In the present study, the
IC50 of FTD alone was 70 µM in the HT29 cell line, which
is a high value and is thus considered ineffective. However, when
AZD6738 was used in combination with FTD, cell viability was
significantly suppressed when FTD was administered at 1 µM or
higher. Thus, the addition of AZD6738 was sufficient to suppress
cell viability, indicating that drugs with poor inhibitory effects
on cell viability can be converted into effective drugs. The
combination therapy of FTD and AZD6738 in BRAF-mutant cell line was
effective. In our previous report on the combination of 5-FU and
AZD6738, 5-FU was administered intraperitoneally (20). By contrast, in the present study,
FTD and AZD6738 were administered orally. The current animal
experiments were able to demonstrate drug efficacy in a way similar
to that performed in actual clinical practice.
The current findings suggested that combining
AZD6738 with various drugs may increase the tumor suppressive
effects of these drugs in colorectal cancer, which is usually
resistant to anticancer drugs and difficult to treat in current
clinical practice. The combination of FTD and AZD6738 was more
effective than FTD alone in vitro. Furthermore, the
combination of TAS-102 and AZD6738 was more effective than TAS-102
alone in vivo. In the future, In the future, if AZD6738 can
be shown to be effective in cell lines resistant to 5-FU, the
mainstay of anticancer drugs, it will make it easier to introduce
AZD6738 into routine clinical practice.
The present study has several limitations. To
accurately compare the effect of FTD with 5-FU on tumor
suppression, these drugs need to be compared in the same cells. In
addition, we cannot confirm that the wild-type p53 genotype of
HCT116 cells contributed to the efficacy of FTD, as other genetic
characteristics may be involved. Knock-in and knockout of the p53
gene in the same cell lines are warranted to confirm the role of
p53. Moreover, only in vitro investigations were conducted
on HCT116 cells; thus, the efficacy of AZD6738 on HCT116 cells, a
wild-type p53 cell line, should be confirmed in vivo.
In conclusion, AZD6738 is effective for inhibiting
colorectal cancer tumor proliferation when combined with FTD. Based
on this finding, future clinical trials should be conducted to
assess combination therapy with AZD6738 and FTD.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms.Seiko Inumaru, a
laboratory assistant, for preparing the experimental reagents and
handling the tumors, and Ms. Ryoko Hara, a laboratory assistant,
for preparing the experimental reagents (both are affiliated with
the Department of Gastroenterology, Nagoya City University Graduate
School of Medicine).
Funding
This research was supported by a Grant-in-Aid for Scientific
Research from the Japan Society for the Promotion of Science
(assignment no. 19K18158).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SH, TS, HT and TH contributed to the conception and
design of this study, analysis and interpretation of the data, and
writing and review of the manuscript. SH, TS, HT, TH, AK, KW, TY,
HU, KS, RO, YM and ST designed the study. SH, TS, AK, KW, TY, HU,
KS, AM and MK performed experiments and obtained data. SH, TS, KW,
TY, HU, KS, RO, AM, MK, YM and ST confirm the authenticity of all
the raw data. SH wrote the manuscript. TS and HT proofread the
manuscript. TS, HT, TH, KS, RO, YM, AM, MK and ST supervised the
study. All authors read and approved the final manuscript, and are
equally responsible for all aspects of the study and guarantee its
completeness and accuracy.
Ethics approval and consent to
participate
In vivo mouse experiments were approved by
the Animal Welfare and Use Committee of the Nagoya City University
Graduate School of Medicine (approval no. 22-006, approved June 06,
2022).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
DDR
|
DNA damage recognition
|
|
ATR
|
ataxia telangiectasia and
Rad3-related
|
|
ATM
|
ataxia telangiectasia mutated
|
|
FBS
|
fetal bovine serum
|
|
FTD
|
trifluridine
|
|
HPMC
|
hydroxypropyl methylcellulose
|
|
HRP
|
horseradish peroxidase
|
|
PARP
|
poly(ADP-ribose) polymerase
|
References
|
1
|
Yoshino T, Oki E, Nozawa H,
Eguchi-Nakajima T, Taniguchi H, Morita S, Takenaka N, Ozawa D and
Shirao K: Rationale and design of the TRUSTY study: A randomised,
multicentre, open-label phase II/III study of
trifluridine/tipiracil plus bevacizumab versus irinotecan,
fluoropyrimidine plus bevacizumab as second-line treatment in
patients with metastatic colorectal cancer progressive during or
following first-line oxaliplatin-based chemotherapy. ESMO Open.
3:e0004112018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirokawa T, Shiotani B, Shimada M, Murata
K, Johmura Y, Haruta M, Tahara H, Takeyama H and Nakanishi M:
CBP-93872 inhibits NBS1-mediated ATR activation, abrogating
maintenance of the DNA double-strand break-specific G2 checkpoint.
