Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide and remains one of the most recalcitrant tumors,
with a poor five-year survival rate. More than 85% of the lung
cancers are non-small cell lung cancers (NSCLCs), mainly composed
of lung adenocarcinoma (LUAD) and lung squamous cell carcinoma
(LUSC) (1). In an Asian population,
over half of the patients with NSCLC carry epidermal growth factor
receptor (EGFR) 19 del (54%) or 21 L858R (43%) mutations,
leading to the recommendations to use EGFR tyrosine kinase
inhibitors (EGFR-TKIs) (2).
Clinically, EGFR-TKIs, such as gefitinib, are still used as
first-line antitumor drugs for patients with NSCLC showing
mutations in the EGFR. According to current evidence,
EGFR-TKIs can improve not only the objective response rate, but
also the progression-free survival rate of affected patients.
However, ~six months to a year after using EGFR-TKIs, the tumor
acquires drug resistance, greatly minimizing the clinical benefit
to patients with NSCLC. Notably, T790M mutations in EGFR
induce first- and second-generation TKI resistance in more than
half of the NSCLC cases. In addition, ~30% of the resistance cases
remain unelucidated (3,4). Hence, overcoming resistance to
EGFR-TKIs has become imperative for the development of
NSCLC-targeted treatment.
Thyroid hormone receptor interactor protein 13
(TRIP13, also known as pachytene checkpoint 2), one of the most
important members of the AAA+ ATPase family, plays
important roles in cell cycle checkpoints, DNA repair, meiotic
recombination and chromosome synapsis (5–10).
Accumulating evidence suggests that TRIP13 is overexpressed in
several human malignancies and is closely associated with poor
prognosis in various diseases, including colorectal cancer,
hepatocellular carcinoma, prostate cancer, glioblastoma, thyroid
cancer and ovarian cancer (11–16).
Regarding the role of TRIP13 in lung cancer, previous research has
focused on its activation of the PI3K/AKT/mTOR signaling pathway
(17,18). A recent study by the authors showed
that TRIP13 could promote lung cancer development by activating the
epithelial-mesenchymal transition and Wnt signaling pathways
(19). Notably, as demonstrated in
previous studies, TRIP13 activation also facilitates chemotherapy
resistance in bladder cancer and multiple myeloma as well as
radiation resistance in head and neck cancer (20–22).
However, the role of TRIP13 in the modulation of EGFR-TKIs
sensitivity in NSCLC remains unclear.
Cell autophagy is a catabolic process that is
required to maintain cell homeostasis. Previous research has shown
that it is associated with several biological processes, including
cell differentiation, development and survival (23–25).
Although the role of autophagy in cell death and survival has not
been thoroughly established, it has been demonstrated that
autophagy is boosted in cancer cells when they are subjected to
harsh conditions, such as anticancer medication therapy, which may
lead to anticancer drug resistance (26–28).
The EGFR signaling pathway is often dysregulated in
various human cancers (29,30). When EGFR binds to a ligand, it
becomes phosphorylated, thereby activating its downstream signaling
pathways. As a result, anti-apoptotic and pro-survival processes
are triggered in cancer cells to promote tumor progression.
Therefore, patients with EGFR mutations could greatly
benefit from EGFR-TKIs, such as gefitinib or erlotinib, which block
the action of this pathway (31,32).
In the present study, resistance to gefitinib and
pro-cancer effects of TRIP13 in NSCLC were identified. The effects
of TRIP13 on autophagy and phosphorylation of the EGFR signaling
pathway were also explored by regulating the expression of TRIP13
in gefitinib-sensitive and -resistant NSCLC cells. Taken together,
the present results suggested that the downregulation of TRIP13
improves the sensitivity of NSCLC cells to gefitinib. TRIP13
expression has the potential to serve as a gefitinib-resistant
biomarker for NSCLC.
Materials and methods
Patients and specimens
A total of 160 NSCLCs and 19 corresponding normal
lung tissue samples were collected from 160 patients (age and sex
distribution are presented in Table
I) who underwent complete surgical excision of LUAD or LUSC at
the First Affiliated Hospital of China Medical University between
May 2018 and August 2020. All tissue specimens were fixed in 4%
neutral formaldehyde at room temperature for 24 h, embedded in
paraffin, and sectioned into 4-µm slices. In addition, a total of
20 pairs of fresh NSCLC tissue samples and the corresponding normal
lung tissue samples were collected and stored at −80°C immediately
after resection. Neither chemotherapy nor neoadjuvant radiotherapy
was performed before the surgery. The World Health Organization
classification of lung tumors (4th edition, 2015) was used to
evaluate histological diagnosis and grade. The eighth edition of
the American Joint Committee on Cancer (AJCC) lung cancer staging
system was used for tumor staging. The present study was approved
[approval no. LS (2021)009] by the Research Ethics Committee of the
China Medical University (Shenyang, China). Written informed
consent was obtained from all patients.
 | Table I.Correlations between TRIP13
expression and clinicopathological factors in NSCLC. |
Table I.
Correlations between TRIP13
expression and clinicopathological factors in NSCLC.
| Clinicopathological
factors | Total number |
TRIP13-negative |
TRIP13-positive | P-value |
|---|
| Tissue |
|
|
| <0.01 |
|
Normal | 19 | 17 | 2 |
|
|
NSCLC | 160 | 45 | 115 |
|
| Pathology type |
|
|
| 0.386 |
|
LUSC | 80 | 21 | 59 |
|
|
LUAD | 80 | 26 | 54 |
|
| Age |
|
|
| 0.318 |
|
<60 | 89 | 29 | 60 |
|
|
≥60 | 71 | 18 | 53 |
|
| Sex |
|
|
| 0.417 |
|
Male | 119 | 37 | 82 |
|
|
Female | 41 | 10 | 31 |
|
| TNM Stage |
|
|
| <0.01 |
| I +
II | 127 | 89 | 38 |
|
| II +
IV | 27 | 9 | 18 |
|
| Tumor status |
|
|
| 0.825 |
| T1 +
T2 | 120 | 36 | 84 |
|
| T3 +
T4 | 30 | 10 | 20 |
|
| Lymph node
metastasis |
|
|
| <0.01 |
| No | 59 | 41 | 18 |
|
|
Yes | 79 | 25 | 54 |
|
| Distant
metastasis |
|
|
| 0.838 |
| No | 149 | 45 | 104 |
|
|
Yes | 11 | 3 | 8 |
|
Bioinformatics analysis
The Kaplan-Meier plotter (http://kmplot.com/analysis/) was used to generate
overall survival curves (examined by the log-rank test) for
patients with lung cancer showing high or low TRIP13 expression.
TRIP13 expression in patients with LUAD and LUSC at various cancer
stages was collected from the Cancer Genome Atlas database (TCGA;
http://cancergenome.nih.gov/). Gene
Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/) was used to perform a
correlation analysis between TRIP13 and EGFR expression in lung
cancers.
Cell culture and transfection
Human lung cancer cell lines HCC827 (with
EGFR 19del) and H1975 (with EGFR L858R and T790M
mutations) were obtained from the Shanghai Cell Bank. Gefitinib
sensitivity is linked to EGFR E746-A750 deletion in exon 19
of the HCC827 cell line. The H1975 cell line carried an EGFR
T790M mutation in exon 20, which is related to gefitinib
resistance.
At 37°C in a 5% CO2 atmosphere, cells
were cultured in the RPMI-1640 medium with 10% fetal bovine serum
and passaged in sterile culture dishes with 0.25% trypsin (all from
Gibco; Thermo Fisher Scientific, Inc.). The gefitinib-acquired
resistance cell line, HCC827GR, was established via a stepwise
increase in the dosage of gefitinib (0.01, 0.1, 1, 5, 10, 20 and 40
µM). After 24 h, the gefitinib-containing medium was replaced with
a regular medium. Each dose was repeated twice until the
half-maximal inhibitory concentration (IC50) of the
HCC827GR cells was satisfactory. To sustain resistance, HCC827GR
cells were cultured in a medium containing 100 nM gefitinib.
