Introduction
Enolase-1 (ENO1) is a multifunctional protein
(1), which mainly acts as a
glycolytic enzyme in the cytosol to catalyze the conversion of
2-phospho-D-glycerate to phosphoenolpyruvate during aerobic
glycolysis (2). Cancer cells
undergo a metabolic shift to an aerobic glycolytic phenotype
(termed the Warburg effect) during tumorigenesis, which provides
cancer cells with survival advantages (3). This metabolic shift can be caused by
an upregulation in ENO1 expression followed by enhanced conversion
of pyruvate into lactate (4,5). ENO1
is also expressed on the cell surface where it acts as a
plasminogen receptor under certain circumstances (6), which helps sequester circulating
plasminogen to facilitate its activation. Resulting from
proteolytic processing from plasminogen, plasmin acts as a potent
extracellular serine protease specialized in the degradation of
extracellular matrix. Owing to this ability, cells armed with
plasminogen receptors would be able to harness the ability of
plasmin for migration or invasion. Moreover, it has been reported
that surface ENO1 promotes infiltration of immune cells (7) and metastasis of cancer cells (8) via its plasminogen-activating ability
(9).
Upregulation of ENO1 has been observed in multiple
cancer types, including multiple myeloma (MM) and prostate, lung
and pancreatic neoplasms, and was often associated with poor
prognosis (10–12). MM is a malignancy of plasma cells
originating in the bone marrow, and is the second most common
hematologic malignancy worldwide and in the United States after
non-Hodgkin lymphoma (13). Despite
the approval of novel therapeutics for MM, such as proteasome
inhibitors (bortezomib) (14),
immunomodulatory drugs (lenalidomide) (15) and monoclonal antibodies
(daratumumab) (16), the treatment
options remain limited for patients with relapsed refractory MM.
Therefore, novel, safe and cost-effective treatments are still in
urgent need. Ray et al (11)
found a significantly higher ENO1 gene expression level in
patients with MM compared with the control subjects. Moreover, the
level of ENO1 gene expression was inversely correlated with
the overall survival of patients with MM. ENO1 protein expression
on the surface of MM cells was also confirmed (11). Since blocking surface ENO1 with
antibodies has been demonstrated to be an effective
anti-invasive/metastasis strategy for tumor progression in lung,
pancreatic and cervical cancer (17–20),
it was hypothesized that ENO1 could be a favorable therapeutic
target for MM using our propriety ENO1-specific monoclonal antibody
(mAb).
It was previously reported by the authors that an
ENO1 mAb could attenuate tumor growth in prostate cancer (PCa)
xenografts via disrupting the tumor microenvironment (TME)
(12). Yu et al (21) also demonstrated that secreted ENO1
in the TME promoted PCa cell migration and metastasis. In addition,
Ray et al (11) reported
that co-culture of MM cells with plasmacytoid dendritic cells
(pDCs) in the TME increased ENO1 expression on the MM cells. After
treatment with an ENO1 inhibitor, pDCs acquired enhanced abilities
of pDC-triggered T and NK cell-mediated anti-MM activity, which
suggested a contribution of ENO1 to MM progression in the bone
marrow TME. In addition, elevated lactate production in MM cells
and the TME also contributes to the survival of MM cells (22). Evidence has also indicated that cell
metabolic reprogramming necessitated cancer cells to upregulate the
expression of key regulators of the glycolytic pathway, such as
glucose transporter 1 (GLUT1), hexokinase 2 (HK2),
phosphofructokinase (PFK) and ENO1 (23).
ENO1 was first recognized as one of the major
regulators of glycolysis and its enzymology has been well studied.
Subsequently, the ‘moonlighting’ functions of surface ENO1 have
been gradually revealed in addition to the ‘main’ function
(glycolysis) of intracellular ENO1, of which ‘moonlighting’ and
‘main’ are defined solely according to the time of discovery
(10). Notably, the multiple
functions of ENO1 have all been suggested to be involved in cancer
development (24). However, to the
best of our knowledge, it has not yet been addressed how ENO1
allocates the ‘main’ and ‘moonlighting’ duties to its intracellular
and extracellular (surface or secreted) forms and how these duties
are regulated. Therefore, in the present study, it was first
determined whether extracellular ENO1 is involved in glycolysis
regulation despite only cytosolic ENO1 being implicated in
glycolysis thus far. Secondly, whether extracellular ENO1 could
regulate glycolysis along with pro-cancer activities in MM cells
was investigated. Lastly, a humanized ENO1-specific antibody was
used to validate the role of extracellular ENO1 in glycolysis and
tumor growth in vivo. To the best of our knowledge, this is
the first study to explore the possibility that extracellular ENO1
(surface or secreted) could regulate glycolysis in cancer cells.
The results of the present study may therefore shed light on the
basic biology of ENO1 and the development of potential therapeutics
for cancer.
Materials and methods
Human tissue array
An MM tissue array was obtained from TissueArray.com (formerly US Biomax, Inc.) (cat. no.
BM483d), which contained 10 cases of plasma cell neoplasms and 11
bone marrow tissue (samples from the non-bone tumors in the chest
surgery), with duplicate cores per case. Full ethical approval was
granted for all aspects of the study and informed patient consent
was obtained before tissue collection.
