Introduction
Epstein-Barr virus (EBV) belongs to the human
γ-herpes virus, which infects ~90% of the human population.
Although the majority of EBV carriers have latent infection with no
significant clinical symptoms (1,2), EBV
infection can also drive human B cell proliferation and
transformation, triggering numerous neoplasm types and autoimmune
diseases, and a compromised immune system worsens the problem
(3). It has been reported that EBV
is associated with a variety of tumors, including Burkitt's
lymphoma, Hodgkin's lymphoma, non-Hodgkin lymphoma [such as natural
killer (NK)/T-cell lymphoma] (4)
and nasopharyngeal carcinoma (2,5),
suggesting that EBV may be a potential driving force for tumor
development. However, the mechanism behind this remains unclear,
and the development of anti-EBV targeting strategies remains
challenging (6,7).
EBV-encoded latent membrane protein 1 (LMP1), which
is a direct target gene of EBV nuclear antigen 2, has been reported
to be an oncogene (8) that plays a
major role in the regulation of tumor-related metabolism (9). LMP1 potentiates tumor growth through
activation of glycolysis (10),
mitochondrial function (11),
hypoxia-inducible factor 1 signaling (12), sterol regulatory element-binding
transcription factor 1 (SREBP1)-mediated lipogenesis (13) and programmed death-ligand 1
upregulation (14). Additionally,
LMP1 activates the nuclear factor-κB (NF-κB) signaling pathway
(15); NF-κB is then translocated
into the nucleus and subsequently activates related target genes
(16). It has been reported that
LMP1 promotes tumorigenesis through NF-κB-mediated peroxisome
proliferator-activated receptor-γ coactivator-1β (PGC1β)
upregulation (17). The PGC1
family, which includes PGC1α and PGC1β (18), plays an important role in
maintaining the balance of glucose and lipids involved in energy
metabolism through regulating various target genes as a
transcriptional coactivator (19).
Previous studies have reported that PGC1β expression triggers
tumorigenesis through mitochondrial metabolism, glycolysis and
redox balance (20–22). Our group has recently reported that
PGC1β promotes tumor growth via lactate dehydrogenase A
(LDHA)-mediated glycolysis (23) in
addition to SREBP1-mediated hexokinase domain component 1 (HKDC1)
upregulation, subsequently increasing mitochondrial metabolism
(24), although the detailed
mechanism for PGC1β-mediated tumorigenesis remains largely
unknown.
Our group has previously reported that LMP1 promotes
tumorigenesis through PGC1β upregulation (17), while numerous forms of lymphoma
exhibit upregulated PGC1β expression in the absence of EBV/LMP1.
The present study aimed to investigate the potential mechanism for
transient EBV/LMP1 exposure-mediated persistent PGC1β activation
through epigenetic changes. The current in vitro findings
revealed that LMP1 knockdown in EBV-positive cells did not result
in a significant change in PGC1β expression or subsequent cell
proliferation.
In the present study, the potential mechanism for
PGC1β-mediated mitochondrial fission by upregulation of protein
dynamin-related protein 1 (DRP1) was evaluated (11). In addition, the potential reason why
transient exposure to either EBV or LMP1 causes persistent PGC1β
expression as well as the subsequent consequence of PGC1β
upregulation on tumor growth was investigated in both in
vitro and in vivo mouse model. The SNK6 cells were used
as tumor cell line to evaluate the potential effect on tumor growth
when the related genes were knocked down, while human primary
hematopoietic stem cells (HSC) were used to evaluate the potential
effect on tumorigenesis when the related genes were overexpressed.
Finally, the potential effects of EBV, LMP1 and PGC1 β on tumor
growth were evaluated through in vivo xenograft mouse model.
It was concluded that transient LMP1 expression caused persistent
PGC1β upregulation and tumor growth through epigenetic
modifications, thus elucidating the potential mechanism for
transient EBV/LMP1 exposure-mediated tumorigenesis while explaining
why numerous forms of lymphoma show absence of EBV/LMP1.
Materials and methods
Reagents and materials
Human HSC were isolated from healthy peripheral
blood mononuclear cells (PBMC) cells using EasySep™ Human
Progenitor Cell Enrichment kit with Platelet Depletion (cat. no.
19356), while human NK cells were isolated from healthy PBMC cells
using EasySep™ Human NK Cell Isolation kit (Stemcell Technologies,
Inc.) according to the manufacturers' instructions. A number of
PBMCs, NK cells or isolated healthy HSC were conditionally
immortalized using hTERT lentivirus vector with an extended life
span to achieve a higher transfection efficiency and experimental
stability (25,26). Tumor cell lines, including B95-8,
Namalwa, HANK1, SNT8 and SNK6, were purchased from American Type
Culture Collection. The B95-8 cell line was used for EBV viral
production and subsequent concentration for the infection of HSC,
PBMCs or NK cells according to a previously described protocol
(27). The EBV LMP1 adenovirus for
LMP1 transient infection and related empty adenovirus control were
kindly provided by Dr Haimou Zhang (Hubei University, China).
Antibodies against CdxA (Cdx1; cat. no. sc-515146), Ets1 (c-Ets;
cat. no. sc-55581), forkhead box D3 (HFH2; cat. no. sc-517206),
GA-binding protein α (GABPα; cat. no. sc-28312), GATA1 (cat. no.
sc-265), GATA2 (cat. no. sc-267), Ki-67 (cat. no. sc-101861),
nuclear respiratory factor 1 (NRF1; cat. no. sc-101102), NF-κB p65
(cat. no. sc-398442) and organic cation transporter 1 (Oct1; cat.
no. sc-8024) were obtained from Santa Cruz Biotechnology, Inc.
Antibodies against DRP1 (cat. no. ab140494), optic atrophy 1 (OPA1;
cat. no. ab157457), mitofusin 2 (MFN2; cat. no. ab56889) and PGC1β
(cat. no. ab240188) were obtained from Abcam. Antibodies against
EBV LMP1 (cat. no. NBP1-79009) and 8-oxo-deoxyguanosine (dG) (cat.
no. 4354-MC-050) were obtained from Novus Biologicals, Ltd. The
small interfering RNA (siRNA) for GABPα and NRF1, and scrambled
control siRNA were purchased from Ambion (Thermo Fisher Scientific,
Inc.), and the target sequences of siRNA were provided in Table SI. The expanded section of
Materials and methods is available in Appendix S1.
Preparation of DRP1 reporter
constructs
The DRP1 gene promoter (2 kb upstream) from Ensembl
gene ID: DNM1L-201 ENST00000266481.10 (for DRP1) (with the
following link: http://uswest.ensembl.org/Homo_sapiens/Transcript/Summary?db=core;g=ENSG00000087470;r=12:32679303-32744350;t=ENST00000266481),
was amplified by PCR from SNK6 genomic DNA, and then subcloned into
the pGL3-basic vector (cat. no. E1751; Promega Corporation) by
using the following primers with restriction sites for MluI
and XhoI, respectively: DRP1 forward, 5′-gcgc-acgcgt-agt tgg
ggc cac agg tat gca-3′ (MluI) and reverse,
5′-atcg-ctcgag-aca gtt cgc ctc ctt cct cct-3′ (XhoI).
Preparation of gene knockdown
lentivirus
The shRNA lentivirus particles for human PGC1β,
human NFκB-p65, EBV LMP1 and non-target control were designed and
purchased from MilliporeSigma, and all the shRNA target sequences
were provided in Table SI. These
lentiviruses were used for infection of tumor cell lines (e.g.
SNK6). The treated cells were cultured in the presence of 10 µg/ml
puromycin, and the gene knockdown efficiency was evaluated by
reverse transcription-quantitative PCR (RT-qPCR) based on an mRNA
decrease of >65% compared with that in the control group. The
primer sequences are shown in Table
SII (23,24).
Immunostaining
Treated SNK6 cells were transferred to coated cover
slips, and then fixed with 3.7% formaldehyde, incubated with 1% BSA
and 0.2% Triton X-100 for 1 h, and blotted with 40 µg/ml primary
antibody against PGC1β, Ki-67 or 8-oxo-dG for 4 h. In
triple-staining experiments, the live cells were stained with
MitoTracker™ Green FM before fixation. After thorough washing, the
cells were incubated with either FITC or Texas Red-labeled
secondary antibody (1:1,000) for 1 h, and the cell nuclei were
further stained with 300 nM DAPI (cat. no. D9542; Sigma-Aldrich;
Merck KGaA). The cells were visualized under a confocal microscope
and the staining was quantitated by ImageJ 1.52v software (National
Institutes of Health).
Mitochondria morphology by electron
microscopy
Treated cells were fixed with 2.5% glutaraldehyde at
room temperature for 15 min, and then dehydrated by using a series
of increasing concentrations of ethanol, and embedded in epoxy
resin and propylene oxide overnight. The 70 nm-thick sample
sections were then stained with lead citrate, and mitochondria
morphology was detected by double-blinded technicians using a
transmission electron microscope (28).
DNA methylation analysis
DNA methylation on the human PGC1β promoter was
evaluated by methylation-specific PCR according to a previously
described method with minor modifications (29–31).
Genomic DNA was extracted and purified from treated cells, and then
treated by bisulfite modification with EpiJET Bisulfite Conversion
kit (cat. no. K1461; Thermo Fisher Scientific, Inc.), and the DNA
was then amplified by using the following primers: Methylated
primers: Forward 5′-ttt tta aag tgt tgg gat tat agg c-3′ and
reverse 5′-acg tta cgt taa cgc taa acg a-3′; and unmethylated
primers: Forward 5′-ttt aaa gtg ttg gga tta tag gtg t-3′ and
reverse 5′-tca cat tac att aac act aaa caa a-3′. The product sizes
were as follows: 142 bp (methylated) with melting temperature (Tm)
66.4°C and 142 bp (unmethylated) with Tm 65.8°C; CpG island size:
197 bp. The methylation results were calculated by normalization of
the unmethylated PCR results (32).