Plasmid transfection was performed using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Subsequent experiments were performed 24 h after plasmid
transfection. The pCMV6-TRIP13 and pCMV6 plasmids were purchased
from Origene Technologies, Inc Co. The plasmid containing the short
hairpin RNA of small interfering RNA of TRIP13 (shTRIP13) and its
negative control plasmid (shNC) were purchased from Shanghai
GeneChem Co., Ltd. 600 µg/ml G418 (Sigma-Aldrich; Merck KGaA) was
used to screen for stably transfected cells. The TRIP13 shRNA
sequences were as follows: shTRIP13-1, 5′-CGATTATGTGATGACAACTTT-3′;
shTRIP13-2, 5′-GUACCGAUAUGGCCAAUUA-3′; shTRIP13-3:
5′-GCAAAUCACUGGGUUCUAC-3′; and shCtrl:
5′-TTCTCCGAACGTGTCACGT-3′.
Gefitinib, an autophagy activator, and EGFR
inhibitor, and the autophagy inhibitor, 3-methyladenin (3-MA) were
purchased from MedChemExpress. Dimethyl sulfoxide (DMSO; Beijing
Solarbio Science & Technology Co., Ltd.) was added for
comparison.
Immunohistochemistry
Briefly, 4-µm thick tissue sections were
deparaffinized in xylene, rehydrated using a gradient alcohol
series, and then autoclaved for 2 min in 0.01 M citrate buffer (pH
6.0). Hydrogen peroxide (0.3%) was used to inhibit endogenous
peroxidase activity, and 5% normal goat serum (Maxin Biotechnology
Co., Ltd.) was added to the mixture to block non-specific binding
at 37°C for 30 min. Next, the tissue sections were incubated with a
rabbit polyclonal anti-TRIP13 antibody at 4°C for 12 h. The TRIP13
staining intensity was graded as follows: 0, no staining; 1, weak;
2, moderate; and 3, high. The percentage score classifications were
1 (1–25%), 2 (26–50%), 3 (51–75%) and 4 (76–100%). Each tumor
sample received a score that was multiplied to generate a final
score ranging from 0 to 12. A cut-off value of 6 was used to divide
patients into positive and negative TRIP13 expression groups. Tumor
samples with scores ≥6 were considered to be positive for TRIP13,
whereas those with scores ≥0 and <6 were considered to be
negative for TRIP13.
Western blot analysis
First, the Bradford method was used to quantify the
total protein isolated from cells using cell lysis buffer (33). Sodium dimethyl sulfoxide
polyacrylamide gel electrophoresis was used to separate 60 µg of
protein, and the concentration (8–12%) of the gels varied with the
molecular weight of the proteins. Next, the proteins were
transferred to a polyvinylidene fluoride membrane (Millipore Sigma)
blocked with 5% non-fat milk at 37°C for 2 h, and incubated with
primary antibodies overnight at 4°C. The primary antibodies used in
the present study are listed in Table
SI. Subsequently, the membranes were then treated for 2 h at
37°C with horseradish peroxidase-conjugated mouse (cat. no.
B900620) or rabbit IgG (cat. no. 30000-0-AP) (both 1:1,000;
ProteinTech Group, Inc.). Finally, the protein bands were
visualized using an electrochemiluminescence substrate (Pierce;
Thermo Fisher Scientific, Inc.) and detected using a bioimaging
system (version 5.2.1; DNR Bio-Imaging Systems Co., Ltd.). GAPDH
(1:1,000; cat. no. 600041Ig; ProteinTech Group, Inc.) was used as a
loading control to determine the relative protein levels.
Cell proliferation assay
Cells were plated in a 96-well plate (~3,000 cells
per well) for 24 h with a medium containing 10% fetal bovine serum
after transfection. After switching to serum-free medium,
3-(4,5-dimethylthiazol-2-yl)-5
-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS;
Dojindo Molecular Technologies, Inc.) was added to each well (20
µl/well) and incubated at 37°C for 2 h. To construct a growth
curve, data were measured spectrophotometrically every 24 h for 5
days at a wavelength of 490 nm. After seeding the cells in 96-well
plates for 24 h, different concentrations of gefitinib were added
and incubated for 72 h to determine the IC50 of
gefitinib. Cell viability was assessed as aforementioned. GraphPad
Prism (version 9.0; GraphPad Software, Inc.) was used to calculate
the IC50 values.
Colony formation assay
After 24 h of transfection, the cells were cultured
in six-well plates (~800 cells/well). After the cells were fully
apposed, gefitinib (0.5 µM for HCC827 cells and 5 µM for H1975 and
HCC827GR cells) was added, and the cells were incubated for 72 h
before being replaced with normal medium. During this experiment,
the culture medium was changed every 3–4 days for 10–15 days. The
cells were then rinsed in phosphate-buffered saline (PBS; Gibco;
Thermo Fisher Scientific, Inc.), fixed for 20 min at 37°C with 4%
paraformaldehyde, and stained for 10 min at 37°C with Giemsa
solution (Beyotime Biotechnology Inc.). Colonies with >50 cells
were counted.
Cell apoptosis assay
Cells were seeded in a 6-cm culture dish, and
gefitinib (0.5 µM for HCC827 cells and 5 µM for H1975 and HCC827GR
cells) was added for 24 h. The cells were then cultured for another
day. The cells from each tube were suspended in binding buffer (0.5
ml of binding buffer and 5 µl of Annexin V-fluorescein
isothiocyanate (Annexin V-FITC) and propidium iodide were added as
required (both from Shanghai GeneChem Co., Ltd.). Apoptosis was
examined using flow cytometry (Accuri™ C6 Plus), and the data was
analyzed using FlowJo (version 10.0; both from Becton Dickinson and
Company Technology Co., Ltd.).
Tumor xenograft mouse models
A total of 14 female BALB/c nude mice (age, four
weeks-old; weight, 22±1.5 g) were acquired from Charles River
Laboratories (Beijing, China). The mice were kept in plastic cages
(three mice/cage), with free access to food and water, and under
controlled lighting (12/12 h light/dark cycle), temperature
(23±2°C) and humidity (50%). The axilla of each mouse was then
subcutaneously injected with 5×106 tumor cells in 0.2 ml
of sterile PBS. The mice were euthanized and autopsied six weeks
after inoculation to observe tumor development. All mice that
reached the study's endpoint were euthanized by cervical
dislocation. The humane endpoints were defined as the following:
the xenograft tumor diameter was >20 mm, the xenograft tumor
reached >20% of the animal body weight, a body weight loss of
>20% happened due to tumor growth, and symptoms of immobility,
inability to eat, ulceration, infection, or necrosis were observed.
The appearance of dilated pupils, as well as the absence of breath
and heartbeat, served as evidence of death. Animal experiments were
approved by The Institutional Animal Research Committee of the
China Medical University. The entire process complied (IACUC issue
no. KT2022488) with the experimental animal ethics guidelines of
China Medical University (Shenyang, China).
Laser confocal immunofluorescence
assay
The NSCLC cells were cultured on coverslips in
24-well plates. After 24 h, the cells were fixed for 20 min with 4%
paraformaldehyde and permeabilized for 10 min with 0.1% Triton
X-100 (Beyotime Institute of Biotechnology). The slides were washed
three times with PBS, blocked with bovine serum albumin (Beijing
Solarbio Science & Technology Co., Ltd.) for 2 h at 25°C, and
incubated overnight at 4°C with anti-TRIP13 (1:50; cat. no.
19602-1-AP; ProteinTech Group, Inc.), or anti-EGFR (1:50; cat. no.
ab52894; Abcam). The cells were then washed again and incubated for
2 h at 25°C with tetramethylrhodamine (TRITC; 1:50; cat. no.
SA00007-1; ProteinTech Group, Inc.) or FITC-conjugated secondary
antibodies (1:50; cat. no. SA00003-8; ProteinTech Group, Inc.), and
cell nuclei were counterstained with DAPI (Beyotime Institute of
Biotechnology) for 10 min. An Olympus FV1000 laser scanning
confocal microscope (Olympus Corporation) was used for the
immunofluorescence analysis.
Co-immunoprecipitation assay
Cells were lysed in a solution of NP-40 (Beyotime
Biotechnology Inc.) and protease inhibitor mixture. The lysates
were centrifuged at 12,000 × g at 4°C for 20 min. The lysates were
then combined and incubated overnight with protein G-agarose beads
(Bimake Biotechnology Co., Ltd.) or appropriate primary antibodies
at 4°C. Western blotting was used to analyze the results.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism (version 9.0; GraphPad Software Inc.). All values from the
studies are expressed as the mean ± standard deviation. Chi-square
test and Spearman's rank correlation were used to examine the
immunohistochemical results. The expression of TRIP13 in tumors and
corresponding normal lung tissues from the same patients were
compared using a paired t-test, while other data from two groups
were compared using an unpaired t-test. Differences between three
or more groups were evaluated using a one-way ANOVA multiple
comparison test, followed by Tukey's post hoc test. The statistical
significance of differences between groups was determined using
P<0.05.