Animal studies
All animal studies were reviewed and approved by The
Ethics Committee of TFBS Bioscience (Taipei, Taiwan). All animal
procedures were performed according to approved protocols from the
Institutional Animal Care and Use Committee (IACUC) of TFBS
Bioscience (IACUC protocol no. TFBS2021-013). A total of 18 male
6–7-week-old BALB/c nude (nu/nu) mice (weighing 17–22 g) were
purchased from BioLASCO Taiwan Co., Ltd. for the present study. The
mice had free access to food and water and were kept on a 12/12-h
light/dark cycle in the chambers under atmospheric conditions of
22°C and 30–60% humidity. Before inoculation, RPMI-8226 cells were
washed with PBS and resuspended in serum-free RPMI-1640 medium and
Matrigel (cat. no. 356231; Corning, Inc.) at a 1:1 ratio. The cells
(5×106/100 µl) were subsequently implanted
subcutaneously into the right flank of the mice. The subcutaneous
tumor volume was determined according to the following formula:
Tumor volume=(shorter diameter2 × longer diameter)/2.
After the tumor size reached >100 mm3, the mice were
randomized into three groups (n=6): Group 1 were administered PBS
as a vehicle control; group 2 were administered a total of nine
doses of ENO1 mAb; and group 3 were administered a total of five
doses of ENO1 mAb. ENO1 mAb (30 mg/kg) was administered twice a
week by intraperitoneal injection. Based on the tumorigenesis
assessment, tumor weight >10% of the animal body weight was
considered an indicator for animal sacrifice. To ameliorate the
suffering of animals observed throughout the experimental studies,
animals were euthanized by CO2 inhalation (30–70% of the
cage volume per min).
Cell culture
The RPMI-8226 [Bioresource Collection and Research
Center (BCRC) no. 60384; RRID: CVCL_0014] and U266 (BCRC no. 60437;
RRID: CVCL_0566) human MM cell lines and the PC-3 human PCa cell
line (BCRC no. 60122, RRID: CVCL_0035) were obtained from the BCRC
(Hsinchu, Taiwan). STR-PCR profiling was also conducted at the
BCRC. The KMS-11 (cat. no. JCRB1179) cell line was obtained from
the Japanese Collection of Research Bioresources Cell Bank (Osaka,
Japan). The cells were cultured in RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) and 50 U/ml
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at
37°C under 5% CO2. For the evaluation of the effects of
recombinant ENO1 on lactate production, lactate dehydrogenase (LDH)
activity and the expression levels of candidate genes, RPMI-8226
cells were cultured in RPMI-1640 containing 2% FBS during
treatment.
Reagents
Genes encoding human ENO1 proteins, including
ENO1-wild-type (WT), ENO1-S40A and ENO1-D245R, were cloned into a
pTrcHis vector, in which the expression of the transgene was
isopropyl-β-D-thiogalactoside-inducible. The expression of
recombinant ENO1 proteins in Escherichia coli and subsequent
purification were performed by Leadgene Biomedical, Inc. (Tainan,
Taiwan). The ENO1 proteins were suspended in 1X PBS containing 7 mM
MgSO4 and 2% trehalose, pH 7.2. Production of the
proprietary ENO1 mAb by HuniLife was previously described (12). The ENO1 mAb was a humanized IgG1
antibody and cross reactive to both human and mouse ENO1, but was
not reactive to ENO2 and ENO3. Human IgG1 (hIgG1; cat. no. HG1K;
Sino Biological, Inc.) and anti-Klebsiella pneumoniae (hIgG1
backbone; provided by Dr Shih-Chong Tsai, Development Center for
Biotechnology, Taipei, Taiwan) antibodies were used as isotype
controls in the in vitro and in vivo studies,
respectively. Recombinant human TNF-α protein (cat. no. 300-01A)
was purchased from PeproTech, Inc. The nuclear factor-κB (NF-κB)
inhibitor, BAY11-7085 (cat. no. sc-202490), was purchased from
Santa Cruz Biotechnology, Inc.
Immunohistochemistry
All tissues were fixed in 10% neutral buffered
formalin at room temperature for 24 h, dehydrated with gradient
ethanol, cleared with xylene, and embedded in paraffin. MM tissue
array sections (5-µm thick) were deparaffinized using two
sequential 5 min washes in fresh xylene at room temperature, then
gradually rehydrated in graded ethanol of 100, 95, 80, 70 and 50%
and distilled water at room temperature for 3 min each.
Heat-induced epitope retrieval was performed with 0.02 M Tris-EDTA
(pH 9.0) using a microwave for 20 min. Endogenous peroxidase
activity was quenched then tissues were stained with primary
antibody against ENO1 (1:500; cat. no. ab227978; Abcam) or isotype
control antibody (1:500; cat. no. ab172730; Abcam) at 4°C
overnight. The tissue slides were subsequently incubated with
HRP-conjugated secondary antibody (1:1,000; cat. no. 7074; Cell
Signaling Technology, Inc.) for 30 min at room temperature. Then,
the DAB reaction was performed until the desired staining was
achieved. The slides were then mounted after tissues were
counterstained with hematoxylin. Whole images of each core were
acquired using a Nikon microscope (Nikon Instruments) and the
positively stained area (%) of each image was quantified using
Nikon NIS-Elements (BR) software (Nikon Instruments Inc.).