In vivo mouse experiments
Tumor cells were treated, and then 2×106
viable cells in 0.1 ml PBS were injected into the lateral tail vein
of mice. The null mice receiving tail vein injections were
separated into the following 4 groups (n=9): i) Group 1 [control
(CTL)], conditionally immortalized HSC at passage #6 after vehicle
lentivirus infection; ii) group 2 (EBV), conditionally immortalized
HSC at passage #6 after EBV infection; iii) group 3 (LMP1↑),
conditionally immortalized HSC at passage #6 after LMP1 adenovirus
infection; and iv) group 4 (EBV/shPGC1β), conditionally
immortalized HSC at passage #6 after EBV infection and shPGC1β
lentivirus infection in the presence of 10 µg/ml puromycin. Mouse
survival was monitored and calculated; the tumor growth was
calculated; the lungs were isolated and stained with hematoxylin
and eosin (H&E); and the gene expression and superoxide anion
(O2−.) release from tumor tissues were
evaluated (7,23,33).
Statistical analysis
The mean level and standard deviation (SD) were
calculated and presented, and each experiment was performed for at
least 4 independent times (n=4) unless otherwise mentioned. The
data was analyzed as normal distribution using Shapiro-Wilk test to
evaluate the normality of the data in SPSS 22 software (34), and the one-way ANOVA (analysis of
variance) together with Tukey-Kramer test were employed to
determine significant difference of different groups. The mouse
survival curve was established by Kaplan-Meier survival analysis
followed by the log-rank test through SPSS 22 software, and
P<0.05 was considered to indicate a statistically significant
difference (33).
Results
LMP1 knockdown in EBV-positive tumor
cells does not reduce PGC1β-mediated tumorigenesis
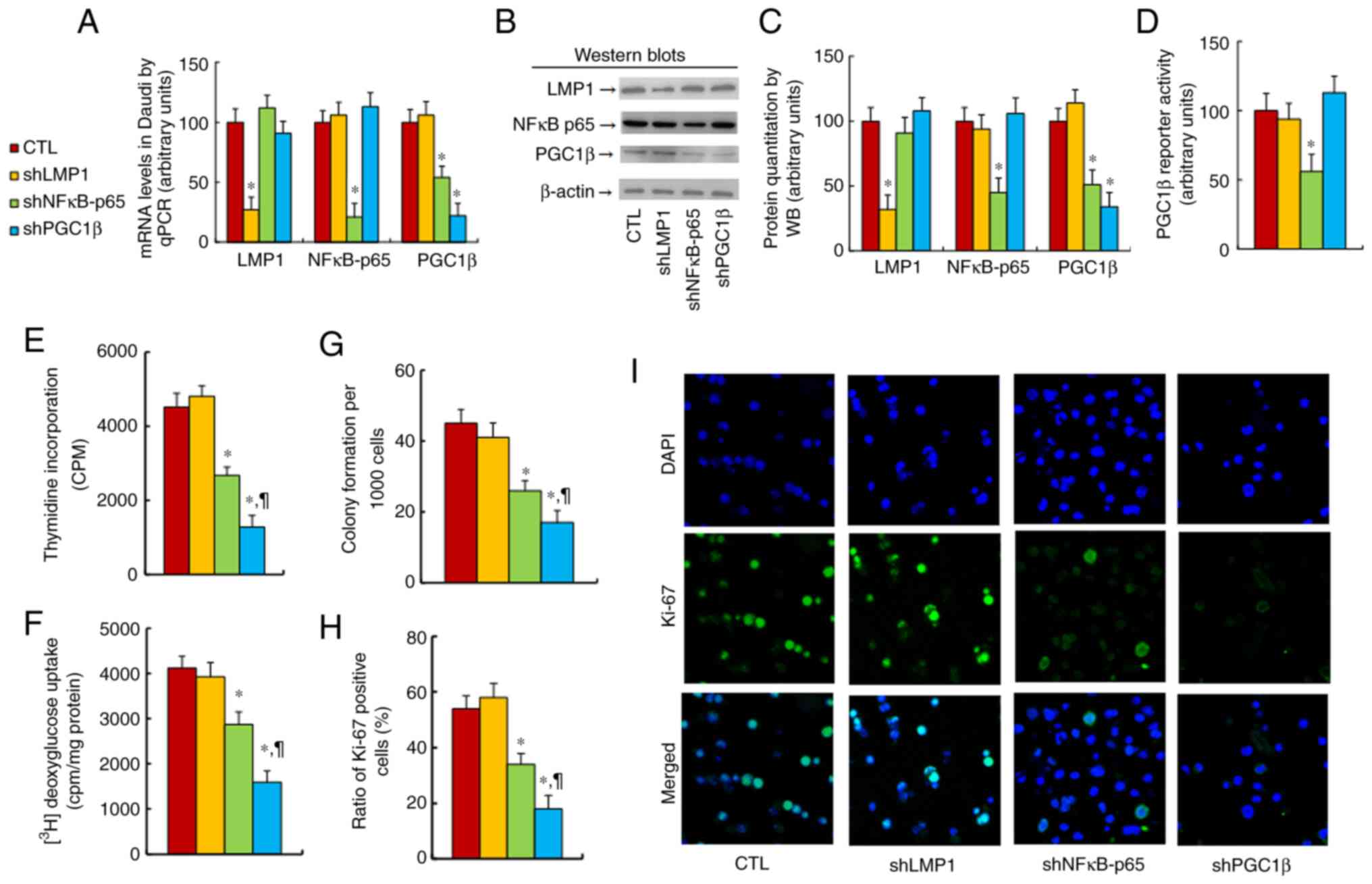
The effect of LMP1 on PGC1β expression was
determined. SNK6 cells were treated with CTL, shLMP1, shNF-κB-p65
or shPGC1β lentivirus before being harvested for biological assays.
It was found that knockdown of either NF-κB-p65 (shNF-κB-p65) or
PGC1β (shPGC1β) significantly reduced PGC1β mRNA levels in SNK6
cells compared with the findings in the CTL group, while LMP1
knockdown (shLMP1) had no effect (Fig.
1A). Next, the levels of proteins corresponding to the LMP1,
NF-κB-p65 and PGC1β genes were measured, and it was revealed that
the protein levels were similar to the mRNA levels (Figs. 1B and C, and S1A). Next, PGC1β reporter activity was
evaluated in the cells, and it was identified that shNF-κB-p65
treatment significantly reduced PGC1β reporter activity, while
shLMP1 and shPGC1β showed no effect (Fig. 1D). Gene expression was also
determined in other EBV-positive cell lines, including HANK1
(Fig. S2A) and SNT8 (Fig. S2B) cells, and it was found that
treatment with either shNF-κB-p65 or shPGC1β significantly reduced
the PGC1β mRNA levels, while shLMP1 showed no effect.
In addition, the potential effects of the treatment
on epigenetic changes on the PGC1β promoter were measured, and it
was observed that none of the treatments had a significant effect
on histone 3 methylation (Fig.
S3A), DNA methylation (Fig.
S3B), histone 3 acetylation (Fig.
S3C) or histone 4 methylation (Fig. S3D). Finally, the effect of
LMP1/PGC1β expression on tumor cell growth was explored, and it was
demonstrated that shLMP1 treatment had no effect, while treatment
with either shNF-κB-p65 or shPGC1β significantly reduced cellular
proliferation, as evaluated by thymidine incorporation (Fig. 1E), glucose uptake (Fig. 1F), colony formation assay (Fig. 1G) and Ki-67-positive cell rate
(Fig. 1H and I). It was concluded
that LMP1 knockdown did not affect PGC1β expression or its
subsequent tumorigenesis in EBV-positive cells.
PGC1β regulates DRP1 expression
through GABPα/NRF1 binding sites on the DRP1 promoter
The current study determined the mechanism behind
PGC1β-mediated DRP1 regulation in immortalized HSC. Different
progressive 5′-promoter deletion constructs for DRP1 were generated
and transfected into HSC together with full-length DRP1 reporter
plasmids. The cells were then infected with either PGC1β or empty
lentivirus (CTL) for luciferase activity assay, and the results
showed that PGC1β-induced DRP1 reporter activity did not change
among the −2,000, −1,600, −1,200, −800, −700, −600, −500 and −400
deletion constructs (numbered according to the Ensembl gene ID:
DNM1L-201 ENST00000266481.10), while PGC1β-induced reporter
activation was partly decreased at −300 deletion and completely
disappeared at the −200-deletion construct. This indicates that the
PGC1β-responsive binding element is located in the range of −400 to
−200 on the DRP1 promoter (Fig.
2A).