Results
Increased TRIP13 expression in
patients with NSCLC is related to the individual cancer stages and
poor prognosis
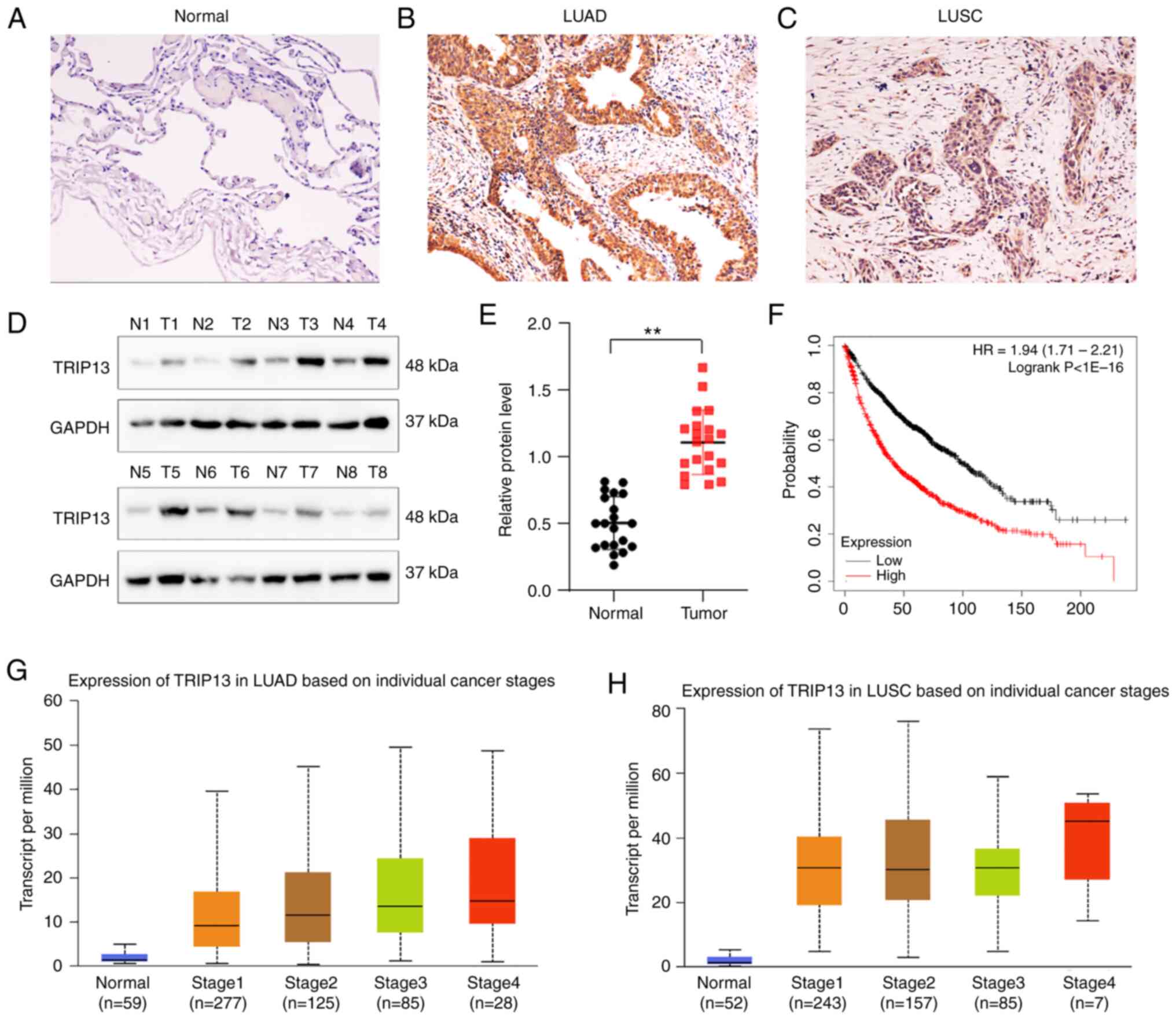
The expression of TRIP13 was examined by
immunohistochemistry in 160 NSCLC and 19 normal lung specimens,
which were collected from paraffin-embedded tissues. TRIP13
positive expression was seldom observed in normal alveolar cells in
corresponding normal lung tissues (10.5%) but was commonly noted in
the cytoplasm of 160 cases (71.8%) of NSCLC (Table I; Fig.
1A-C). Western blot analysis confirmed that the expression
levels of TRIP13 were significantly higher in lung cancer tissues
than in normal lung samples, both of which were collected from
fresh tissues (n=20) (Fig. 1D and
E). As indicated in Table I,
the expression of TRIP13 was correlated with the TNM stage and
lymph node metastasis of NSCLC, but was not significantly
correlated with patient age, sex, tumor status or distant
metastasis.
Kaplan-Meier survival analysis indicated that
patients with lung cancer showing higher TRIP13 levels had a poorer
overall survival rate (Fig. 1F).
Based on the findings from the TCGA database, TRIP13 was also
overexpressed in different individual lung cancer stages (Fig. 1G and H) as well as in the
histological subtypes LUADs and LUSCs (Fig. S1A and D). In addition, analyses of
TCGA database revealed that TRIP13 expression was linked to nodal
metastasis status and patient age (Fig. S1B, C, E, and F).
Increased expression of TRIP13 induces
gefitinib resistance in NSCLC cells
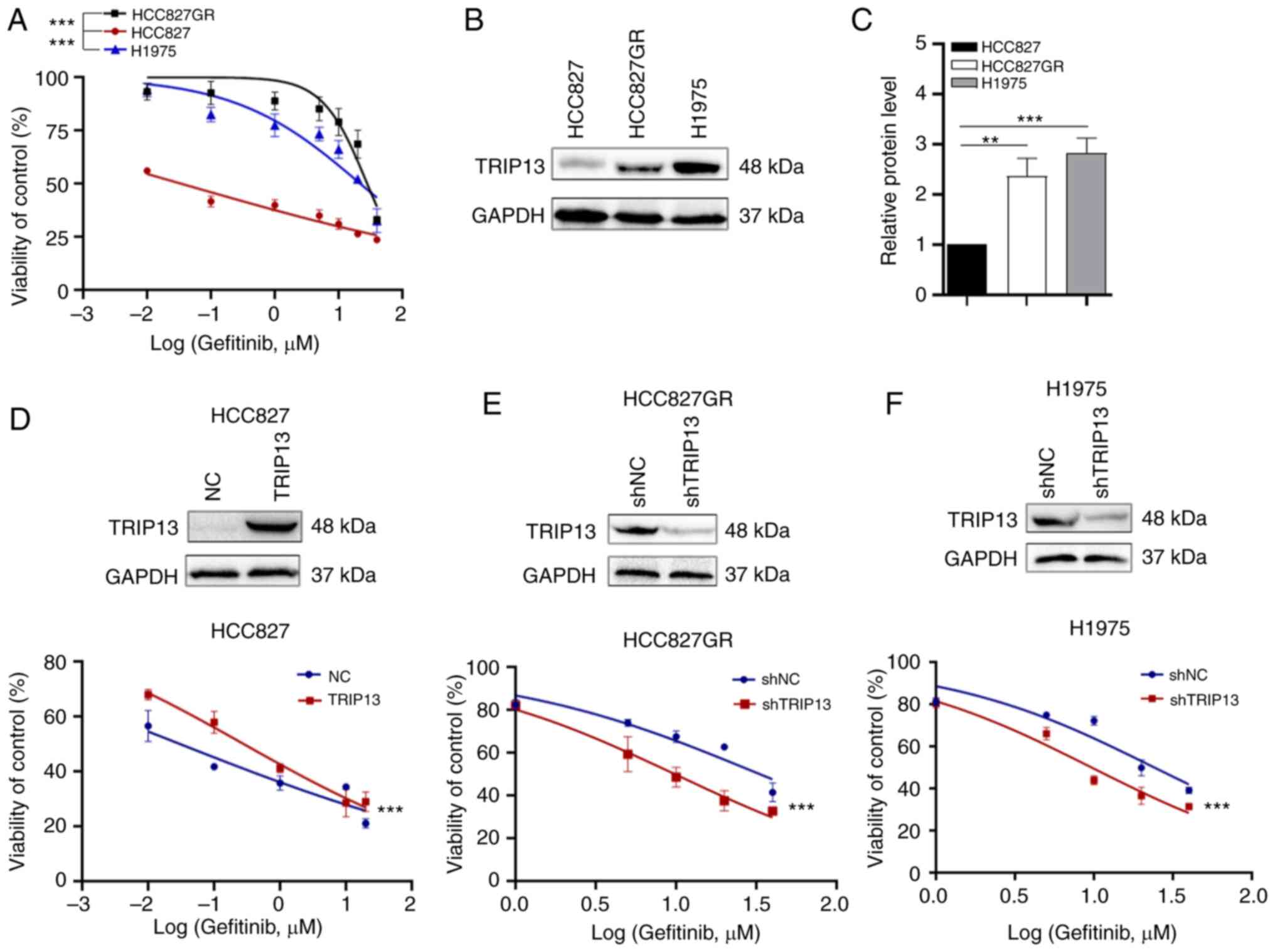
HCC827 cells were used as gefitinib-sensitive NSCLC
cells, and H1975 and HCC827GR cells as natural and acquired
gefitinib-resistant NSCLC cells, respectively. As indicated in the
MTS assays, both HCC827GR and H1975 cells had higher
IC50 values for gefitinib compared with HCC827 cells
(Fig. 2A). To confirm the
correlation between TRIP13 expression and tolerance of NSCLC cells
to gefitinib, the protein levels of TRIP13 in these cells were
detected by western blotting. TRIP13 expression was significantly
higher in HCC827GR and H1975 cells than in HCC827 cells (Fig. 2B and C). TRIP13 expression in NSCLC
cells was upregulated or knocked down to determine the relationship
between TRIP13 expression and gefitinib sensitivity. As revealed in
Fig. 2, the IC50 of
gefitinib was increased by upregulating the expression of TRIP13 in
HCC827 cells (Fig. 2D) and
decreased by the knockdown of TRIP13 in HCC827GR and H1975 cells
(Fig. 2E and F), which verified our
hypothesis that TRIP13 desensitizes NSCLC cells to gefitinib.