RNA interference (RNAi)
The ENO1 (Shanghai GenePharma Co., Ltd.) and HIF-1α
(cat. no. AM51331; Invitrogen; Thermo Fisher Scientific, Inc.)
siRNA sequences were as follows: si-ENO1 #1 sense,
5′-CGUACCGCUUCCUUAGAACUUTT-3′ and antisense,
5′-AAGUUCUAAGGAAGCGGUACGTT-3′; si-ENO1 #2 sense
5′-GAAUGUCAUCAAGGAGAAAUATT-3′ and antisense,
5′-UAUUUCUCCUUGAUGACAUUCTT-3′; si-HIF1A sense,
5′-GGGUAAAGAACAAAACACA-3′ and antisense, 5′-UGUGUUUUGUUCUUUACCC-3′;
and scramble siRNA sense 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. Cells were transfected with 50 nM
dsRNA duplexes using RNAiMAX (cat. no. 13778; Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, RPMI 8226 (5×105), U266
(5×105) and PC-3 (2×105) cells were plated in
2.5 ml of culture medium per well of six-well plates, and then were
transfected with dsRNA duplexes (50 nM) using RNAiMAX for 72 h at
37°C under 5% CO2, and then the ENO1-knockdown cells
were used for subsequent experiments. For knockdown of HIF-1α, RPMI
8226 cells were transfected again for another 24 h following the
first transfection using the same protocol as aforementioned.
ENO1 stable overexpression
The ENO1 gene was inserted into the
expression vector, pLenti-C-Myc-DDK-P2A-Puro (cat. no. RC205494L3;
OriGene Technologies, Inc.). KMS-11 cells were plated at
8×105 cells in 2 ml of culture medium per well of
six-well plates, and then were transfected with ENO1 expressing
plasmid (4 µg) using Avalanche Transfection Reagent (cat. no.
EZT-RPMI-1; EZ Biosystems™) for 5 h at 37°C under 5%
CO2, followed by adding 0.5 ml of culture medium. Those
cells stably overexpressing ENO1 (Myc/DDK-tagged) were selected
with puromycin (2 µg/ml) for >2 weeks. Mock cells treated with
transfection reagent alone were used as the control group. The
efficacy of stable ENO1 overexpression was detected by
immunoblotting analysis.
Flow cytometry detection of cell
surface ENO1
ENO1-knockdown RPMI-8226 and U266 cells were stained
for 30 min at room temperature with LIVE/DEAD Fixable Near-IR (775)
(cat. no. L34975; Invitrogen; Thermo Fisher Scientific, Inc.), and
then washed twice in PBS at 300 × g for 5 min. Cells were further
incubated with Fc-block receptor (cat. no. 130-059901; Miltenyi
Biotech, Inc.) for 10 min at 4°C and then washed once in cold Stain
Buffer (cat. no. 554656; BD Biosciences) at 300 × g for 5 min.
Cells were stained with ENO1 (1:50; cat. no. H00002023-M01; Abnova)
or isotype-matched mouse IgG1 (1:50; cat. no. 401402; Biolegend,
Inc.) antibodies for 30 min at 4°C, then washed twice in cold Stain
Buffer at 300 × g for 5 min. Cells were then incubated with PE goat
anti-mouse IgG (1:20; cat. no. 405307; Biolegend, Inc.) for 30 min
at 4°C, followed by washing twice in cold Stain Buffer at 300 × g
for 5 min. After staining, the samples were resuspended in cold
Stain Buffer and analyzed by a CytoFlex flow cytometer (Beckman
Coulter, Inc.). Data were acquired using CytExpert 2.4 software and
analyzed using Kaluza Analysis 2.1 Software (Beckman Coulter,
Inc.). For each sample, 1×104 cells were acquired. For
analysis, the dead cells were first excluded with LIVE/DEAD Fixable
Near-IR (775) Dead Cell Stain. Quantification of the surface ENO1
level [in mean fluorescence intensity (MFI)] was obtained by
subtracting the MFI of the samples from the isotype control.
Antibody labelling assay for detection
of cell surface ENO1
The method was as previously described (25), with modification. Briefly,
ENO1-knockdown RPMI-8226 (1×105) and PC-3
(0.5×105) cells were harvested and washed twice with
cold PBS, fixed with 100 µl of 1% paraformaldehyde for 10 min on
ice, and blocked with 200 µl of 1% BSA (cat. no. A9418; Sigma
Aldrich; Merck KGaA) in PBS for 2 h at room temperature. After
blocking, the cells were probed with ENO1 antibody (1:200; cat. no.
H00002023-M01; Abnova) at 37°C for 1 h followed by HRP-conjugated
anti-mouse secondary antibody (1:7,500; cat. no. 2076; Cell
Signaling Technology, Inc.) at 37°C for 30 min. Final detection was
performed using TMB substrate (cat. no. 5120; KPL; SeraCare; LGC
Clinical Diagnostics), and the absorbance at 450 nm was determined
using a microplate reader.
Immunoblotting analysis
RPMI-8226 or PC-3 lysates were prepared and
subjected to immunoblotting analysis according to standard
protocol. Briefly, cells were lysed in RIPA buffer (25 mM Tris-HCl,
pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS;
cat. no. 89900; Thermo Fisher Scientific, Inc.) containing the 1X
protease inhibitor cocktail (cat. no. 78340; Thermo Fisher
Scientific, Inc.) on ice for 30 min, followed by centrifugation
(13,500 × g for 10 min at 4°C). The protein concentrations were
determined using a BCA protein assay kit (cat. no. 23225; Pierce;
Thermo Fisher Scientific, Inc.). Proteins (30 µg) were resolved on
a 10% SDS-PAGE gel, and then transferred to PVDF membranes (cat. no
10600023; Cytiva Life Sciences). The membranes were blocked in TBST
(0.1% Tween-20 in TBS) with 5% non-fat dry milk for 1 h at room
temperature, and then incubated overnight at 4°C with diluted
primary antibodies in TBST with 5% non-fat dry milk. The membranes
were then washed three times for 10 min each with TBST at room
temperature on a gel rocker, and incubated with secondary
antibodies coupled to HRP in TBST with 5% non-fat dry milk for 1 h
at room temperature, followed by washing three times for 10 min
each with TBST at room temperature on a gel rocker. The
immunoreactive bands were detected with the ECL™ Select Western
Blotting Detection Reagent (cat. no GERPN2235; Cytiva). The primary
antibodies were as follows: ENO1 (1:2,000; cat. no. ab190365;
Abcam), HIF-1α (1:1,000; cat. no. 610958; BD Biosciences), HK2
(1:2,000; cat. no. sc-130358; Santa Cruz Biotechnology, Inc.),
6-phosphofructo-2-kinase/fructose-2,6 biphosphatase 3 (PFKFB3;
1:2,000; cat. no. ab96699; Abcam), GLUT1 (1:5,000; cat. no.