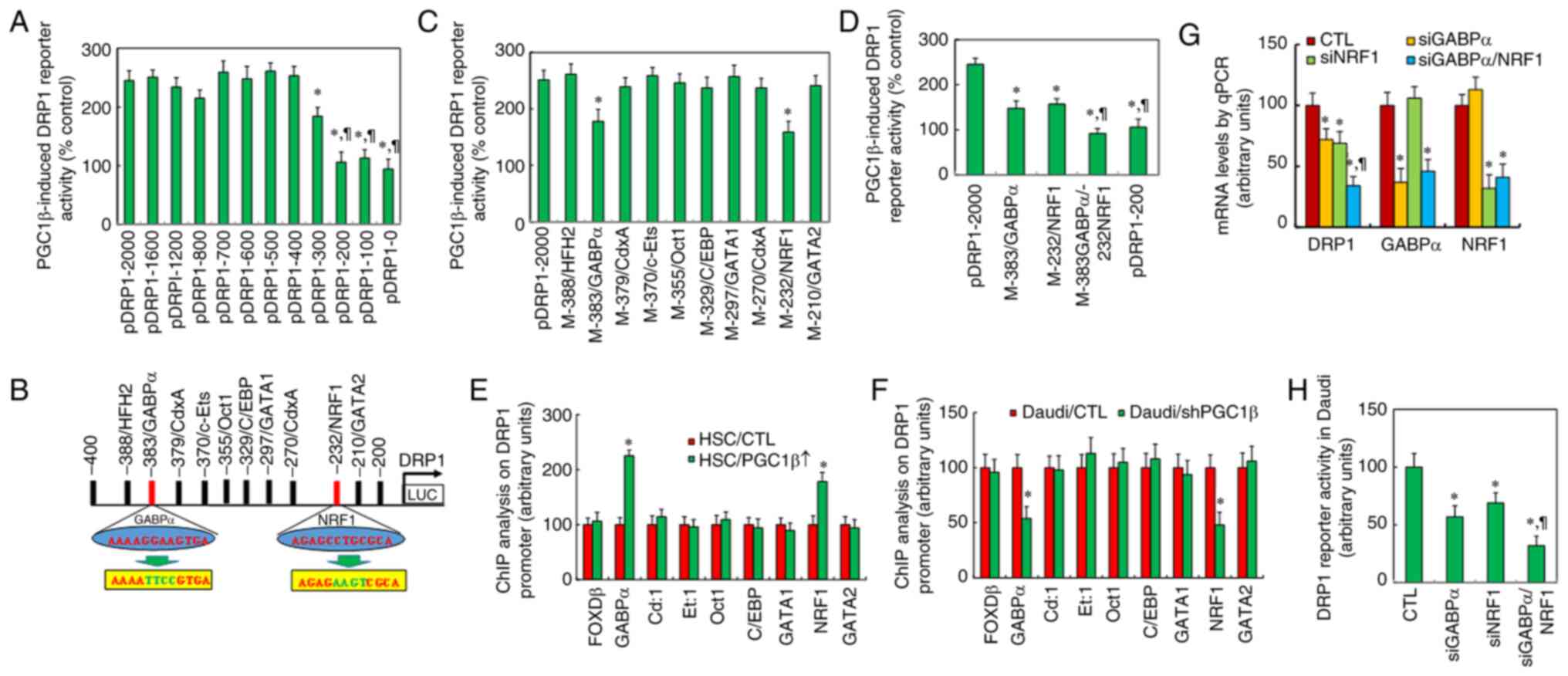 | Figure 2.Peroxisome proliferator-activated
receptor-γ coactivator-1β regulates DRP1 expression through
GABPα/NRF1 binding sites on the DRP1 promoter. (A) Luciferase
activities for the indicated deletion reporter constructs in
immortalized HSC. *P<0.05 vs. pDRP1-2,000 group;
¶P<0.05 vs. pDRP1-300 group (n=5). (B) Schematic
model for potential transcriptional binding motif in the range of
−400 to −200 on the DRP1 promoter, with binding motif for GABPα and
NRF1 in red and related mutation sites in green. (C) Luciferase
activity for full-length DRP1 (pDRP1-2000) or the indicated
mutation reporter constructs. *P<0.05 vs. pDRP1-2,000 group
(n=5). (D) Luciferase activities for single or double mutants for
GABPα (−383) and/or NRF1 (−232). *P<0.05 vs. pDRP1-2,000 group;
¶P<0.05 vs. M-383/GABPα group (n=5). (E) ChIP
analysis (n=5). *P<0.05 vs. HSC/CTL group. (F) ChIP analysis
(n=5). *P<0.05 vs. SNK6/CTL group. (G and H) Small interfering
RNA was used to knockdown either GABPα or NRF1 in SNK6 cells for
further analysis. (G) mRNA levels were determined by quantitative
PCR (n=4). (H) DRP1 reporter activity (n=5). *P<0.05 vs. CTL
group. DRP1, dynamin-related protein 1; GABPα, GA-binding protein
α; NRF1, nuclear respiratory factor 1; CTL, control; HSC,
hematopoietic stem cells; ChIP, chromatin immunoprecipitation. |
All the potential transcription factor binding sites
in the range of −400 to −200 were analyzed from the database, and
the following motifs were identified: HFH2, GABPα (marked in red),
CdxA, c-Ets, Oct1, CCAAT-enhancer-binding protein, GATA1, NRF1
(marked in red) and GATA2 (Fig.
2B). All the single mutations were then generated on the full
length of the DRP1 reporter construct (p-DRP1-2000) for these
binding motifs for the luciferase reporter assay, and the results
showed that only single mutants for GABPα (from ggaa to ttcc, in
green at −383) and NRF1 (from cctg to aagt, in green at −232)
showed a significant reduction in reporter activity, indicating
that GABPα and NRF1 may be responsible for PGC1β-induced DRP1
activation (Fig. 2C). Next, GABPα
or NRF1 single or double mutants (M-383GABPα/-232NRF1) were
transfected for the reporter assay, and it was found that both
single mutants partly reduced, while the double mutants completely
reduced, the PGC1β-induced DRP1 reporter activation (Fig. 2D).
The binding abilities were then determined through
chromatin immunoprecipitation analysis on the DRP1 promoter. The
results revealed that both GABPα and NRF1 increased the binding
ability after PGC1β infection in HSC compared with that of the CTL
group, while other transcriptional factors had no effect (Fig. 2E). Additionally, both GABPα and NRF1
showed reduced binding ability after shPGC1β lentivirus infection
in SNK6 cells compared with that in the CTL group, while other
transcriptional factors had no effect (Fig. 2F).
Finally, the potential effect of GABPα and NRF1 on
DRP1 expression in SNK6 cells was determined by siRNA techniques,
and it was observed that GABPα and NRF1 were knocked down
successfully by siGABP and siNRF1 respectively. Additionally, the
DRP1 mRNA level was significantly decreased by single knockdown of
either GABPα or NRF1 compared with that of the CTL group, and the
DRP1 mRNA level was further decreased by double knockdown
(siGABPα/NRF1) compared with that of the single siGABPα group
(Fig. 2G). The DRP1 reporter
activity was also determined, and it was found to be similar to the
DRP1 mRNA levels (Fig. 2H). It was
concluded that DRP1 expression was regulated by PGC1β through GABPα
and NRF1 binding sites on the DRP1 promoter.
EBV/LMP1 exposure causes persistent
epigenetic modifications on the PGC1β promoter
The present study evaluated the potential effect of
LMP1 expression on epigenetic changes on the PGC1β promoter.
Conditionally immortalized HSC were treated with CTL, transient EBV
infection, transient LMP1 infection by LMP1 adenovirus (LMP↑), or
EBV infection together with permanent PGC1β knockdown through
shPGC1β lentivirus in the presence of 10 µg/ml puromycin
(EBV/shPGC1β). The cells were continuously cultured from passage #1
to #6 before being harvested for analysis on different passages.
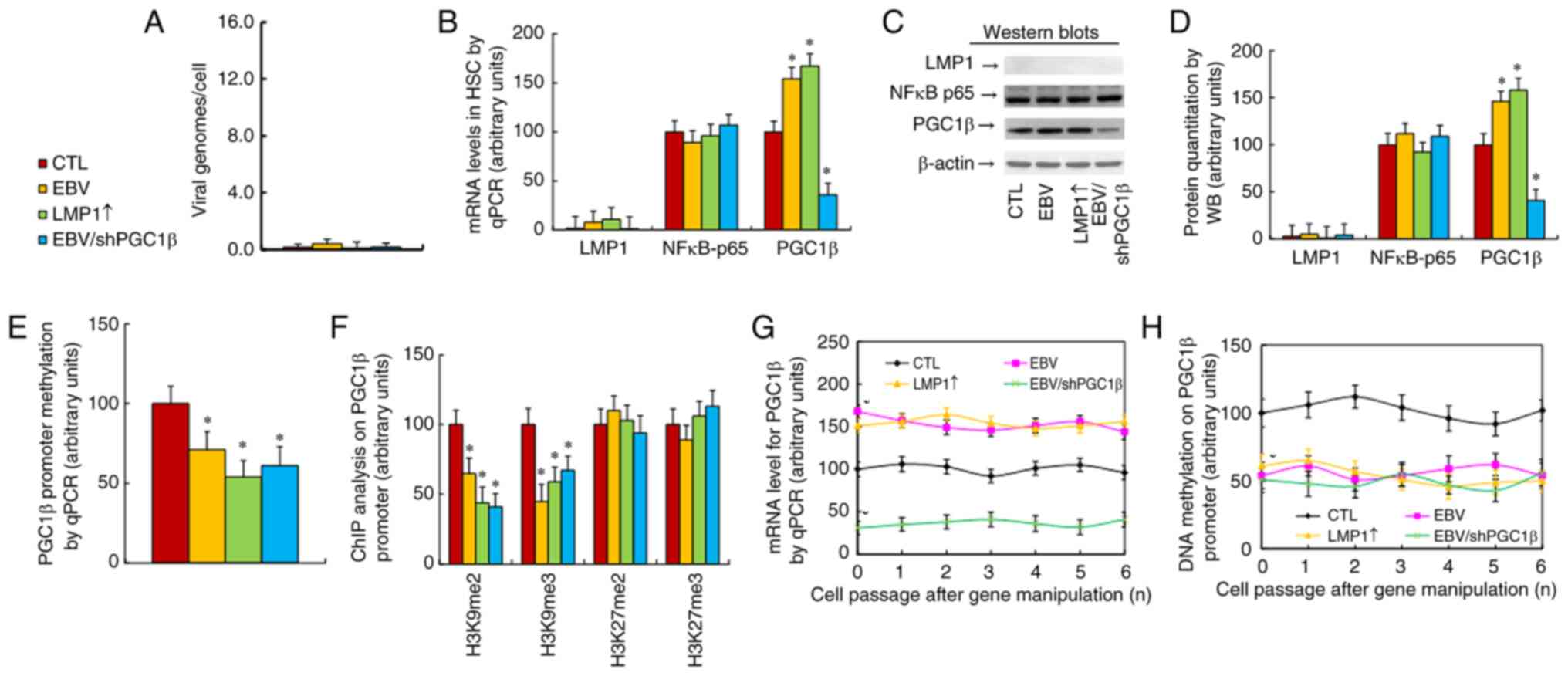
The properties of cells at passage #1 were first evaluated, and the
results revealed that EBV infection, including EBV and EBV/shPGC1β
treatments, significantly increased the quantity of EBV genome
copies compared with those of the CTL group (Fig. S4A). Additionally, treatments with
EBV, LMP1↑ and EBV/shPGC1β significantly increased LMP1 mRNA
levels. Moreover, treatments with EBV and LMP1↑ significantly
increased, while EBV/shPGC1β treatment significantly decreased, the
PGC1β mRNA levels. None of treatments had a significant effect on
NF-κB-p65 expression compared with that of the CTL group (Fig. S4B). The effect on epigenetic
changes on the PGC1β promoter at passage #1 was also determined,
and it was found that treatments with EBV, LMP1↑ and EBV/shPGC1β
significantly decreased the epigenetic modifications on H3K9me2 and
H3K9me3 (Fig. S4C) in addition to
DNA methylation (Fig. S4D). These
treatments had no effect on histone 4 methylation (Fig. S4E) or histone 3 acetylation
(Fig. S4F) compared with the CTL
group.