Cancer Res. 74:3880–3889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harper JW and Elledge SJ: The DNA damage
response: Ten years after. Mol Cell. 28:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Foote KM, Nissink JWM, McGuire T, Turner
P, Guichard S, Yates JWT, Lau A, Blades K, Heathcote D, Odedra R,
et al: Discovery and characterization of AZD6738, a potent
inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR)
kinase with application as an anticancer agent. J Med Chem.
61:9889–9907. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bradbury A, Hall S, Curtin N and Drew Y:
Targeting ATR as cancer therapy: A new era for synthetic lethality
and synergistic combinations? Pharmacol Ther. 207:1074502020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yap TA, Tan DSP, Terbuch A, Caldwell R,
Guo C, Goh BC, Heong V, Haris NRM, Bashir S, Drew Y, et al:
First-in-human trial of the oral ataxia telangiectasia and
RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced
solid tumors. Cancer Discov. 11:80–91. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yap TA, O'Carrigan B, Penney MS, Lim JS,
Brown JS, de Miguel Luken MJ, Tunariu N, Perez-Lopez R, Rodrigues
DN, Riisnaes R, et al: Phase I trial of first-in-class ATR
inhibitor M6620 (VX-970) as monotherapy or in combination with
carboplatin in patients with advanced solid tumors. J Clin Oncol.
38:3195–3204. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kwok M, Davies N, Agathanggelou A, Smith
E, Petermann E, Yates E, Brown J, Lau A and Stankovic T: Synthetic
lethality in chronic lymphocytic leukaemia with DNA damage response
defects by targeting the ATR pathway. Lancet. 385 (Suppl
1):S582015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yap TA, Krebs MG, Postel-Vinay S,
El-Khouiery A, Soria JC, Lopez J, Berges A, Cheung SYA,
Irurzun-Arana I, Goldwin A, et al: Ceralasertib (AZD6738), an oral
ATR kinase inhibitor, in combination with carboplatin in patients
with advanced solid tumors: A phase I study. Clin Cancer Res.
27:5213–5224. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wallez Y, Dunlop CR, Johnson TI, Koh SB,
Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A,
Richards FM and Jodrell DI: The ATR inhibitor AZD6738 synergizes
with gemcitabine in vitro and in vivo to induce pancreatic ductal
adenocarcinoma regression. Mol Cancer Ther. 17:1670–1682. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M,
Zhou P, Guo Z, Wang J, Wang H, Dai W, et al: ATR inhibitor AZD6738
enhances the antitumor activity of radiotherapy and immune
checkpoint inhibitors by potentiating the tumor immune
microenvironment in hepatocellular carcinoma. J Immunother Cancer.
8:e0003402020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nam AR, Jin MH, Park JE, Bang JH, Oh DY
and Bang YJ: Therapeutic targeting of the DNA damage response using
an ATR inhibitor in biliary tract cancer. Cancer Res Treat.
51:1167–1179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Min A, Im SA, Jang H, Kim S, Lee M, Kim
DK, Yang Y, Kim HJ, Lee KH, Kim JW, et al: AZD6738, a novel oral
inhibitor of ATR, induces synthetic lethality with ATM deficiency
in gastric cancer cells. Mol Cancer Ther. 16:566–577. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smith J, Tho LM, Xu N and Gillespie DA:
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and
cancer. Adv Cancer Res. 108:73–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Toczyski DP, Galgoczy DJ and Hartwell LH:
CDC5 and CKII control adaptation to the yeast DNA damage
checkpoint. Cell. 90:1097–1106. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan S, Smith ML, Rivet DJ II, Duba D, Zhan
Q, Kohn KW, Fornace AJ Jr and O'Connor PM: Disruption of p53
function sensitizes breast cancer MCF-7 cells to cisplatin and
pentoxifylline. Cancer Res. 55:1649–1654. 1995.PubMed/NCBI
|
|
17
|
Reaper PM, Griffiths MR, Long JM, Charrier
JD, Maccormick S, Charlton PA, Golec JM and Pollard JR: Selective
killing of ATM-or p53-deficient cancer cells through inhibition of
ATR. Nat Chem Biol. 7:428–430. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pabla N, Huang S, Mi QS, Daniel R and Dong
Z: ATR-Chk2 signaling in p53 activation and DNA damage response
during cisplatin-induced apoptosis. J Biol Chem. 283:6572–6583.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iacopetta B: TP53 mutation in colorectal
cancer. Hum Mutat. 21:271–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suzuki T, Hirokawa T, Maeda A, Harata S,
Watanabe K, Yanagita T, Ushigome H, Nakai N, Maeda Y, Shiga K, et
al: ATR inhibitor AZD6738 increases the sensitivity of colorectal
cancer cells to 5-fluorouracil by inhibiting repair of DNA damage.