ab115730; Abcam), IκBα (1:2,000; cat. no. 9242; Cell Signaling
Technology, Inc.), PHD2 (1:1,000; cat. no. sc-271835; Santa Cruz
Biotechnology, Inc.) and GAPDH (1:5,000; cat. no. sc-32233; Santa
Cruz Biotechnology, Inc.). The secondary antibodies were as
follows: HRP-conjugated anti-mouse secondary antibody (1:5,000;
cat. no. 2076; Cell Signaling Technology, Inc.) and HRP-conjugated
anti-rabbit secondary antibody (1:5,000; cat. no. 7074; Cell
Signaling Technology, Inc.).
Lactate and LDH assays
The levels of lactate in cell culture supernatant
and intracellular LDH activity were measured using colorimetric
lactate (cat. no. MET-5012; Cell Biolabs, Inc.) and LDH (cat. no.
CK12-20; Dojindo Laboratories, Inc.) assay kits, respectively,
according to the manufacturer's instructions. LDH activity results
were normalized to the protein concentrations of the cell lysates,
and the protein concentrations were determined using a BCA protein
assay kit.
Cell viability and migration
assays
Cell viability was measured using Cell Counting
Kit-8 (cat. no. CK04-20; Dojindo Laboratories, Inc.) according to
the manufacturer's instructions. Briefly, RPMI 8226
(2×104), U266 (2×104) and PC-3
(1×104) cells were plated in 100 µl of culture medium
per well of 96-well plates. At 0, 24, 48 and 72 h, 10 µl of the
Cell Counting Kit-8 solution was added to each well and incubated
at 37°C for 2 h, and the absorbance at 450 nm was measured using a
microplate reader. For the migration assay, cells were resuspended
in medium supplemented with 2% FBS in the absence or presence of
various concentrations of ENO1 mAb or control hIgG1. After adding
900 µl of medium containing 10% FBS to the bottom chamber of the
migration plates, the coating-free insert (8-µm pores; cat. no.
3464; Corning, Inc.) was placed and the upper chamber was seeded
with the treated cells. The cells were then allowed to migrate at
37°C for 18 h. The remaining cells in the upper chamber were
removed and the cells in the bottom chamber were fixed with
methanol for 10 min at room temperature, followed by staining with
1% crystal violet for an additional 2 h or overnight at room
temperature. The insert was gently washed with PBS and dried. The
bound crystal violet was eluted with 33% acetic acid and the
absorbance at 590 nm was measured using a microplate reader.
Cytokine ELISA and measurement of
enolase activity
Measurement of cytokine levels was performed using
ELISA kits, including human vascular endothelial growth factor
(VEGF; cat. no. DY293B-05; R&D Systems, Inc.), human
interleukin 6 (IL-6; cat. no. DY206-05; R&D Systems, Inc.) and
human transforming growth factor-β (TGF-β; cat. no. DY240-05;
R&D Systems, Inc.), according to the manufacturer's
instructions. The enolase activity of cell lysates or recombinant
ENO1 (10 ng) were determined using an enolase activity colorimetric
assay kit (cat. no. K691; BioVision, Inc.), according to the
manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA in the treated cells was extracted using
an rSYNC RNA Isolation kit (cat. no. RS300; Geneaid Biotech Ltd.),
and RT of the RNA was performed using an iScript™ cDNA Synthesis
kit (cat. no. 1708891; Bio-Rad Laboratories, Inc.), according to
the manufacturer's instructions. The primer sequences used to
amplify the resulting cDNA were as follows: HIF-1α sense,
5′-AATTCTCAACCACAGTGCATTGTATGT-3′ and antisense,
5′-CTTTGGTGAATAGCTGAGTCATTTTCA-3′; HK2 sense,
5′-GAGTTTGACCTGGATGTGGTTGC-3′ and antisense,
5′-CCTCCATGTAGCAGGCATTGCT-3′; PFKFB3 sense,
5′-GGCAGGAGAATGTGCTGGTCAT-3′ and antisense,
5′-CATAAGCGACAGGCGTCAGTTTC-3′; GLUT1 sense,
5′-TTGCAGGCTTCTCCAACTGGAC-3′ and antisense,
5′-CAGAACCAGGAGCACAGTGAAG-3′; ENO1 sense,
5′-AGTCAACCAGATTGGCTCCGTG-3′ and antisense,
5′-CACAACCAGGTCAGCGATGAAG-3′; c-Myc sense,
5′-CCTGGTGCTCCATGAGGAGAC-3′ and antisense,
5′-CAGACTCTGACCTTTTGCCAGG-3′ and ACTB sense,
5′-CACCATTGGCAATGAGCGGTTC-3′ and antisense,
5′-AGGTCTTTGCGGATGTCCACGT-3′. qPCR was performed on a BioRad CFX
384 using the PowerUp™ SYBR™ Green Master Mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The following thermal cycling
conditions were used: 50°C for 2 min; 95°C for 2 min; 95°C for 15
sec; and 60°C for 1 min for 40 cycles. The fold change in the
expression of target genes were calculated by the relative
quantification method (26). ΔCq
values were obtained as follows: ΔCq=Cq of ACTB-Cq of the
target gene. ΔΔCq values were then obtained as follows: ΔΔCq=ΔCq of
the treated group-ΔCq of the control group. Fold change was
calculated as 2−ΔΔCq, with control groups as 1-fold.