Next, the properties of cells at passage #6 were
determined. The results showed that the EBV genome was almost
non-detectable upon treatment with EBV, LMP1↑ or EBV/shPGC1β after
continuous culturing for 6 passages (Fig. 3A). Additionally, LMP1 mRNA was not
detectable, while the NF-κB-p65 mRNA levels showed no changes. On
the other hand, PGC1β mRNA remained increased after EBV and LMP1↑
treatment, while it remained decreased upon EBV/shPGC1β treatment
compared with that in the CTL group (Fig. 3B). The protein levels were also
measured, and the protein expression was similar to the mRNA levels
(Figs. 3C and D, and S1B). Epigenetic changes on the PGC1β
promoter were then determined, and it was found that treatments
with EBV, LMP1↑ and EBV/shPGC1β significantly decreased DNA
methylation (Fig. 3E) in addition
to epigenetic modifications on H3K9me2 and H3K9me3 (Fig. 3F), while having no effect on histone
3 acetylation (Fig. S5A) or
histone 4 methylation (Fig. S5B)
compared with the findings in the CTL group, where the epigenetic
modifications were similar to those noted in cells at passage
#1.
Finally, the PGC1β levels in cells from passage #1
to #6 were measured, and it was found that treatments with EBV and
LMP1↑ significantly increased, while EBV/shPGC1β decreased, PGC1β
mRNA. This remained persistent from passage #1 to #6 (Fig. 3G). The present study also measured
DNA methylation on the PGC1β promoter, and found that treatments
with EBV, LMP1↑ or EBV/shPGC1β significantly decreased DNA
methylation, which remained persistent from passage #1 to #6
(Fig. 3H). The potential effect of
LMP1 expression on PGC1β expression in other cells was also
determined, and it was revealed that treatments with EBV and LMP1↑
significantly increased, while EBV/shPGC1β treatment significantly
decreased, PGC1β expression in both PBMCs (Fig. S6A) and NK cells (Fig. S6B); this remained persistent from
passage #1 to #6. Thus, it was concluded that transient EBV/LMP1
exposure caused persistent epigenetic modifications on the PGC1β
promoter, which subsequently resulted in persistent PGC1β
upregulation even in the absence of EBV/LMP1.
EBV/LMP1 exposure ameliorates
oxidative stress, while PGC1β knockdown reverses this effect
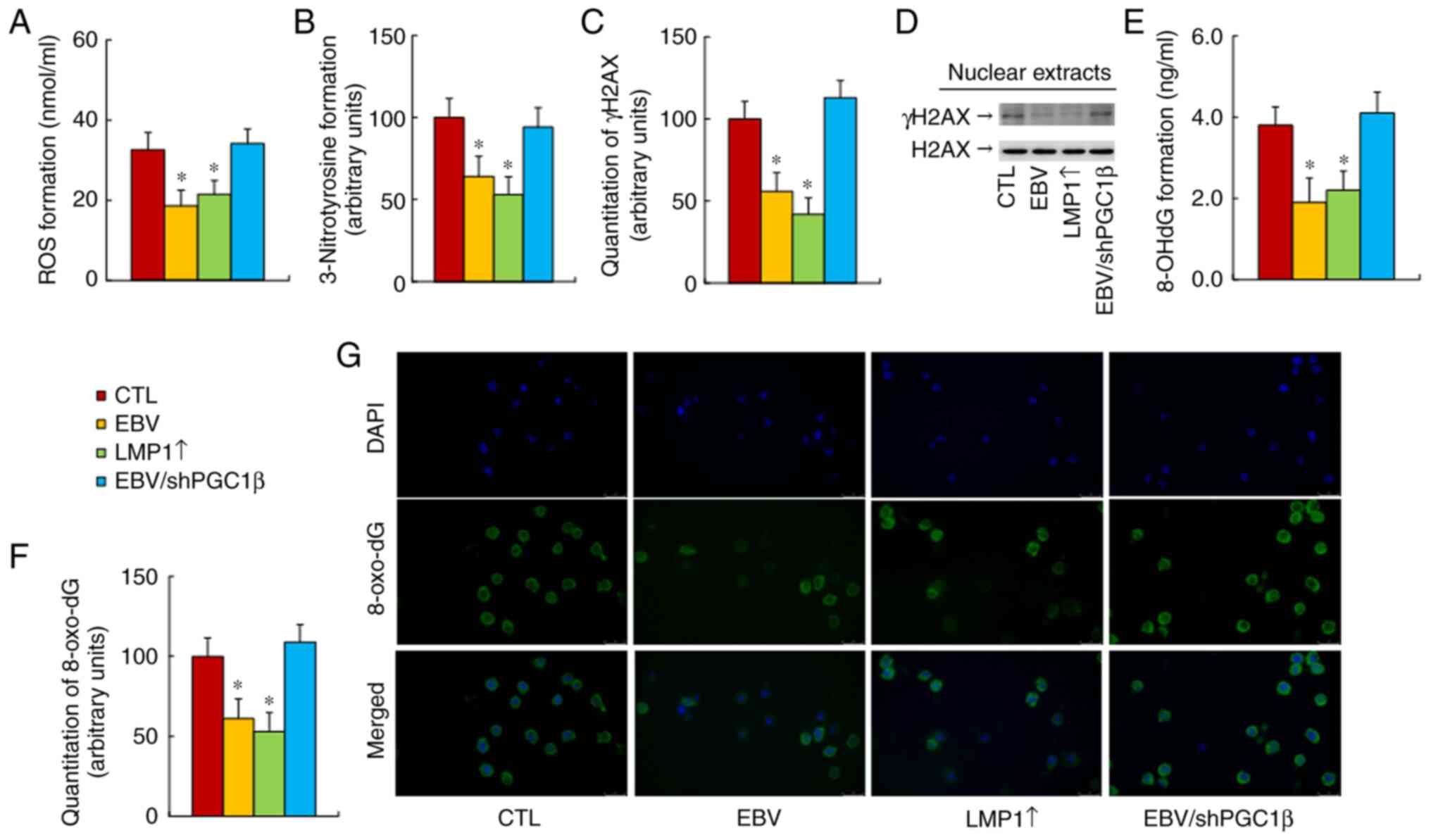
The current study evaluated the effect of EBV/LMP
expression on oxidative stress in treated cells at passage #6, and
found that both EBV and LMP1↑ treatments significantly decreased
reactive oxygen species (ROS) (Fig.
4A) and 3-nitrotyrosine formation (Fig. 4B) formation compared with those of
the CTL group, while PGC1β knockdown (EBV/shPGC1β) completely
reversed this effect. The oxidative stress-mediated DNA damage was
also determined, and the results showed that both EBV and LMP1↑
treatments significantly decreased γH2AX (Figs. 4C and D, and S1C), 8-OHdG (Fig. 4E) and 8-oxo-dG (Fig. 4F and G) formation, while PGC1β
knockdown completely reversed this effect. Thus, it was concluded
that EBV/LMP1 exposure-mediated PGC1β expression ameliorated
oxidative stress.