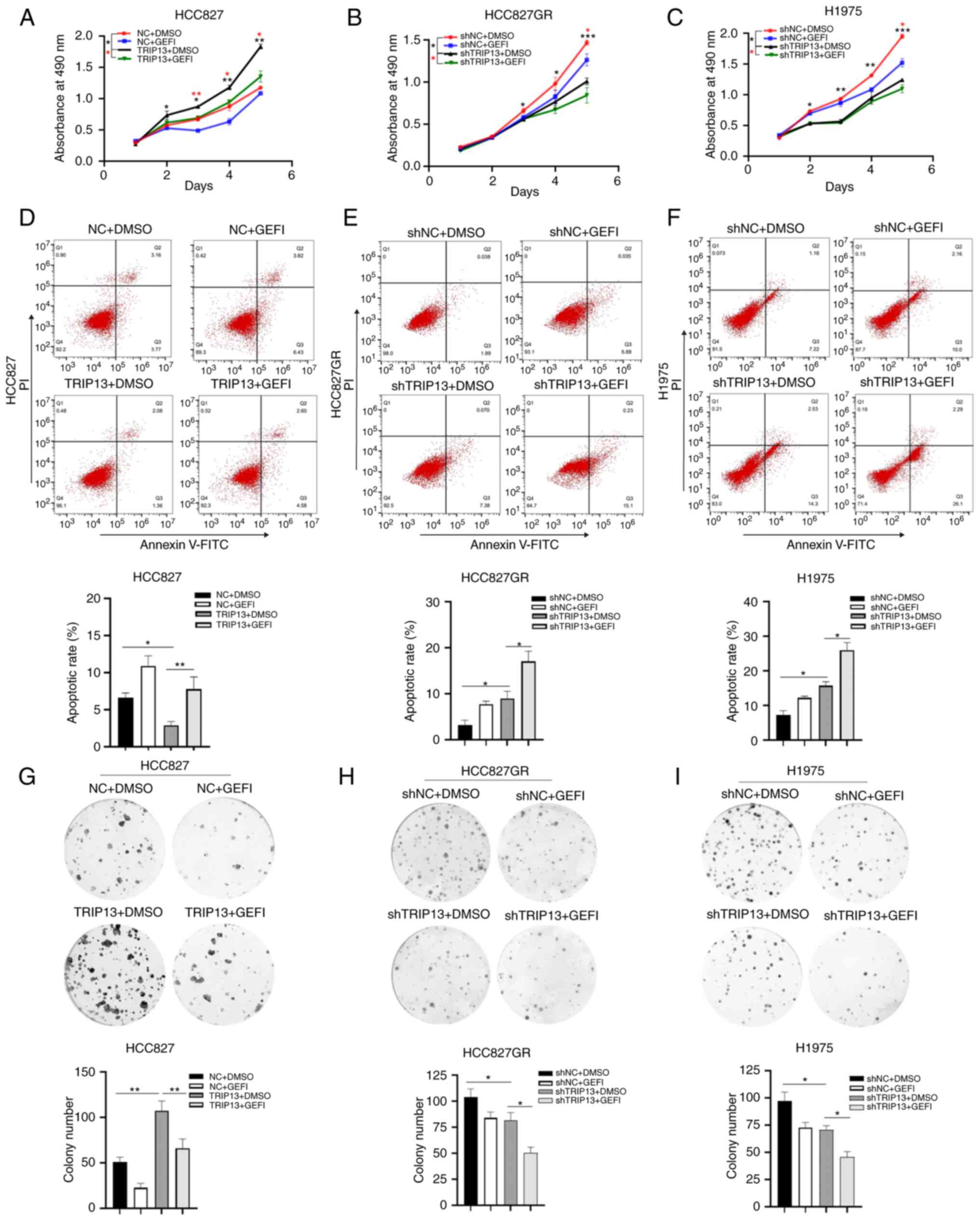
TRIP13 promotes the proliferation and
colony formation while inhibiting the apoptosis of NSCLC cells in
vitro
To further evaluate the role of TRIP13 in gefitinib
sensitivity, the TRIP13 levels in cells (Fig. S2A and B) were adjusted and
different functional experiments related to drug resistance were
performed. All NSCLC cells were treated with gefitinib to
investigate whether TRIP13 induced drug resistance following
gefitinib treatment. The results demonstrated that upregulation of
TRIP13 expression enhanced the proliferation and colony formation
of HCC827 cells (Fig. 3A and G),
whereas downregulation of TRIP13 expression inhibited the
proliferation and colony formation of HCC827GR (Fig. 3B and H) and H1975 (Fig. 3C and I) cells. Overexpression of
TRIP13 decreased the apoptotic rate of HCC827 cells (Fig. 3D), whereas low TRIP13 expression
increased the apoptosis of HCC827GR (Fig. 3E) and H1975 (Fig. 3F) cells.
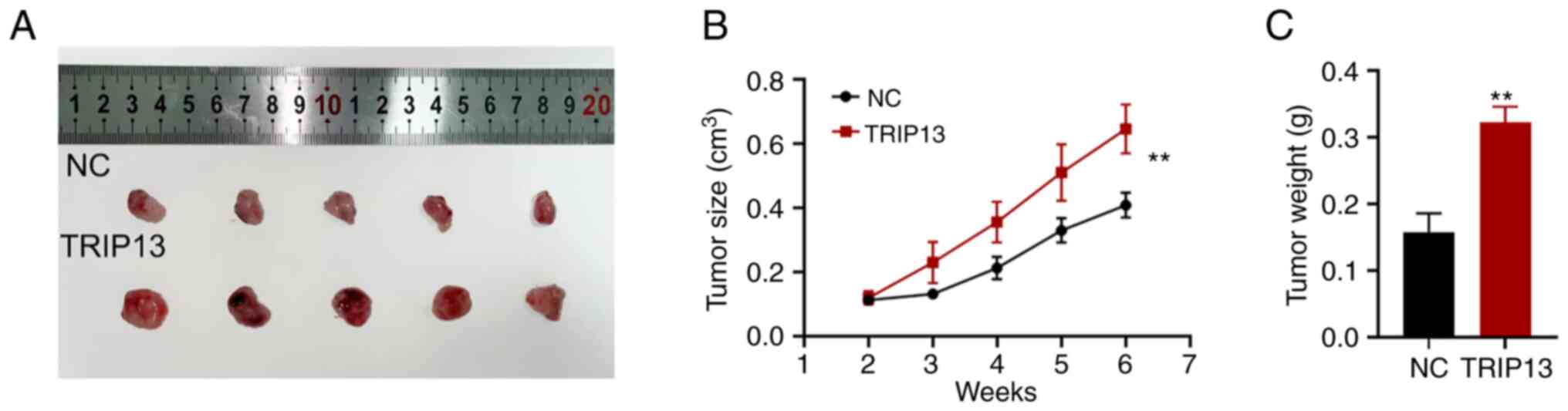
TRIP13 promotes NSCLC cells
proliferation in vivo
To evaluate the biological function of TRIP13 in
vivo, HCC827 cells stably expressing TRIP13 and control HCC827
cells were subcutaneously injected into nude mice. The tumorigenic
rates of the two groups were comparable (5/7, 71.4%), but the
tumorigenic volume and weight were higher in the nude mice in the
highly TRIP13-expressing group than in the control group (Fig. 4A-C). These findings indicated that
TRIP13 plays a crucial regulatory role in fostering NSCLC cell
proliferation in vivo.