Measurement of glucose uptake
To evaluate glucose uptake, a colorimetric assay kit
(cat. no. ab136955; Abcam) was used according to the manufacturer's
instructions. Briefly, RPMI-8226 and U266 cells were treated with
different concentrations of ENO1 mAb (1, 10 and 100 µg/ml) or hIgG1
(100 µg/ml) in RPMI-1640 media supplemented with 2% FBS for 48 h.
Cells were then washed twice with PBS and cultured in glucose-free
RPMI (Gibco; Thermo Fisher Scientific, Inc.) for 1 h. Subsequently,
the cells were incubated in PBS for 40 min and then with 10 mM
2-deoxy-D-glucose (2-DG) for 20 min. Next, cells were washed three
times with PBS to remove exogenous 2-DG, lysed with extraction
buffer, freeze/thawed once and heated at 85°C for 40 min to degrade
endogenous NAD(P), followed by centrifugation at 300 × g for 2 min
at 4°C. The resulting supernatant was analyzed for
2-deoxy-D-glucose-6-phosphate content using a microplate reader at
412 nm. Cells treated with 100 µM phloretin were used as a positive
control.
Measurement of plasminogen receptor
activity of ENO1
RPMI-8226 and U266 cells were harvested and washed
twice with PBS at 300 × g for 2 min, resuspended in PBS at
1×106 cells/ml and preincubated with various
concentrations of ENO1 mAb (1, 10 and 100 µg/ml) or hIgG1 (100
µg/ml) at 37°C for 1 h, followed by treatment with 40 mM human
Glu-plasminogen (cat. no. 528180; Sigma-Aldrich; Merck KGaA) for 1
h. After incubation, the cells were washed with PBS three times and
resuspended in 100 µl PBS containing 1.5 µM tissue plasminogen
activator (tPA) (cat. no. 612200; Sigma Aldrich; Merck KGaA) and
0.1 mM plasmin chromogenic substrate, Chromogenix S-2251 (cat. no.
S820332; DiaPharma), at 37°C for 2.5 h. The plasminogen receptor
activity (plasmin activation) was determined by measuring the
absorbance at 405 nm.
Statistical analysis
All statistical analyses were performed using SPSS
18.0 (SPSS, Inc.). The data are presented as the mean ± SEM from at
least three separate experiments. Differences between two
individual experiments were compared using two-tailed unpaired
Student's t-tests (Fig. 1B) or
two-tailed paired Student's t-tests (Fig. S5B-F). Comparisons of multiple
groups were performed by using one-way analysis of variance
(ANOVA), and subsequent comparisons of individual groups were
performed using Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
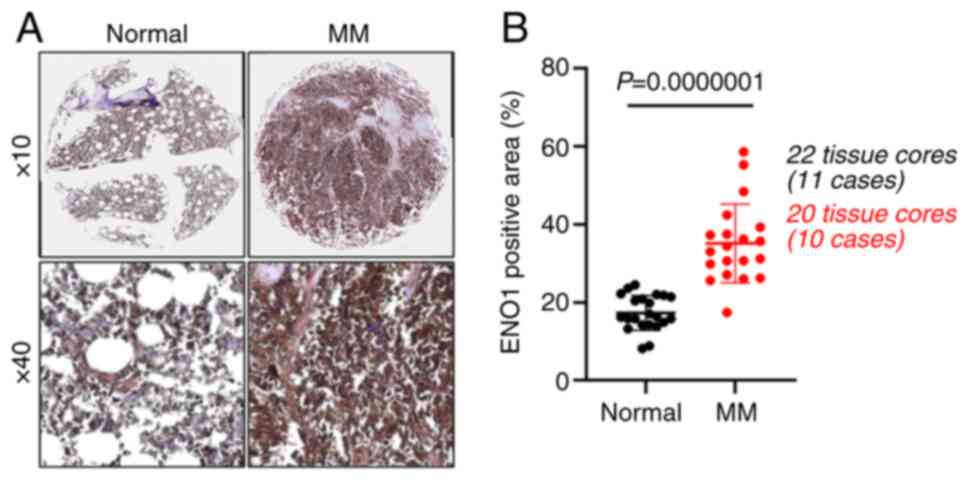
ENO1 is upregulated in the tumor
tissues from patients with MM
A previous study demonstrated that ENO1 gene
expression was significantly higher in patients with MM compared
with control subjects (11).
Therefore, in the present study, ENO1 protein expression was
further examined using a human MM tissue array. The tissue samples
included in tissue array (#BM483d) are summarized in Table SI. As revealed in Fig. 1, ENO1 protein expression was
significantly higher in the bone marrow tissues from patients with
MM compared with normal bone marrow tissues. These results were in
agreement with the authors' previous study, in which upregulation
of ENO1 in advanced grade human PCa tissues was also observed
(12). These data led to a
hypothesis that ENO1 may be involved in the pathogenesis of MM and
PCa.