Oncol Rep. 47:782022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsuoka K, Iimori M, Niimi S, Tsukihara
H, Watanabe S, Kiyonari S, Kiniwa M, Ando K, Tokunaga E, Saeki H,
et al: Trifluridine induces p53-dependent sustained G2 phase arrest
with its massive misincorporation into DNA and Few DNA strand
breaks. Mol Cancer Ther. 14:1004–1013. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka N, Sakamoto K, Okabe H, Fujioka A,
Yamamura K, Nakagawa F, Nagase H, Yokogawa T, Oguchi K, Ishida K,
et al: Repeated oral dosing of TAS-102 confers high trifluridine
incorporation into DNA and sustained antitumor activity in mouse
models. Oncol Rep. 32:2319–2326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohashi S, Kikuchi O, Nakai Y, Ida T, Saito
T, Kondo Y, Yamamoto Y, Mitani Y, Nguyen Vu TH, Fukuyama K, et al:
Synthetic lethality with trifluridine/tipiracil and checkpoint
kinase 1 inhibitor for esophageal squamous cell carcinoma. Mol
Cancer Ther. 19:1363–1372. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mayer RJ, Van Cutsem E, Falcone A, Yoshino
T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero
J, Komatsu Y, et al: Randomized trial of TAS-102 for refractory
metastatic colorectal cancer. N Engl J Med. 372:1909–1919. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukushima M, Suzuki N, Emura T, Yano S,
Kazuno H, Tada Y, Yamada Y and Asao T: Structure and activity of
specific inhibitors of thymidine phosphorylase to potentiate the
function of antitumor 2′-deoxyribonucleosides. Biochem Pharmacol.
59:1227–1236. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Emura T, Suzuki N, Fujioka A, Ohshimo H
and Fukushima M: Potentiation of the antitumor activity of alpha,
alpha, alpha-trifluorothymidine by the co-administration of an
inhibitor of thymidine phosphorylase at a suitable molar ratio
in vivo. Int J Oncol. 27:449–455. 2005.PubMed/NCBI
|
|
28
|
Iwata T, Uchino T, Koyama A, Johmura Y,
Koyama K, Saito T, Ishiguro S, Arikawa T, Komatsu S, Miyachi M, et
al: The G2 checkpoint inhibitor CBP-93872 increases the sensitivity
of colorectal and pancreatic cancer cells to chemotherapy. PLoS
One. 12:e01782212017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Emura T, Suzuki N, Yamaguchi M, Ohshimo H
and Fukushima M: A novel combination antimetabolite, TAS-102,
exhibits antitumor activity in FU-resistant human cancer cells
through a mechanism involving FTD incorporation in DNA. Int J
Oncol. 25:571–578. 2004.PubMed/NCBI
|
|
30
|
Vendetti FP, Lau A, Schamus S, Conrads TP,
O'Connor MJ and Bakkenist CJ: The orally active and bioavailable
ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of
cisplatin to resolve ATM-deficient non-small cell lung cancer in
vivo. Oncotarget. 6:44289–44305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Checkley S, MacCallum L, Yates J, Jasper
P, Luo H, Tolsma J and Bendtsen C: Bridging the gap between in
vitro and in vivo: Dose and schedule predictions for the ATR
inhibitor AZD6738. Sci Rep. 5:135452015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dillon MT, Barker HE, Pedersen M, Hafsi H,
Bhide SA, Newbold KL, Nutting CM, McLaughlin M and Harrington KJ:
Radiosensitization by the ATR inhibitor AZD6738 through generation
of acentric micronuclei. Mol Cancer Ther. 16:25–34. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kataoka Y, Iimori M, Niimi S, Tsukihara H,
Wakasa T, Saeki H, Oki E, Maehara Y and Kitao H: Cytotoxicity of
trifluridine correlates with the thymidine kinase 1 expression
level. Sci Rep. 9:79642019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rothkamm K, Christiansen S, Rieckmann T,
Horn M, Frenzel T, Brinker A, Schumacher U, Stein A, Petersen C and
Burdak-Rothkamm S: Radiosensitisation and enhanced tumour growth
delay of colorectal cancer cells by sustained treatment with
trifluridine/tipiracil and X-rays. Cancer Lett. 493:179–188. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wilson Z, Odedra R, Wallez Y, Wijnhoven
PWG, Hughes AM, Gerrard J, Jones GN, Bargh-Dawson H, Brown E, Young
LA, et al: ATR inhibitor AZD6738 (Ceralasertib) exerts antitumor
activity as a monotherapy and in combination with chemotherapy and
the PARP inhibitor olaparib. Cancer Res. 82:1140–1152. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goto H, Izawa I, Li P and Inagaki M: Novel
regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good
candidate for anti-cancer therapy? Cancer Sci. 103:1195–1200. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bernabe-Ramirez C, Patel R, Chahal J and
Saif MW: Treatment options in BRAF-mutant metastatic colorectal
cancer. Anticancer Drugs. 31:545–557. 2020. View Article : Google Scholar : PubMed/NCBI
|