TRIP13 contributes to autophagy in
NSCLC cells
Immunocytochemical results identified that when
TRIP13 was overexpressed in HCC827 cells, the number of
LC3B-positive puncta (autophagosome-like structures) increased,
indicating that autophagy was stimulated. Gefitinib acts as an
autophagy agonist, improving the ability of TRIP13 to promote
autophagy, while the autophagy inhibitor 3-MA diminished this
effect (Fig. 5A and B). TRIP13
overexpression increased gefitinib-induced LC3B accumulation in
HCC827 cells, whereas TRIP13 downregulation decreased
gefitinib-induced LC3B accumulation in HCC827GR and H1975 cells.
Meanwhile, 3-MA-induced P62 accumulation was enhanced by TRIP13
downregulation in HCC827GR and H1975 cells and reduced by TRIP13
upregulation in HCC827 cells (Fig.
5C-K).
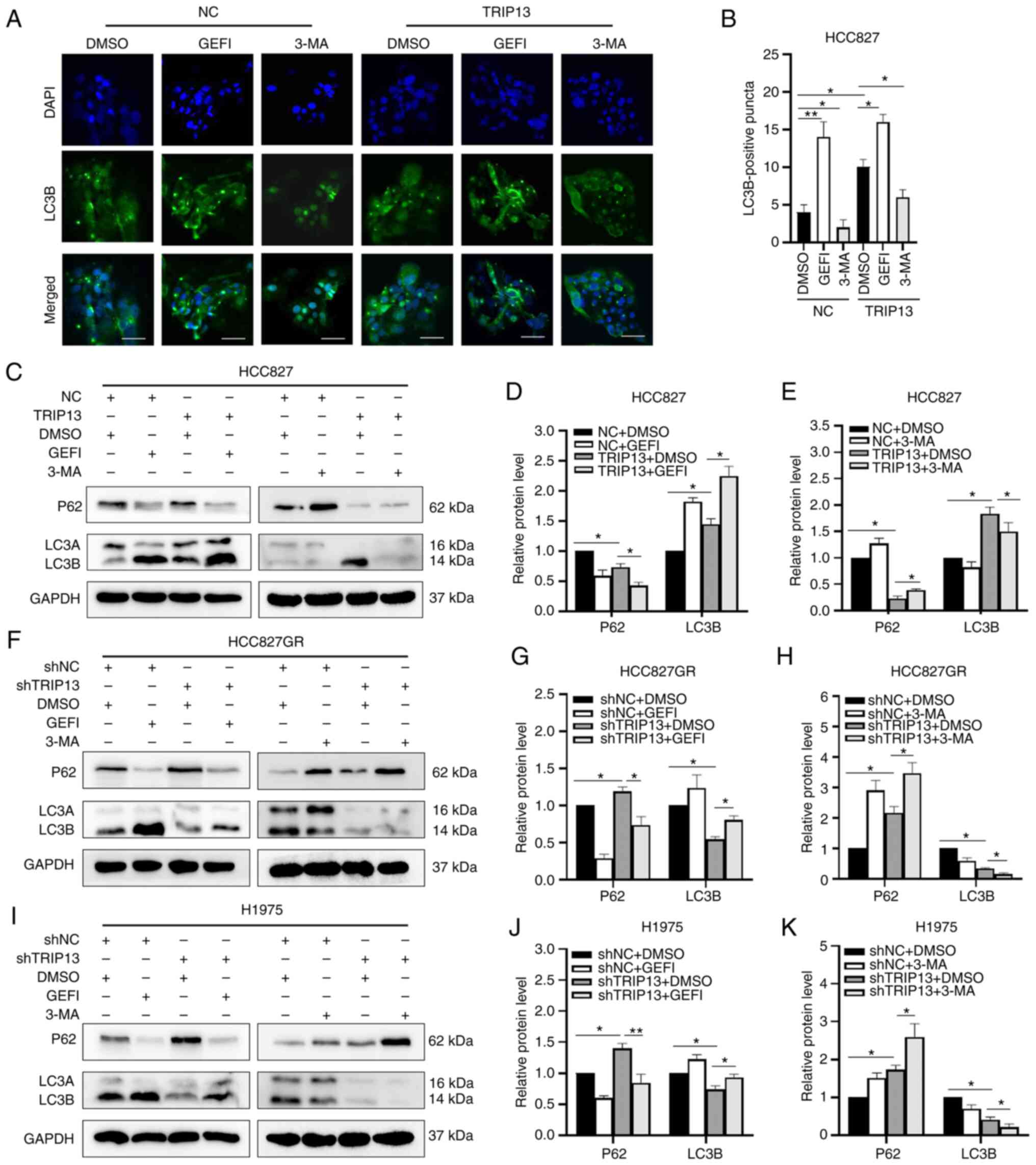 | Figure 5.Effect of TRIP13 on autophagy of
non-small cell lung cancer cells. (A) Representative
immunofluorescence images of HCC827 cells with or without TRIP13
upregulation following treatment with gefitinib (0.5 µM for 12 h)
or 3-MA (10 mM for 12 h). Scale bar, 10 µm. (B) Numbers of
LC3B-positive puncta (autophagosome-like structures) in HCC827
cells with or without TRIP13 upregulation. A total of 20 cells were
counted from every independent experiment, which was repeated
thrice and calculated. (C) Expression levels of LC3 and P62
proteins in HCC827 cells following TRIP13 overexpression and
treatment with gefitinib (0.5 µM for 12 h, left) or 3-MA (10 mM for
12 h, right). (F and I) LC3 and P62 protein expression levels in
H1975 and HCC827GR cells after TRIP13 knockdown and treatment with
gefitinib (5 µM for 12 h, left) or 3-MA (10 mM for 12 h, right).