EBV/LMP1 exposure potentiates
mitochondrial function, while PGC1β knockdown reverses this
effect
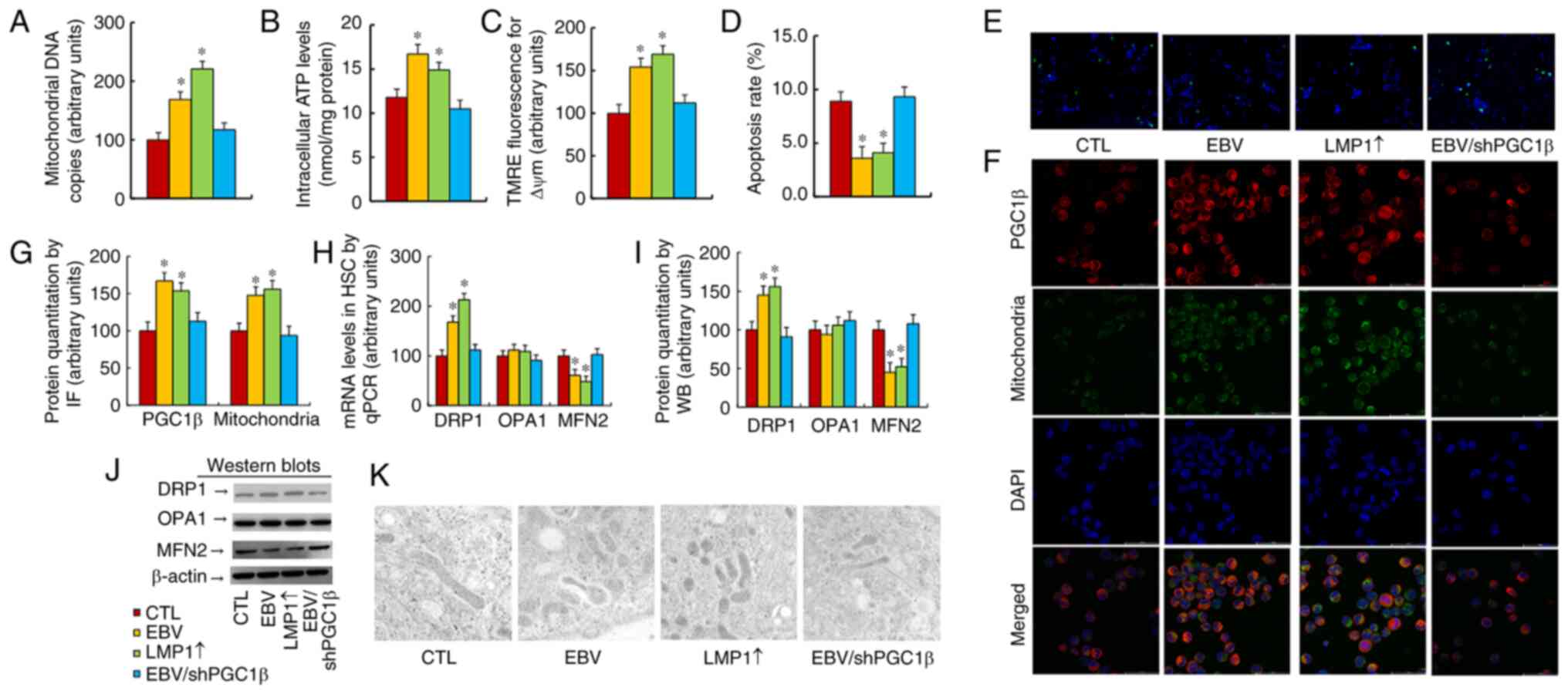
The present study evaluated the effect of EBV/LMP1
expression on mitochondrial function, and found that both EBV and
LMP1↑ treatments significantly increased mitochondrial DNA copies
(Fig. 5A), intracellular ATP level
(Fig. 5B) and mitochondrial
membrane potential (Fig. 5C), in
addition to decreasing the apoptotic rate (Figs. 4D and 5E) compared with the findings in the CTL
group. Notably, PGC1β knockdown completely reversed this
effect.
Mitochondrial mass and gene expression were then
evaluated, and the results revealed that both EBV and LMP1↑
treatments significantly increased PGC1β expression and
mitochondrial mass by immunostaining (Fig. 5F and G) compared with those of the
CTL group. Both EBV and LMP1↑ treatments significantly increased
DRP1 expression and decreased MFN2 expression, but had no effect on
OPA1 expression (Figs. 5H-J and
S1D). Finally, changes in
mitochondrial morphology were evaluated by using an electron
microscope, and it was found that both EBV and LMP1↑ treatments
significantly increased mitochondrial fission compared with that of
the CTL group, while PGC1β knockdown completely reversed this
effect (Fig. 5K). Therefore, it was
concluded that EBV/LMP1 exposure-mediated PGC1β expression
potentiated mitochondrial function by upregulation of mitochondrial
fission.
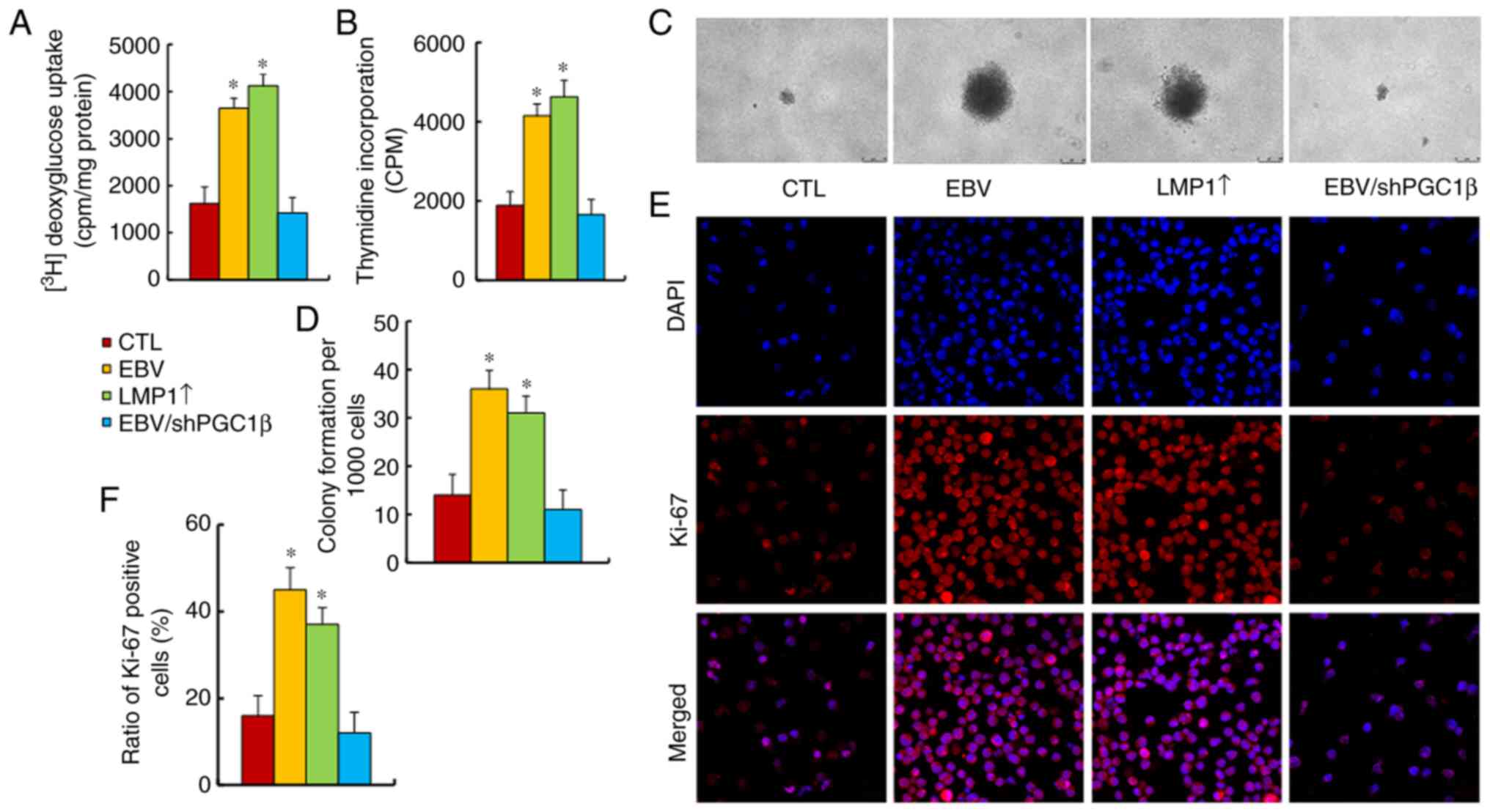
EBV/LMP1 exposure potentiates tumor
cell proliferation, while PGC1β knockdown reverses this effect
The present study evaluated the effect of EBV/LMP1
exposure on tumor cell proliferation, and found that both EBV and
LMP1↑ treatments significantly increased
[3H]-2-deoxyglucose uptake (Fig. 6A), cellular proliferation by
thymidine incorporation (Fig. 6B),
colony formation (Fig. 6C and D)
and Ki-67 positive cells (Fig. 6E and
F), compared with those in the CTL group. Of note, PGC1β
knockdown completely reversed this effect. Thus, it was concluded
that EBV/LMP1 exposure-mediated PGC1β expression potentiated tumor
cell proliferation.
EBV/LMP1 exposure potentiates tumor
growth in vivo in mice, while PGC1β knockdown reverses this
effect
The current study determined the potential effect of
EBV/LMP1 expression on in vivo tumor growth. Treated cells
at passage #6 were harvested for tail vein injection to monitor the
process of tumor growth. Gene expression was first determined in
the tumor tissues, and it was found that cells that received
treatment with either EBV or LMP1↑ showed significantly increased
mRNA levels of PGC1β and DRP1 but decreased mRNA levels of MFN2
compared with those of the CTL group. PGC1β knockdown completely
reversed this effect (Fig. 7A).