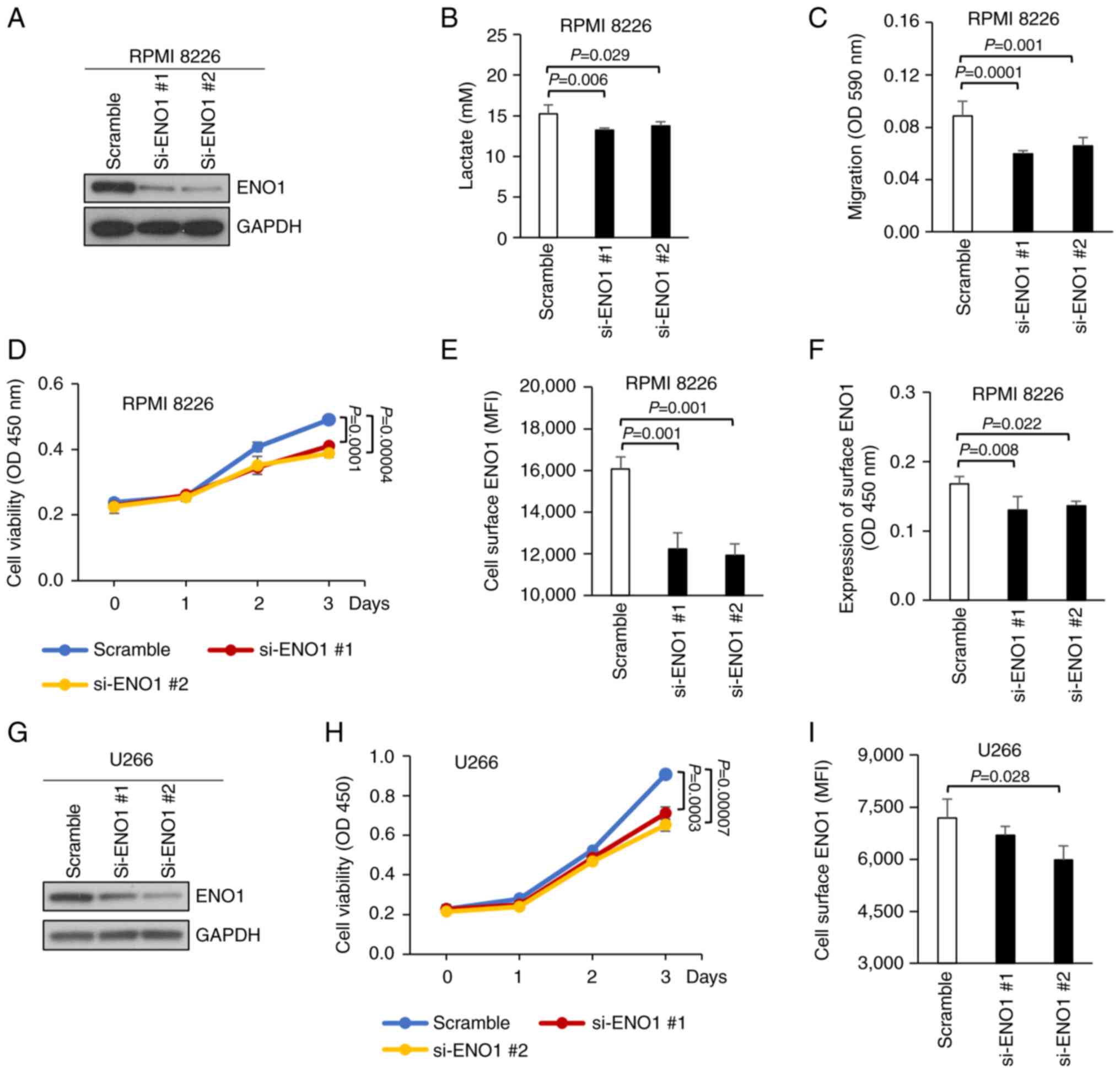
ENO1 knockdown attenuates lactate
production, cell migration and cell viability
Upregulation of ENO1 has been found in various
cancer types, where it enhances glycolysis and cell growth,
migration and invasion (5,18,27,28),
which are generally considered pro-cancer activities. However, the
pathological roles of ENO1 in MM remain largely unknown. Therefore,
in the present study, RNAi-mediated knockdown of ENO1 in human MM
cells (RPMI-8226) was performed. This RNAi approach (by si-ENO1 #1
or #2) successfully reduced the expression of total cellular ENO1
protein in RPMI-8226 MM cells (Fig.
2A). In addition, a significant decrease in the levels of
secreted lactate (the endpoint of glycolysis following the Warburg
effect) from the ENO1-knockdown RPMI-8226 MM cells was observed
(Fig. 2B). Moreover, knocking down
ENO1 expression also inhibited cell migration (Fig. 2C) and cell viability (Fig. 2D). In another MM cell line (U266),
cell viability was also reduced following knockdown of cellular
ENO1 protein expression (Fig. 2G and
H). In addition to MM, PCa cells have also been previously
reported to exhibit the Warburg effect, where glycolysis was
preferentially used for cell growth (29,30).
Therefore, the same RNAi-mediated knockdown of ENO1 expression was
applied to the PC-3 human PCa cells (Fig. S1A). Consistently, ENO1 knockdown
also decreased lactate production (Fig. S1B), migration (Fig. S1C) and viability (Fig. S1D) of PC-3 cells. Taken together,
these results suggested that ENO1 was required for glycolysis,
motility and viability of MM and PCa cells.