(D, E, G, H, J and K) Relative expression levels of proteins for
each group are displayed in the histograms. DMSO and GAPDH were
used as drug control and internal control, respectively. The data
obtained were expressed as the mean ± SD. *P<0.05 and
**P<0.01. TRIP13, thyroid hormone receptor interactor 13; LC3A,
microtubule-associated protein 1 light chain 3 alpha; LC3B,
microtubule-associated protein 1 light chain 3 beta; P62, also
known as SQSTM1, Sequestosome 1; DMSO, dimethyl sulfoxide; GEFI,
gefitinib; 3-MA, 3-Methyladenine; NC, negative control cells; sh-,
short hairpin. |
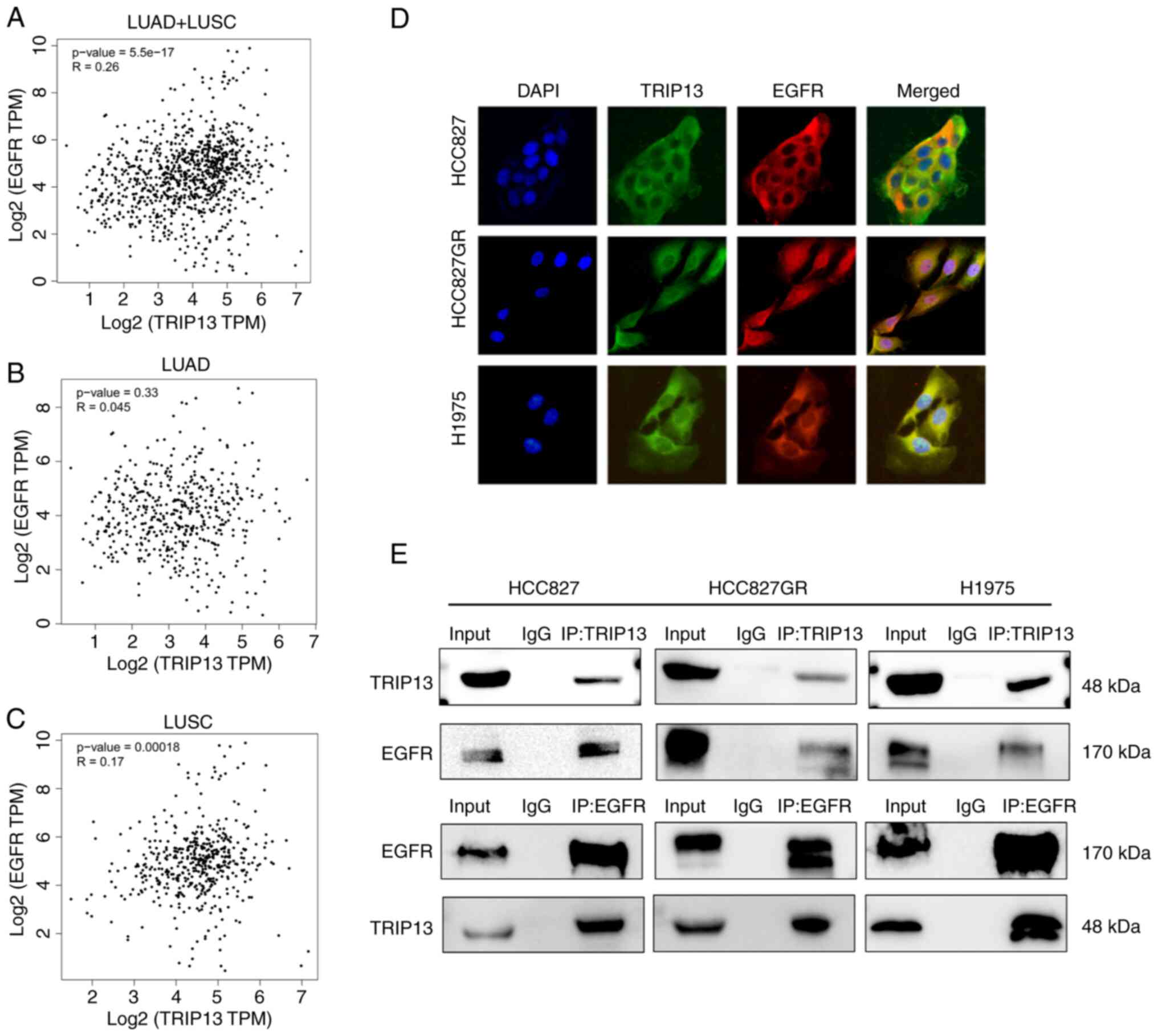
TRIP13 interacts with EGFR in NSCLC
cells
Given that EGFR plays a significant role in EGFR-TKI
resistance, the function of TRIP13 in gefitinib resistance was
further investigated by using the GEPIA database to predict the
relationship between TRIP13 and EGFR. However, according to the
database, TRIP13 expression did not correlate significantly with
EGFR expression in NSCLC cells (Fig.
6A-C). Using laser confocal immunofluorescence, it was observed
that TRIP13 and EGFR were primarily colocalized in the cytoplasm of
NSCLC cells (Fig. 6D). Finally,
co-immunoprecipitation experiments confirmed that TRIP13 could bind
to EGFR in both gefitinib-sensitive and gefitinib-resistant NSCLC
cells (Fig. 6E).
TRIP13 activates the phosphorylation
of EGFR and its downstream signaling pathways in NSCLC cells
TRIP13 was discovered to activate the PI3K/AKT/mTOR
signaling pathway in lung cancers, which was likewise in line with
the data from TCGA database (Fig.
S2C). However, the relationship between TRIP13 and the EGFR
pathway in lung cancer has not been clarified. To further study the
effect of TRIP13 on EGFR, it was examined whether the EGFR
inhibitor gefitinib affected TRIP13 expression in the three cell
lines. According to the results of western blotting, TRIP13
expression decreased with increasing gefitinib concentration
(Fig. 7A-C). The expression levels
of TRIP13 were then changed in HCC827, HCC827GR and H1975 cells and
the alterations in the total and phosphorylated (p-)EGFR (Tyr1068)
at the protein level were examined. P-EGFR (Tyr1068) levels were
increased along with TRIP13 overexpression in HCC827 cells and
decreased after TRIP13 downregulation in HCC827GR and H1975 cells.
TRIP13 upregulation also enhanced total EGFR levels in HCC827
cells, whereas TRIP13 knockdown reduced its levels in HCC827GR and
H1975 cells. Notably, the level of p-EGFR was significantly altered
compared with that of total EGFR. Alterations in downstream
phosphorylated key proteins of the EGFR pathway, including p-JAK
(Tyr1007 and Tyr1008), p-STAT3 (Tyr705), p-PI3K (Tyr 199), p-AKT
(Ser473), p-MEK (Ser217 and Ser221) and p-ERK (Thr202 and Tyr187),
were consistent with those of p-EGFR (Tyr1068). However, despite
changes in the expression of TRIP13, the total protein levels of
the abovementioned downstream proteins were not significantly
altered. As an EGFR inhibitor, gefitinib reversed the effects of
TRIP13 on EGFR and its downstream proteins (Fig. 7D-G).
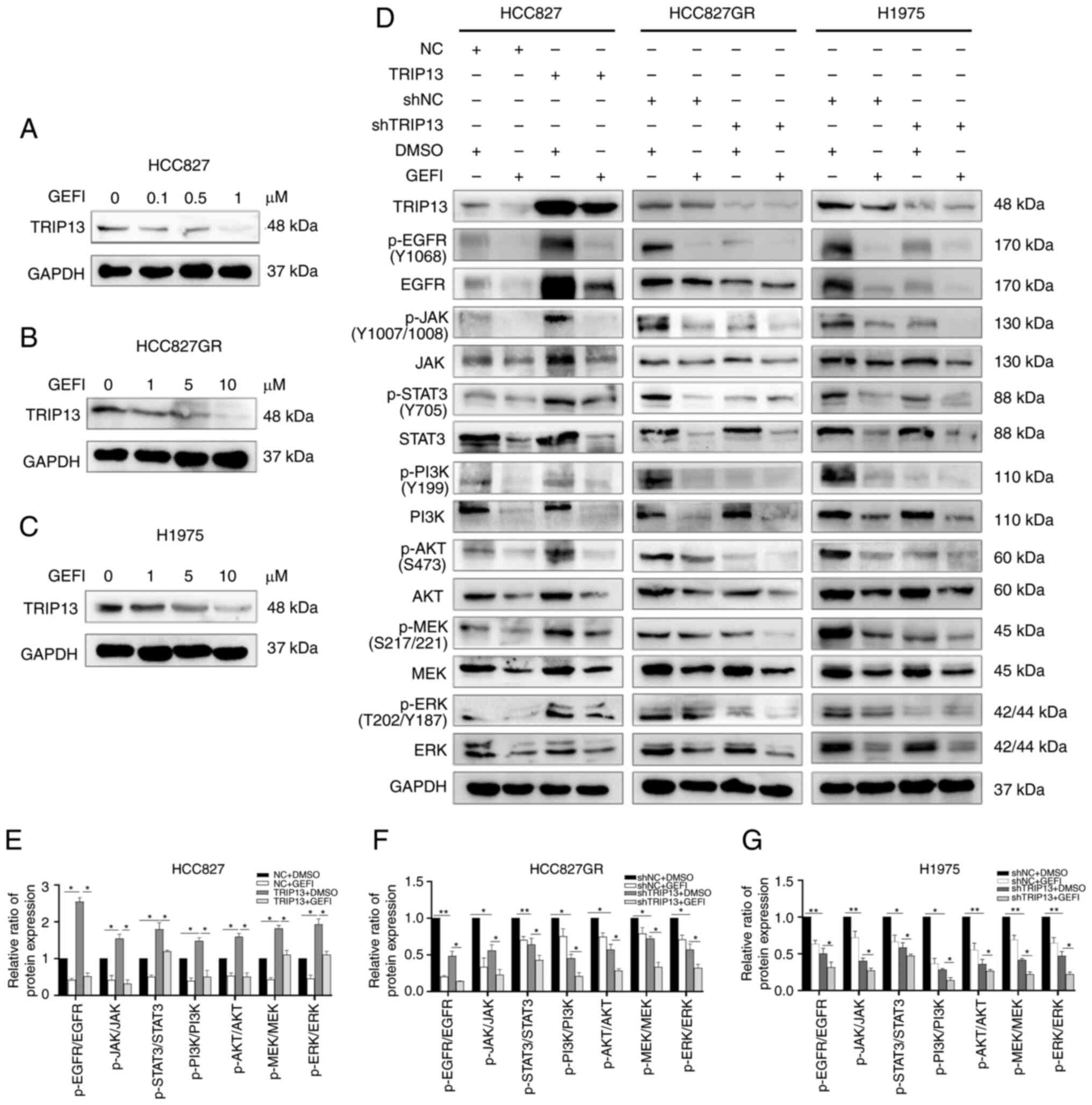 | Figure 7.TRIP13 increases the expression of
key EGFR pathway proteins in NSCLC cells. (A-C) Western blot
analysis showing the dose-dependent decrease of TRIP13 levels
induced by gefitinib at 12 h in HCC827, HCC827GR and H1975 NSCLC
cells. (D) Expression of key EGFR pathway proteins in HCC827
(treated with 0.5 µM gefitinib for 12 h), H1975 and HCC827GR cells
(treated with 5 µM gefitinib for 12 h) after TRIP13 overexpression
or knockdown. (E-G) Relative expression levels of proteins for each
group determined via western blotting. DMSO and GAPDH were used as
drug control and internal control, respectively. The data obtained
were expressed as the mean ± SD. *P<0.05 and **P<0.01.