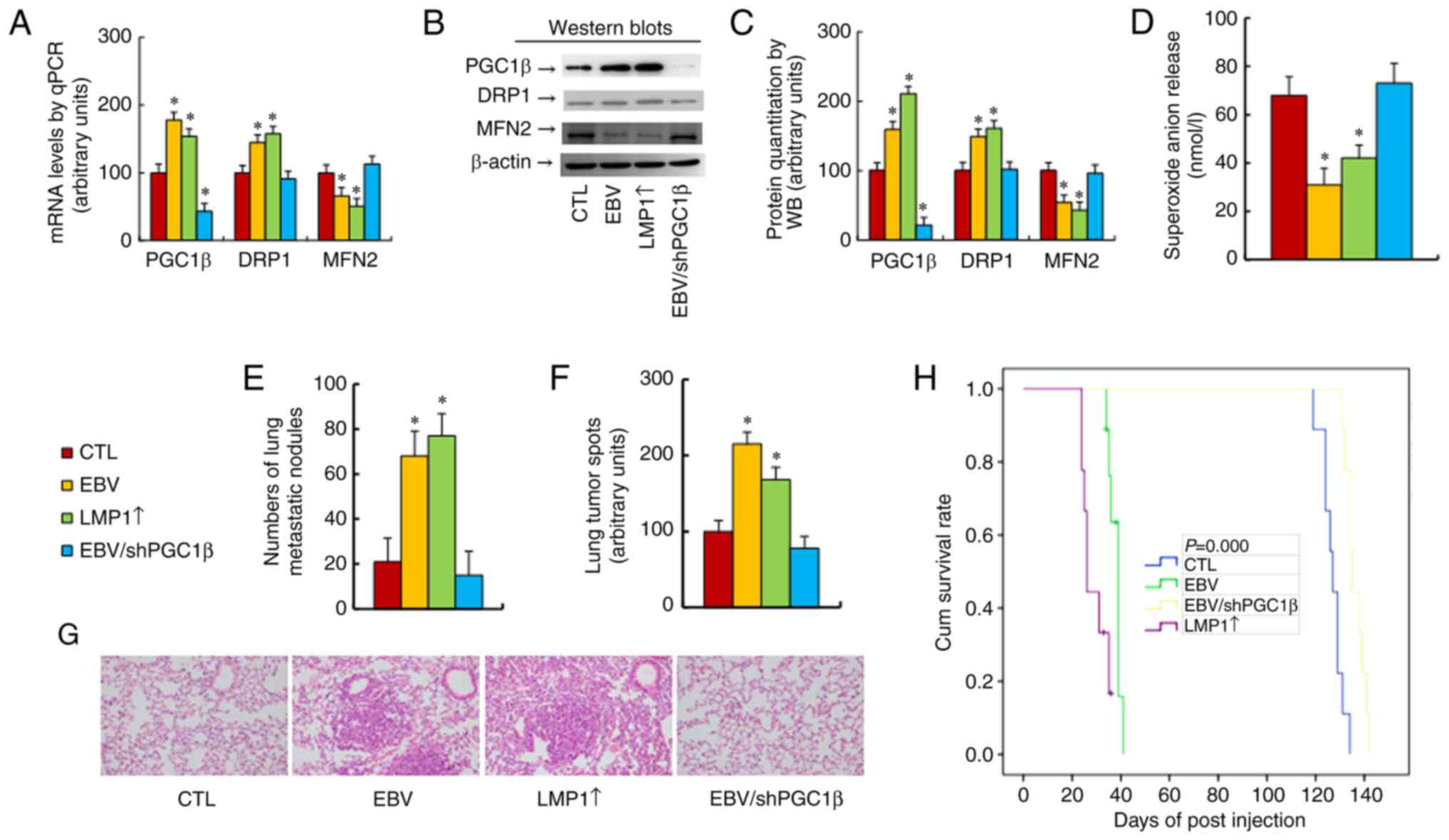 | Figure 7.EBV/LMP1 exposure potentiates tumor
growth in vivo in mice, while PGC1β knockdown reverses this
effect. Conditionally immortalized hematopoietic stem cells were
treated with CTL, EBV, LMP1 adenovirus (LMP1↑) or EBV together with
shPGC1β lentivirus in the presence of 10 µg/ml puromycin
(EBV/shPGC1β). The cells were continuously cultured, and cells at
passage #6 were harvested for tail vein injection to monitor the
process of tumor growth in mice. (A-D) Isolated tumor tissues for
biological assays. (A) mRNA levels, as determined by quantitative
PCR (n=4). (B) Representative western blot bands. (C) Protein
quantitation of panel B (n=5). (D) Superoxide anion release (n=5).
(E) Tumor colony formation in lungs (n=9). (F) Quantification of
lung tumor spots (n=5). (G) Representative image of hematoxylin and
eosin staining for panel F. (H) Kaplan-Meier analysis of mouse
survival (n=9). *P<0.05 vs. CTL group. EBV, Epstein-Barr virus;
LMP1, latent membrane protein 1; PGC1β, peroxisome
proliferator-activated receptor-γ coactivator-1β; CTL, control; sh,
short hairpin. |
Next, the protein levels were determined, and they
were identified to be similar to the mRNA levels (Figs. 7B and C, and S1E). Furthermore, both EBV and LMP1↑
treatments significantly decreased O2−.
release (Fig. 7D) and increased the
number of lung metastatic nodules (Fig.
7E) and lung tumor spots (Fig. 7F
and G) compared with the CTL group. In addition, both EBV and
LMP1↑ treatments significantly decreased mouse survival compared
with that of the CTL group (Fig.
7H). PGC1β knockdown completely reversed this effect. It was
concluded that EBV/LMP1 exposure-mediated PGC1β expression
significantly potentiated tumor growth in vivo and shortened
mouse survival.
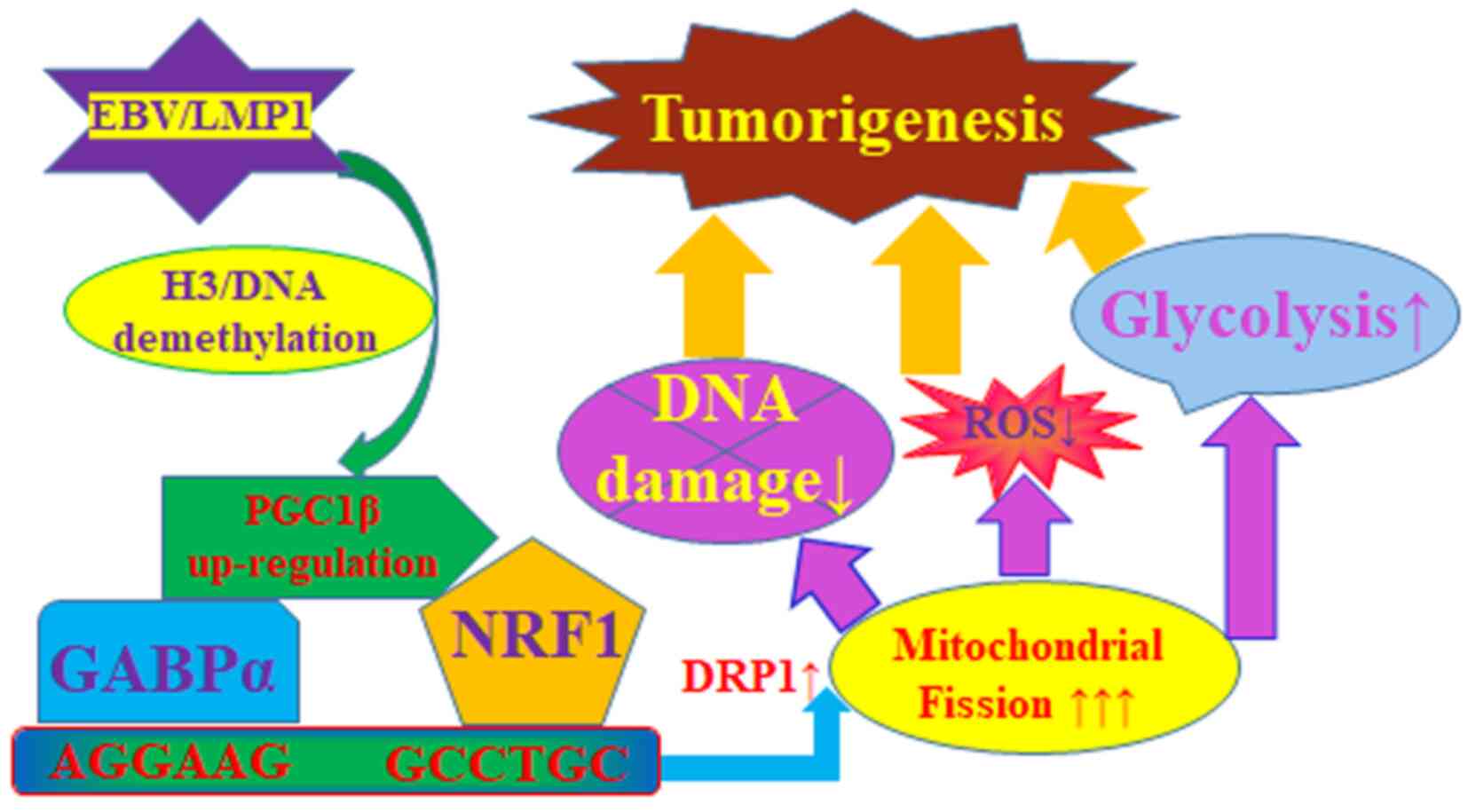
Schematic model of EBV/LMP1-mediated
epigenetic modifications and tumorigenesis through upregulation of
PGC1β and DRP1
The current study established a schematic model for
EBV/LMP1-mediated tumorigenesis through persistent epigenetic
changes on the PGC1β promoter and subsequent upregulation of PGC1β
and DRP1: Briefly, transient EBV/LMP1 exposure triggers epigenetic
changes, including decreased histone 3 methylation and DNA
methylation on the PGC1β promoter, resulting in persistent PGC1β
upregulation. Increased PGC1β expression upregulates DRP1
expression through the transcription factors GABPα and NRF1,
resulting in mitochondrial fission with potentiated glycolysis,
minimized oxidative stress and DNA damage. Taken together,
transient EBV/LMP1 exposure triggers persistent PGC1β upregulation
and tumorigenesis (Fig. 8).