Extracellular ENO1 enhances glycolysis
and pro-cancer activities
The enzymatic role of cytosolic ENO1 in glycolysis
is well-known. Therefore, the aforementioned reduced lactate levels
and biological effects of ENO1 knockdown, which reduced the total
cellular expression of ENO1, were not surprising (Figs. 2A, G and S1A). However, knockdown of ENO1 also
simultaneously reduced the levels of surface (or membrane bound)
ENO1 in MM and PCa cells, as detected by flow cytometry (Fig. 2E and I) and antibody labelling
assays (Figs. 2F and S1E). Therefore, it was of interest to
determine whether extracellular ENO1 (surface or secreted) was
involved in the reduced glycolysis and related biological
activities observed in the knockdown studies, since ENO1 enzymatic
activity had been considered to be exclusive to the cytosolic form
thus far. To determine this, cells were treated with recombinant
ENO1-WT. Notably, extracellular ENO1 dose-dependently increased the
level of lactate secreted from RPMI-8226 cells, and this increase
was ENO1-specific since it was attenuated by co-treatment with an
ENO1-specific mAb (Fig. 3A).
Consistently, recombinant ENO1 also significantly promoted
intracellular LDH activity (P=0.028; Fig. 3A), which catalyzed the conversion of
pyruvate to lactate in glycolysis. This enhanced LDH activity was
reduced upon ENO1 mAb co-treatment, although this reduction was not
statistically significant (Fig.
3A).
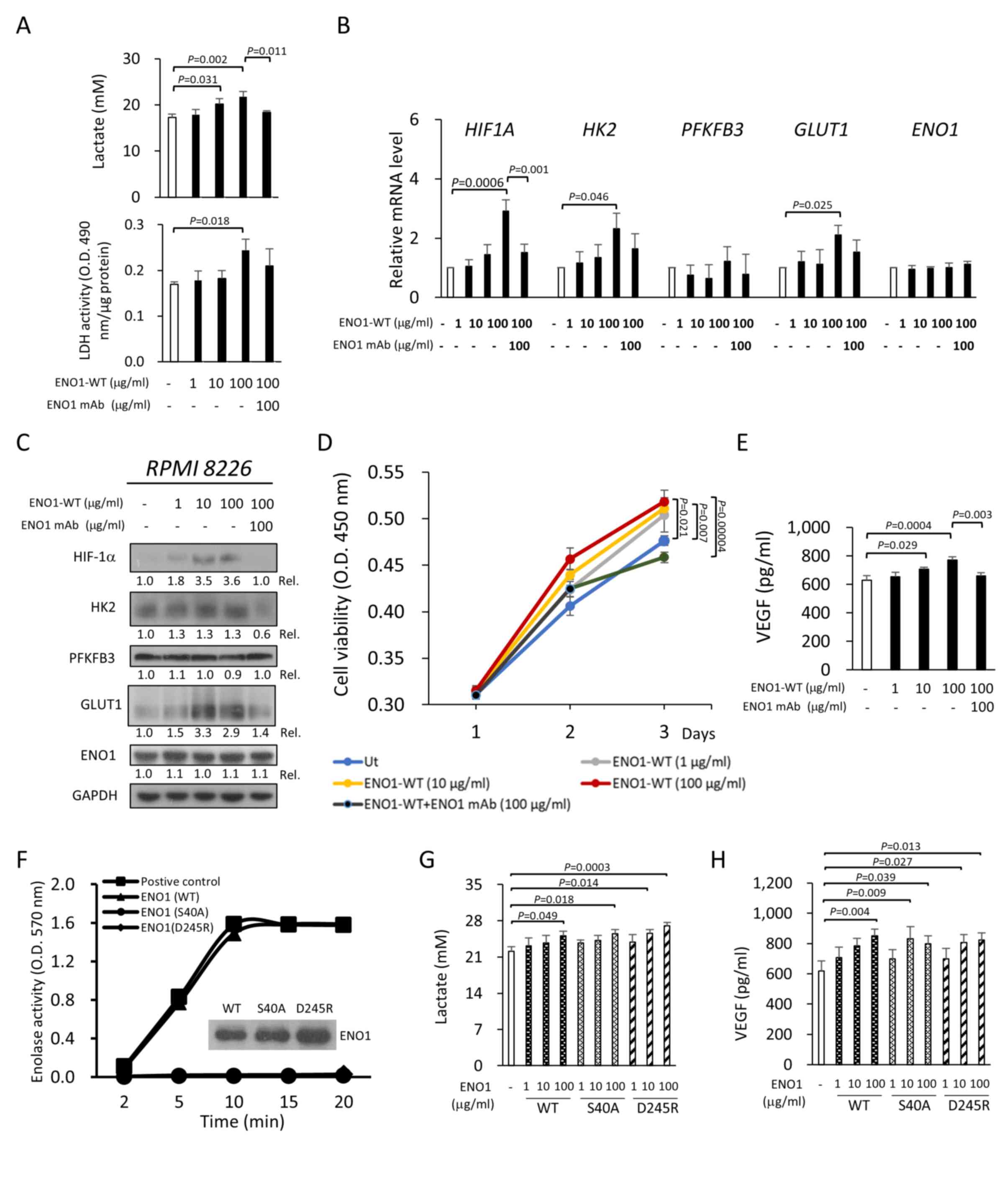 | Figure 3.Extracellular ENO1 enhances
glycolysis and pro-cancer activities. RPMI-8226 cells were treated
with the indicated concentrations of recombinant ENO1-WT. The
studies were conducted in the presence or absence of 100 µg/ml ENO1
mAb (also termed HuL001). (A) The lactate concentration in the
culture medium (upper panel) and intracellular LDH activity (lower
panel) were measured 48 h after ENO1-WT treatment. (B) The
HIF1A, HK2, PFKFB3, GLUT1 and ENO1 mRNA levels were
quantified by reverse transcription-quantitative PCR after 6 h of
ENO1-WT treatment. (C) The HIF-1α, HK2, PFKFB3, GLUT1 and ENO1
protein levels were analyzed by immunoblotting after 24 h of
ENO1-WT treatment. The amounts of studied proteins were first
normalized with GAPDH, and the Rel was then calculated by comparing
with the levels in the untreated cells, of which the value is set
to 1.0. (D) Cell viability was measured using Cell Counting Kit-8.