TRIP13, thyroid hormone receptor interactor 13; EGFR, epidermal
growth factor receptor; NSCLC, non-small cell lung cancer; EGFR,
epidermal growth factor receptor (Tyr1068); JAK, Janus kinase
(Tyr1007/1008); STAT3, signal transducer and activator of
transcription 3 (Tyr705); PI3K,
phosphatidylinositol-4,5-bisphosphate 3-kinase (Tyr199); AKT, V-akt
murine thymoma viral oncogene homolog (Ser473); MEK,
mitogen-activated protein kinase (Ser217/221); ERK,
mitogen-activated protein kinase (Thr202/Tyr187); DMSO, dimethyl
sulfoxide; GEFI, gefitinib; p-, phosphorylated; NC, negative
control cells; sh-, short hairpin. |
Discussion
In lung cancer, TRIP13 has been found to promote
carcinogenesis by activating the PI3K/AKT/mTOR signaling pathway,
which is also consistent with the TCGA data. However, the
underlying mechanisms remain unknown (17,18).
The role of TRIP13 in tumor drug resistance has markedly received
attention in recent years. TRIP13 induces cisplatin and doxorubicin
resistance in bladder cancer cells and desensitizes head and neck
cancers to radiation and cetuximab. In addition, TRIP13 molecular
inhibitors have a beneficial synergistic effect against multiple
myeloma (20–22). However, the relationship between
TRIP13 and gefitinib resistance in NSCLC has not been reported.
In the present study, HCC827 (with EGFR
19del) and H1975 (with EGFR L858R and T790M mutations) cells
were used as gefitinib-sensitive and gefitinib-resistant NSCLC
cells, respectively. HCC827GR cells were used to investigate the
role of TRIP13 in the development of gefitinib resistance in NSCLC.
TRIP13 expression was higher in gefitinib-resistant cells than in
gefitinib-sensitive cells. It was hypothesized that both
EGFR T790M mutation and continuous gefitinib treatment would
increase TRIP13 expression, leading to the development of a
gefitinib-resistant phenotype. Alterations in the IC50
values of NSCLC cells were correlated with changes in TRIP13
protein levels. As revealed by colony formation, proliferation and
apoptosis assays in vitro, TRIP13 overexpression lowered the
susceptibility of HCC827 cells to gefitinib, whereas TRIP13
knockdown restored gefitinib sensitivity in both H1975 and HCC827GR
cells. A possible explanation for the aforementioned results is
that TRIP13 expression was enhanced in gefitinib-resistant cells
compared with that in gefitinib-sensitive cells, promoting cell
proliferation and colony formation and inhibiting apoptosis,
ultimately leading to gefitinib resistance. Taken together, the
aforementioned findings indicated that TRIP13 plays a critical role
in gefitinib resistance and suggested that TRIP13 may be a
potential biomarker for gefitinib resistance in NSCLC.
Considering that patients with advanced lung cancer
develop TKI-induced autophagy following long-term gefitinib
treatment and that autophagy promotes gefitinib resistance
(34,35), the relationship between TRIP13 and
autophagy was investigated using DMSO as a control, gefitinib as an
autophagy agonist and 3-MA as an autophagy inhibitor.
Immunofluorescence results suggested that TRIP13 increased the
number of LC3B-positive puncta in the NSCLC cells. Moreover, TRIP13
overexpression increased LC3B and decreased P62 expression levels
in HCC827 cells. TRIP13 downregulation had the opposite effects in
H1975 and HCC827GR cells. All aforementioned effects were improved
by gefitinib treatment and reversed by 3-MA. Therefore, TRIP13 may
induce gefitinib resistance in NSCLC cells by enhancing autophagy.
Notably, TRIP13 promoted lung carcinogenesis by triggering the
PI3K/AKT/mTOR pathway. However, the activation of this pathway
suppresses autophagy, which contradicts the findings of the present
findings (36).
The authors' hypothesis concerning this
contradiction was that TRIP13 overexpression inhibited autophagy by
triggering the PI3K/AKT/mTOR pathway. It also enhanced autophagy
via other pathways, such as promoting drug resistance-related
pathways or directly regulating key autophagy-related proteins,
which had a greater impact on improving autophagy than the
PI3K/AKT/mTOR pathway. TRIP13 generally promotes autophagy.
Although the GEPIA database did not demonstrate a
significant correlation between TRIP13 and EGFR in NSCLC cells,
immunofluorescence and co-immunoprecipitation assays confirmed that
TRIP13 colocalized with EGFR in the cytoplasm and underwent protein
interactions in NSCLC cells. Notably, EGFR was found in both the
cell membrane and cytoplasm of HCC827 cells but was mostly located
in the nucleus and less in the cytoplasm of HCC827GR cells.
Long-term gefitinib therapy in sensitive cells resulted in a shift
in EGFR location from the cytoplasm to the nucleus. Indeed, the
change in EGFR location in cells is also one of the reasons why
EGFR-TKI resistance arises, as demonstrated in a previous study
(37). There is a positive protein
interaction between TRIP13 and EGFR, thus the expression of TRIP13
will decrease along with EGFR after gefitinib is added. When EGFR
interacts with ligands, it activates various downstream signaling
pathways via phosphorylation. The main pathways implicated in
EGFR-TKI resistance include the MEK/ERK, JAK/STAT3 and
PI3K/Akt/mTOR signaling pathways. All three pathways are downstream
of EGFR and have been implicated in several malignancies (29–32).
In contrast to the PI3K/AKT/mTOR pathway, which suppresses
autophagy, both the MEK/ERK and JAK/STAT3 pathways increase
autophagy. It was identified that TRIP13 upregulation enhanced the
protein phosphorylation levels of EGFR downstream pathways, whereas
TRIP13 downregulation had the opposite effect. TRIP13 contributes
to the phosphorylation of the EGFR signaling pathway in NSCLC,
resulting in gefitinib resistance.
However, the present study has not examined the
expression alterations of TRIP13 in NSCLC patients before and after
gefitinib treatment, and patients with gefitinib-resistant, which
will help to confirm the present results. The specific mechanism by
which TRIP13 induces autophagy and gefitinib resistance in NSCLC
remains unclear. The specific role of TRIP13 in second- and
third-generation TKIs including afatinib and osimertinib also needs
to be confirmed. In addition, it is crucial to determine whether
TRIP13 is involved in pathways other than EGFR for gefitinib
resistance in NSCLC. Further studies are needed to understand the
mechanisms underlying this phenomenon and confirm the therapeutic
applicability of these drugs.