Discussion
The present study demonstrated that LMP1 knockdown
in EBV-positive tumor cells did not suppress PGC1β expression or
tumor cell proliferation. However, transient LMP1 expression in HSC
caused epigenetic changes on the PGC1β promoter, resulting in
persistent PGC1β upregulation with potentiated DRP1 expression and
mitochondrial function, thus contributing to PGC1β-mediated
tumorigenesis.
As a potential oncogene (8), LMP1 transforms B cells and activates
the NF-κB signaling pathway through two domains of transformation
effector sites that are located at the C-terminus, subsequently
resulting in target gene expression and tumorigenesis (15–17).
The present study found that LMP1 expression triggered epigenetic
changes by demethylation of both histone 3 and DNA on the PGC1β
promoter, subsequently contributing to PGC1β upregulation. Our
preliminary study showed that EBV/LMP1 exposure could change the
expression of either DNA methyltransferases or histone
methyltransferases (35,36), subsequently contributing to
epigenetic changes. Currently, investigation on the potential
mechanism concerning EBV/LMP1-mediated epigenetic modifications is
underway in our laboratory. Notably, LMP1 expression-mediated
epigenetic changes and PGC1β upregulation were persistent even in
the absence of LMP1 after the cells were cultured from passage #1
to #6, indicating that LMP1-mediated tumorigenesis may be
independent from LMP1. This may explain why numerous forms of
lymphoma show absence of EBV/LMP1; thus, the current study
elucidated a novel mechanism and pathway for EBV/LMP1-mediated
tumorigenesis through epigenetic changes (37,38).
It has been reported that EBV/LMP1 can infect and transform B
lymphocytes (16), while the
present study showed that HSC could also be infected and
transformed, since numerous HSC could differentiate into either NK
or T cells, which may explain why EBV is associated with NK/T-cell
lymphoma (39).
It has been reported that DRP1 is responsible for
mitochondrial fission (11), while
both OPA1 and MFN2 are responsible for mitochondrial fusion
(40). The present study found that
EBV/LMP1 exposure induced DRP1 upregulation and MFN2
downregulation, but showed no effect on OPA1. This effect was
completely reversed by PGC1β knockdown, indicating that
EBV/LMP1-mediated mitochondrial fission/fusion gene expression was
mediated by PGC1β (41). Further
experiments showed that DRP1 was regulated by PGC1β through GABPα
and NRF1 binding sites located on the DRP1 promoter; however, the
potential mechanism for PGC1β-mediated MFN2 downregulation remains
unknown. Moreover, the present results revealed that
EBV/LMP1-mediated PGC1β expression significantly changed
mitochondrial morphology together with potentiated mitochondrial
function, including increased mitochondria DNA copies,
intracellular ATP levels and mitochondrial membrane potential, as
well as decreased apoptotic rate. Notably, EBV/LMP1-mediated PGC1β
expression significantly ameliorated oxidative stress and
subsequent DNA damage; this may be due to the fact that increased
mitochondrial replication partly diminishes ROS generation
(42).
PGC1β is a transcriptional coactivator that
regulates genes involved in mitochondrial function, and energy
balance and metabolism (19). It
has been reported that PGC1β contributes to tumorigenesis through
regulation of glycolysis and mitochondrial metabolism (20–22,43,44).
Our group has recently found that PGC1β regulates tumor growth
through LDHA-mediated glycolysis and HKDC1-mediated mitochondrial
function (23,24). In the present study, it was found
that PGC1β contributed to tumorigenesis through DRP1-mediated
mitochondrial fission. Additionally, increased PGC1β expression
upregulates the expression of LDHA (23), HKDC1 (24) and OGG1 (45), resulting in potentiated glycolysis,
and minimized oxidative stress and DNA damage.
It has benn previously reported by the authors that
LMP1 upregulates the PGC1β expression through activation of nuclear
factor-κB (NFκB) pathway (17), but
did not show the reason as why and/or how NFκB is activated in the
presence of LMP1. The present study revealed that knockdown of
either LMP1, NFκB or PGC1β has no effect, while transient EBV/LMP1
exposure induces persistent demethylation on both histone 3 and DNA
on the PGC1β promoter, indicating that LMP1-mediated NFκB
activation is probably due to LMP1-mediated demethylation and
subsequently potentiated binding ability of NFκB on the PGC1β
promoter. Our future work involves investigating the potential
mechanism of LMP1-mediated persistent epigenetic changes with the
aim of addressing the following questions: i) How could
LMP1-mediated epigenetic modifications be reversed? ii) do
LMP1-mediated epigenetic changes only happen on the PGC1β promoter,
or do they also occur on other genes?; and iii) how could it be
determined whether upregulated PGC1β is due to historical LMP1
infection in EBV/LMP1 absent tumors? These questions are part of
our ongoing investigation.
In conclusion, the present study demonstrated that
PGC1β modulated mitochondrial fission and glycolysis through DRP1
expression, and that EBV/LMP1 transient exposure triggered
persistent epigenetic changes with subsequent PGC1β upregulation
and tumor growth. These findings suggest a potential mechanism for
transient EBV/LMP1 exposure-mediated tumor growth through
persistent PCG1β expression and provide a hypothesis to explain why
numerous forms of lymphoma exhibit absence of EBV/LMP1.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors are grateful for the kind generous gift
from Dr Haimou Zhang (Hubei University, China) for providing EBV
LMP1 adenovirus.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82170191, 82100210 and 82201739),
the Natural Science Foundation of Guangdong (grant no.
2021A1515012185), the Shenzhen Municipal Science and Technology
Innovation (grant nos. JCYJ20160428172335984 and
JCYJ20220530160214031), the Research Foundation of Peking
University Shenzhen Hospital (grant nos. JCYJ2020017, JCYJ2021007
and KYQD202100X), the General Program for Clinical Research at
Peking University Shenzhen Hospital (grant no. LCYJ2021037), the
Sanming Project of Medicine in Shenzhen (grant ns. SZSM201612071),
the Shenzhen Key Medical Discipline Construction Fund (grant ns.
SZXK078) and the Cell Technology Center and Transformation Base,
Innovation Center of Guangdong-Hong Kong-Macao Greater Bay Area,
Ministry of Science and Technology of China [grant no.
YCZYPT(2018)03-1].
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or from the corresponding author
on reasonable request.
Authors' contributions
PY wrote the paper and designed the primers. PY and
HZ designed the experiments, analyzed the data, interpreted the
results and confirm the authenticity of all the raw data. JF and RL
performed part of the gene analysis and IHC staining experiments.
JC and WZ performed part of animal operation and biomedical
analysis. SC and PZ performed the remaining experiments. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocols for animal study (approval no.
2021-092) and human subjects (approval no. 2020-050) were approved
by the Committee from Peking University Shenzhen Hospital
(Shenzhen, China) and the written consents for human subjects were
given on the base of the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DRP1
|
dynamin-related protein 1
|
|
EBV
|
Epstein-Barr virus
|
|
GABPα
|
GA-binding protein α
|
|
HKDC1
|
hexokinase domain component 1
|
|
HSC
|
hematopoietic stem cells
|
|
LDHA
|
lactate dehydrogenase A
|
|
LMP1
|
latent membrane protein 1
|
|
MFN2
|
mitofusin 2
|
|
MMP
|
mitochondrial membrane potential
|
|
NF-κB
|
nuclear factor-κB
|
|
NRF1
|
nuclear respiratory factor 1
|
|
NKTCL
|
natural killer/T-cell lymphoma
|
|
O2.-
|
superoxide anion
|
|
OGG1
|
8-oxoguanine DNA glycosylase 1
|
|
OPA1
|
optic atrophy 1
|
|
PGC1β
|
peroxisome proliferator-activated
receptor-γ coactivator-1β
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Wirtz T, Weber T, Kracker S, Sommermann T,
Rajewsky K and Yasuda T: Mouse model for acute Epstein-Barr virus
infection. Proc Natl Acad Sci USA. 113:13821–13826. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai MH, Lin X, Shumilov A, Bernhardt K,
Feederle R, Poirey R, Kopp-Schneider A, Pereira B, Almeida R and
Delecluse HJ: The biological properties of different Epstein-Barr
virus strains explain their association with various types of
cancers. Oncotarget. 8:10238–10254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fujimoto A and Suzuki R: Epstein-barr
virus-associated Post-transplant lymphoproliferative disorders
after hematopoietic stem cell transplantation: Pathogenesis, risk
factors and clinical outcomes. Cancers (Basel). 12:3282020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang Y, Zhu Y, Cao JZ, Zhang YJ, Xu LM,
Yuan ZY, Wu JX, Wang W, Wu T, Lu B, et al: Risk-adapted therapy for
early-stage extranodal nasal-type NK/T-cell lymphoma: Analysis from
a multicenter study. Blood. 126:1424–1432. 15172015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lai J, Tan WJ, Too CT, Choo JA, Wong LH,
Mustafa FB, Srinivasan N, Lim AP, Zhong Y, Gascoigne NR, et al:
Targeting Epstein-Barr virus-transformed B lymphoblastoid cells
using antibodies with T-cell receptor-like specificities. Blood.
128:1396–1407. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nam YS, Im KI, Kim N, Song Y, Lee JS, Jeon
YW and Cho SG: Down-regulation of intracellular reactive oxygen
species attenuates P-glycoprotein-associated chemoresistance in
Epstein-Barr virus-positive NK/T-cell lymphoma. Am J Transl Res.
11:1359–1373. 2019.PubMed/NCBI
|
|
7
|
Zhang H, Lu J, Jiao Y, Chen Q, Li M, Wang
Z, Yu Z, Huang X, Yao A, Gao Q, et al: Aspirin inhibits Natural
Killer/T-cell lymphoma by modulation of VEGF expression and
mitochondrial function. Front Oncol. 8:6792019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chou YC, Lin SJ, Lu J, Yeh TH, Chen CL,
Weng PL, Lin JH, Yao M and Tsai CH: Requirement for LMP1-induced
RON receptor tyrosine kinase in Epstein-Barr virus-mediated B-cell
proliferation. Blood. 118:1340–1349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang LW, Jiang S and Gewurz BE:
Epstein-barr virus LMP1-mediated oncogenicity. J Virol.