In conclusion, the present study indicated that
TRIP13 could induce gefitinib resistance in NSCLC by promoting
autophagy and phosphorylation of the EGFR signaling pathway. Thus,
TRIP13 can be used as a biomarker and therapeutic target for
gefitinib-resistant NSCLC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Liaoning (grant no. 2020-MS-179).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZXX designed the study, carried out the experiments,
collected and analyzed the data and wrote the manuscript. MXL and
XQZ made recommendations and helped with data analysis. XZR and HTX
were in charge of the conception and supervision of the study. All
authors revised draft versions and read and approved the final
manuscript. ZXX and HTX confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The present study was approved [approval no. LS
(2021)009] by the Research Ethics Committee of the China Medical
University. The animal research protocol was reviewed and approved
(IACUC Issue no. KT2022488) by the Institutional Animal Research
Committee of China Medical University (Shenyang, China). Written
informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
EGFR-TKIs
|
epidermal growth factor receptor
tyrosine kinase inhibitors
|
|
TRIP13
|
thyroid hormone receptor interactor
13
|
|
LUAD
|
lung adenocarcinoma
|
|
LUSC
|
lung squamous cell carcinoma
|
|
IC50
|
half-maximal inhibitory
concentration
|
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shigematsu H, Lin L, Takahashi T, Nomura
M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et
al: Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J Natl
Cancer Inst. 97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Majem M and Remon J: Tumor heterogeneity:
Evolution through space and time in EGFR mutant non small cell lung
cancer patients. Transl Lung Cancer Res. 2:226–237. 2013.PubMed/NCBI
|
|
4
|
Westover D, Zugazagoitia J, Cho BC, Lovly
CM and Paz-Ares L: Mechanisms of acquired resistance to first- and
second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 29
(Suppl_1):i10–i19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clairmont CS, Sarangi P, Ponnienselvan K,
Galli LD, Csete I, Moreau L, Adelmant G, Chowdhury D, Marto JA and
D'Andrea AD: TRIP13 regulates DNA repair pathway choice through
REV7 conformational change. Nat Cell Biol. 22:87–96. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma HT and Poon RY: TRIP13 functions in the
establishment of the spindle assembly checkpoint by replenishing
O-MAD2. Cell Rep. 22:1439–1450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye Q, Kim DH, Dereli I, Rosenberg SC,
Hagemann G, Herzog F, Tóth A, Cleveland DW and Corbett KD: The AAA+
ATPase TRIP13 remodels HORMA domains through N-terminal engagement
and unfolding. EMBO J. 36:2419–2434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alfieri C, Chang L and Barford D:
Mechanism for remodelling of the cell cycle checkpoint protein MAD2
by the ATPase TRIP13. Nature. 559:274–278. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yost S, de Wolf B, Hanks S, Zachariou A,
Marcozzi C, Clarke M, de Voer R, Etemad B, Uijttewaal E, Ramsay E,
et al: Biallelic TRIP13 mutations predispose to Wilms tumor and
chromosome missegregation. Nat Genet. 49:1148–1151. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Raina VB and Vader G: Homeostatic control
of meiotic prophase checkpoint function by Pch2 and Hop1. Curr
Biol. 30:4413–4424.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li C, Xia J, Franqui-Machin R, Chen F, He
Y, Ashby TC, Teng F, Xu H, Liu D, Gai D, et al: TRIP13 modulates
protein deubiquitination and accelerates tumor development and
progression of B cell malignancies. J Clin Invest. 131:e1468932021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sheng N, Yan L, Wu K, You W, Gong J, Hu L,
Tan G, Chen H and Wang Z: TRIP13 promotes tumor growth and is
associated with poor prognosis in colorectal cancer. Cell Death
Dis. 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang G, Zhu Q, Fu G, Hou J, Hu X, Cao J,
Peng W, Wang X, Chen F and Cui H: TRIP13 promotes the cell
proliferation, migration and invasion of glioblastoma through the
FBXW7/c-MYC axis. Br J Cancer. 121:1069–1078. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou XY and Shu XM: TRIP13 promotes
proliferation and invasion of epithelial ovarian cancer cells
through notch signaling pathway. Eur Rev Med Pharmacol Sci.
23:522–529. 2019.PubMed/NCBI
|
|
15
|
Dong L, Ding H, Li Y, Xue D, Li Z, Liu Y,
Zhang T, Zhou J and Wang P: TRIP13 is a predictor for poor
prognosis and regulates cell proliferation, migration and invasion
in prostate cancer. Int J Biol Macromol. 121:200–206. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu L, Xiao Y, Zhou X, Wang J, Chen S, Peng
T and Zhu X: TRIP13 interference inhibits the proliferation and
metastasis of thyroid cancer cells through regulating TTC5/p53
pathway and epithelial-mesenchymal transition related genes
expression. Biomed Pharmacother. 120:1095082019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cai W, Ni W, Jin Y and Li Y: TRIP13
promotes lung cancer cell growth and metastasis through
AKT/mTORC1/c-Myc signaling. Cancer Biomark. 30:237–248. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li W, Zhang G, Li X, Wang X, Li Q, Hong L,
Shen Y, Zhao C, Gong X, Chen Y and Zhou J: Thyroid hormone receptor
interactor 13 (TRIP13) overexpression associated with tumor
progression and poor prognosis in lung adenocarcinoma. Biochem
Biophys Res Commun. 499:416–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li ZH, Lei L, Fei LR, Huang WJ, Zheng YW,
Yang MQ, Wang Z, Liu CC and Xu HT: TRIP13 promotes the
proliferation and invasion of lung cancer cells via the Wnt
signaling pathway and epithelial-mesenchymal transition. J Mol
Histol. 52:11–20. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Huang J, Li B, Xue H, Tricot G, Hu
L, Xu Z, Sun X, Chang S, Gao L, et al: A small-molecule inhibitor
targeting TRIP13 suppresses multiple myeloma progression. Cancer
Res. 80:536–548. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Banerjee R, Liu M, Bellile E, Schmitd LB,
Goto M, Hutchinson MN, Singh P, Zhang S, Damodaran DP, Nyati MK, et
al: Phosphorylation of TRIP13 at Y56 induces radiation resistance
but sensitizes head and neck cancer to cetuximab. Mol Ther.
30:468–484. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu S, Guo M, Fan Z, Chen Y, Shi X, Gu C
and Yang Y: Elevated TRIP13 drives cell proliferation and drug
resistance in bladder cancer. Am J Transl Res. 11:4397–4410.
2019.PubMed/NCBI
|
|
23
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dikic I and Elazar Z: Mechanism and
medical implications of mammalian autophagy. Nat Rev Mol Cell Biol.
19:349–364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu M and Zhang P: EGFR-mediated autophagy
in tumourigenesis and therapeutic resistance. Cancer Lett.
469:207–216. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Amaravadi RK, Kimmelman AC and Debnath J:
Targeting autophagy in cancer: Recent advances and future
directions. Cancer Discov. 9:1167–81. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sigismund S, Avanzato D and Lanzetti L:
Emerging functions of the EGFR in cancer. Mol Oncol. 12:3–20. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng WL, Feng PH, Lee KY, Chen KY, Sun
WL, Van Hiep N, Luo CS and Wu SM: The role of EREG/EGFR pathway in
tumor progression. Int J Mol Sci. 22:128282021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pao W and Miller VA: Epidermal growth
factor receptor mutations, small-molecule kinase inhibitors, and
non-small-cell lung cancer: Current knowledge and future
directions. J Clin Oncol. 23:2556–2568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu Q, Yu S, Zhao W, Qin S, Chu Q and Wu
K: EGFR-TKIs resistance via EGFR-independent signaling pathways.
Mol Cancer. 17:532018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li YY, Lam SK, Mak JC, Zheng CY and Ho JC:
Erlotinib-induced autophagy in epidermal growth factor receptor
mutated non-small cell lung cancer. Lung Cancer. 81:354–361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gremke N, Polo P, Dort A, Schneikert J,
Elmshäuser S, Brehm C, Klingmüller U, Schmitt A, Reinhardt HC,
Timofeev O, et al: mTOR-mediated cancer drug resistance suppresses
autophagy and generates a druggable metabolic vulnerability. Nat
Commun. 11:46842020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rong X, Liang Y, Han Q, Zhao Y, Jiang G,
Zhang X, Lin X, Liu Y, Zhang Y, Han X, et al: Molecular mechanisms
of tyrosine kinase inhibitor resistance induced by
membranous/cytoplasmic/nuclear translocation of epidermal growth
factor receptor. J Thorac Oncol. 14:1766–1783. 2019. View Article : Google Scholar : PubMed/NCBI
|