91:e01718–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang
M, Chen L, Yang L, Tao Y, Jiang Y, et al: Targeting Epstein-Barr
virus oncoprotein LMP1-mediated glycolysis sensitizes
nasopharyngeal carcinoma to radiation therapy. Oncogene.
33:4568–4578. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pal AD, Basak NP, Banerjee AS and Banerjee
S: Epstein-Barr virus latent membrane protein-2A alters
mitochondrial dynamics promoting cellular migration mediated by
Notch signaling pathway. Carcinogenesis. 35:1592–1601. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wakisaka N, Kondo S, Yoshizaki T, Murono
S, Furukawa M and Pagano JS: Epstein-Barr virus latent membrane
protein 1 induces synthesis of hypoxia-inducible factor 1 alpha.
Mol Cell Biol. 24:5223–5234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lo AK, Lung RW, Dawson CW, Young LS, Ko
CW, Yeung WW, Kang W, To KF and Lo KW: Activation of sterol
regulatory element-binding protein 1 (SREBP1)-mediated lipogenesis
by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1)
promotes cell proliferation and progression of nasopharyngeal
carcinoma. J Pathol. 246:180–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ,
Xia ZJ, Huang HQ, Jiang WQ, Zhang YJ and Wang L: PD-L1 is
upregulated by EBV-driven LMP1 through NF-κB pathway and correlates
with poor prognosis in natural killer/T-cell lymphoma. J Hematol
Oncol. 9:1092016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Higuchi M, Izumi KM and Kieff E:
Epstein-Barr virus latent-infection membrane proteins are
palmitoylated and raft-associated: Protein 1 binds to the
cytoskeleton through TNF receptor cytoplasmic factors. Proc Natl
Acad Sci USA. 98:4675–4680. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dudziak D, Kieser A, Dirmeier U,
Nimmerjahn F, Berchtold S, Steinkasserer A, Marschall G,
Hammerschmidt W, Laux G and Bornkamm GW: Latent membrane protein 1
of Epstein-Barr virus induces CD83 by the NF-kappaB signaling
pathway. J Virol. 77:8290–8298. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feng J, Chen Q, Zhang P, Huang X, Xie W,
Zhang H and Yao P: Latent membrane Protein 1 promotes tumorigenesis
through upregulation of PGC1β signaling pathway. Stem Cell Rev Rep.
17:1486–1499. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin J, Puigserver P, Donovan J, Tarr P and
Spiegelman BM: Peroxisome proliferator-activated receptor gamma
coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription
coactivator associated with host cell factor. J Biol Chem.
277:1645–1648. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin J, Handschin C and Spiegelman BM:
Metabolic control through the PGC-1 family of transcription
coactivators. Cell Metab. 1:361–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deblois G, Chahrour G, Perry MC,
Sylvain-Drolet G, Muller WJ and Giguere V: Transcriptional control
of the ERBB2 amplicon by ERRalpha and PGC-1beta promotes mammary
gland tumorigenesis. Cancer Res. 70:10277–10287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bellafante E, Morgano A, Salvatore L,
Murzilli S, Di Tullio G, D'Orazio A, Latorre D, Villani G and
Moschetta A: PGC-1β promotes enterocyte lifespan and tumorigenesis
in the intestine. Proc Natl Acad Sci USA. 111:E4523–E4531. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Piccinin E, Villani G and Moschetta A:
Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The
role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol.
16:160–174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Li L, Chen Q, Li M, Feng J, Sun
Y, Zhao R, Zhu Y, Lv Y, Zhu Z, et al: PGC1β regulates multiple
myeloma tumor growth through LDHA-mediated glycolytic metabolism.
Mol Oncol. 12:1579–1595. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Lv Y, Sun Y, Zhang H, Xie W, Zhong
L, Chen Q, Li M, Li L, Feng J, et al: PGC1β regulates breast tumor
growth and metastasis by SREBP1-mediated HKDC1 expression. Front
Oncol. 9:2902019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bodnar AG, Ouellette M, Frolkis M, Holt
SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S and
Wright WE: Extension of life-span by introduction of telomerase
into normal human cells. Science. 279:349–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kong D, Zhan Y, Liu Z, Ding T, Li M, Yu H,
Zhang L, Li H, Luo A, Zhang D, et al: SIRT1-mediated ERβ
suppression in the endothelium contributes to vascular aging. Aging
Cell. 15:1092–1102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lan K, Verma SC, Murakami M, Bajaj B and
Robertson ES: Epstein-Barr Virus (EBV): Infection, propagation,
quantitation, and storage. Curr Protoc Microbiol. Chapter 14: Unit
14E.2. 2007. View Article : Google Scholar
|
|
28
|
Gong LJ, Wang XY, Gu WY and Wu X:
Pinocembrin ameliorates intermittent hypoxia-induced
neuroinflammation through BNIP3-dependent mitophagy in a murine
model of sleep apnea. J Neuroinflammation. 17:3372020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogino S, Kawasaki T, Brahmandam M, Cantor
M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ,
Laird PW, Loda M and Fuchs CS: Precision and performance
characteristics of bisulfite conversion and real-time PCR
(MethyLight) for quantitative DNA methylation analysis. J Mol
Diagn. 8:209–217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C, Shibata D, Danenberg PV and Laird PW: MethyLight: A
high-throughput assay to measure DNA methylation. Nucleic Acids
Res. 28:E322000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nosho K, Irahara N, Shima K, Kure S,
Kirkner GJ, Schernhammer ES, Hazra A, Hunter DJ, Quackenbush J,
Spiegelman D, et al: Comprehensive biostatistical analysis of CpG
island methylator phenotype in colorectal cancer using a large
population-based sample. PLoS One. 3:e36982008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou Y, Lu Q, Zheng D, Chu Z, Liu Z, Chen
H, Ruan Q, Ge X, Zhang Z, Wang X, et al: Prenatal levonorgestrel
exposure induces autism-like behavior in offspring through ERβ
suppression in the amygdala. Mol Autism. 8:462017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang H, Li L, Li M, Huang X, Xie W, Xiang
W and Yao P: Combination of betulinic acid and chidamide inhibits
acute myeloid leukemia by suppression of the HIF1α pathway and
generation of reactive oxygen species. Oncotarget. 8:94743–94758.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ramezani G, Norouzi A, Arabshahi SKS,
Sohrabi Z, Zazoli AZ, Saravani S and Pourbairamian G: Study of
medical students' learning approaches and their association with
academic performance and problem-solving styles. J Educ Health
Promot. 11:2522022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vargas-Ayala RC, Jay A, Manara F, Maroui
MA, Hernandez-Vargas H, Diederichs A, Robitaille A, Sirand C,
Ceraolo MG, Romero-Medina MC, et al: Interplay between the
Epigenetic Enzyme Lysine (K)-Specific demethylase 2B and
Epstein-Barr Virus Infection. J Virol. 93:e00273–19. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L,
Liu X, Tang M, Luo X, Fan J, et al: EBV(LMP1)-induced metabolic
reprogramming inhibits necroptosis through the hypermethylation of
the RIP3 promoter. Theranostics. 9:2424–2438. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martin KA, Lupey LN and Tempera I:
Epstein-Barr Virus oncoprotein LMP1 mediates epigenetic changes in
host gene expression through PARP1. J Virol. 90:8520–8530. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leonard S, Wei W, Anderton J, Vockerodt M,
Rowe M, Murray PG and Woodman CB: Epigenetic and transcriptional
changes which follow Epstein-Barr virus infection of germinal
center B cells and their relevance to the pathogenesis of Hodgkin's
lymphoma. J Virol. 85:9568–9577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Z, Liu W, Zhang W, Ye Y, Guan P, Gao
L and Zhao S: Chronic active Epstein-Barr Virus infection of
T/NK-cell type mimicking classic hodgkin lymphoma:
Clinicopathologic and genetic features of 8 cases supporting a
variant with ‘Hodgkin/Reed-Sternberg-like’ cells of NK phenotype.
Am J Surg Pathol. 43:1611–1621. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rodrigues T and Ferraz LS: Therapeutic
potential of targeting mitochondrial dynamics in cancer. Biochem
Pharmacol. 182:1142822020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kelly DP and Scarpulla RC: Transcriptional
regulatory circuits controlling mitochondrial biogenesis and
function. Genes Dev. 18:357–368. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu J, Ji L, Ruan Y, Wan Z, Lin Z, Xia S,
Tao L, Zheng J, Cai L, Wang Y, et al: UBQLN1 mediates sorafenib
resistance through regulating mitochondrial biogenesis and ROS
homeostasis by targeting PGC1β in hepatocellular carcinoma. Signal
Transduct Target Ther. 6:1902021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang CY, Kazmin D, Jasper JS, Kunder R,
Zuercher WJ and McDonnell DP: The metabolic regulator ERRalpha, a
downstream target of HER2/IGF-1R, as a therapeutic target in breast
cancer. Cancer Cell. 20:500–510. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Deblois G, St-Pierre J and Giguere V: The
PGC-1/ERR signaling axis in cancer. Oncogene. 32:3483–3490. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen Q, Feng J, Wu J, Yu Z, Zhang W, Chen
Y, Yao P and Zhang H: HKDC1 C-terminal based peptides inhibit
extranodal natural killer/T-cell lymphoma by modulation of
mitochondrial function and EBV suppression. Leukemia. 34:2736–2748.
2020. View Article : Google Scholar : PubMed/NCBI
|