(E) Secretion of VEGF was measured by ELISA after 48 h of ENO1-WT
treatment. (F) The enolase activity of ENO1-WT and two
catalytically dead mutants, ENO1-S40A and ENO1-D245R, was measured.
RPMI-8226 cells were treated with the indicated concentrations of
ENO1-WT, ENO1-S40A and ENO1-D245R for 48 h. The (G) lactate and (H)
VEGF concentrations in the culture medium were measured. All
results are presented as the mean ± SD of three independent
experiments. P-values were calculated with one-way ANOVA (with
Tukey's post hoc test). ENO1, enolase-1; ENO1-WT, wild-type ENO1;
GLUT1, glucose transporter 1; HIF1A or HIF-1α, hypoxia-inducible
factor 1-α; HK2, hexokinase 2; LDH, lactate dehydrogenase; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6 biphosphatase 3; Rel.,
relative ratio; Ut, untreated; VEGF, vascular endothelial growth
factor. |
Next, the possible mechanism of this enhanced
aerobic glycolysis by extracellular ENO1 was investigated. Gene
expression levels of HIF-1α and glycolysis-related genes (GRGs)
after treatment of cells with ENO1 protein were analyzed using
RT-qPCR. Compared with the control, ENO1-treated RPMI-8226 cells
had higher HIF1A, HK2 and GLUT1 mRNA levels, which
was not observed for PFKFB3 and ENO1 (Fig. 3B). Furthermore, the effects of
extracellular ENO1 on the transcription of glycolysis-related genes
were attenuated by ENO1 mAb (Fig.
3B). Consistently, extracellular ENO1 also enhanced HIF-1α, HK2
and GLUT1 protein expression in RPMI-8226 cells, but not the
protein expression of PFKFB3 and ENO1 (Fig. 3C). This increase in ENO1-induced
protein expression was reduced by ENO1 mAb co-treatment. Since
aerobic glycolysis is a hallmark of cancer, cell viability and the
production of VEGF (a key mediator of angiogenesis) were also
measured in MM cells following treatment with extracellular ENO1.
Cell viability and VEGF production in RPMI-8226 cells were
increased following ENO1 treatment, and were abrogated by ENO1 mAb
co-treatment (Fig. 3D and E).
Similar results were also obtained following treatment of PC-3
cells. ENO1 also increased the lactate production (Fig. S2A), intracellular LDH activity
(Fig. S2B) and cell migration
(Fig. S2C) of PC-3 cells, and
these effects were attenuated by ENO1 mAb treatment. These data,
for the first time (to the best of our knowledge), demonstrated
that extracellular ENO1 enhanced glycolysis, possibly through a
mechanism controlling the expression of HIF-1α and
glycolysis-related proteins. The increase of glycolysis by
extracellular ENO1 simultaneously enhanced cell viability,
migration and VEGF production.
Although it was demonstrated that extracellular ENO1
enhanced glycolysis, it was of interest to determine whether its
enzymatic activity was essential for extracellular ENO1-mediated
glycolysis in the cytoplasm. For this purpose, two catalytically
inactive ENO1 mutants (ENO1-S40A and ENO1-D245R) without enolase
activity were generated (Fig. 3F).
Subsequently, it was observed that mutant ENO1 proteins induced
production of lactate (both ENO1-S40A and ENO1-D245R; Fig. 3G) and VEGF (both ENO1-S40A and
ENO1-D245R; Fig. 3H) in RPMI-8226
cells, similar to ENO1-WT. In parallel, increased cell migration
was also observed in PC-3 cells treated with ENO1-S40A and
ENO1-D245R (Fig. S2D). Thus, the
catalytic activity of extracellular ENO1 was not required for the
enhancement of glycolysis and pro-cancer activities.
Extracellular ENO1 enhances glycolysis
and pro-cancer activities via HIF-1α
It has been reported that HIF-1α regulates the
expression of glycolysis-related genes, leading to lactate
production and tumor progression (31). Therefore, whether extracellular ENO1
regulates glycolysis and pro-cancer activities through HIF-1α was
further investigated. It was demonstrated that extracellular ENO1
upregulated the mRNA (Fig. 4A) and
protein (Fig. 4B) levels of HIF-1α,
HK2 and GLUT1 in RPMI-8226 cells, which was abrogated by knockdown
of HIF-1α expression (with si-HIF1A) but not by control siRNA.
Notably, knockdown of HIF-1α expression also inhibited
extracellular ENO1-induced lactate secretion (Fig. 4C) and pro-cancer activities, such as
increased IL-6 (a growth factor for MM; Fig. 4D), VEGF (Fig. 4E), cell viability (Fig. 4F) and cell migration (Fig. 4G). Although the reductions in
ENO1-induced lactate secretion (P=0.067) and cell viability
(P=0.425) were not statistically significant, a trend of reduction
was observed. Taken together, these results suggested that the
extracellular ENO1/HIF-1α axis enhanced glycolysis and pro-cancer
activities in MM cells.
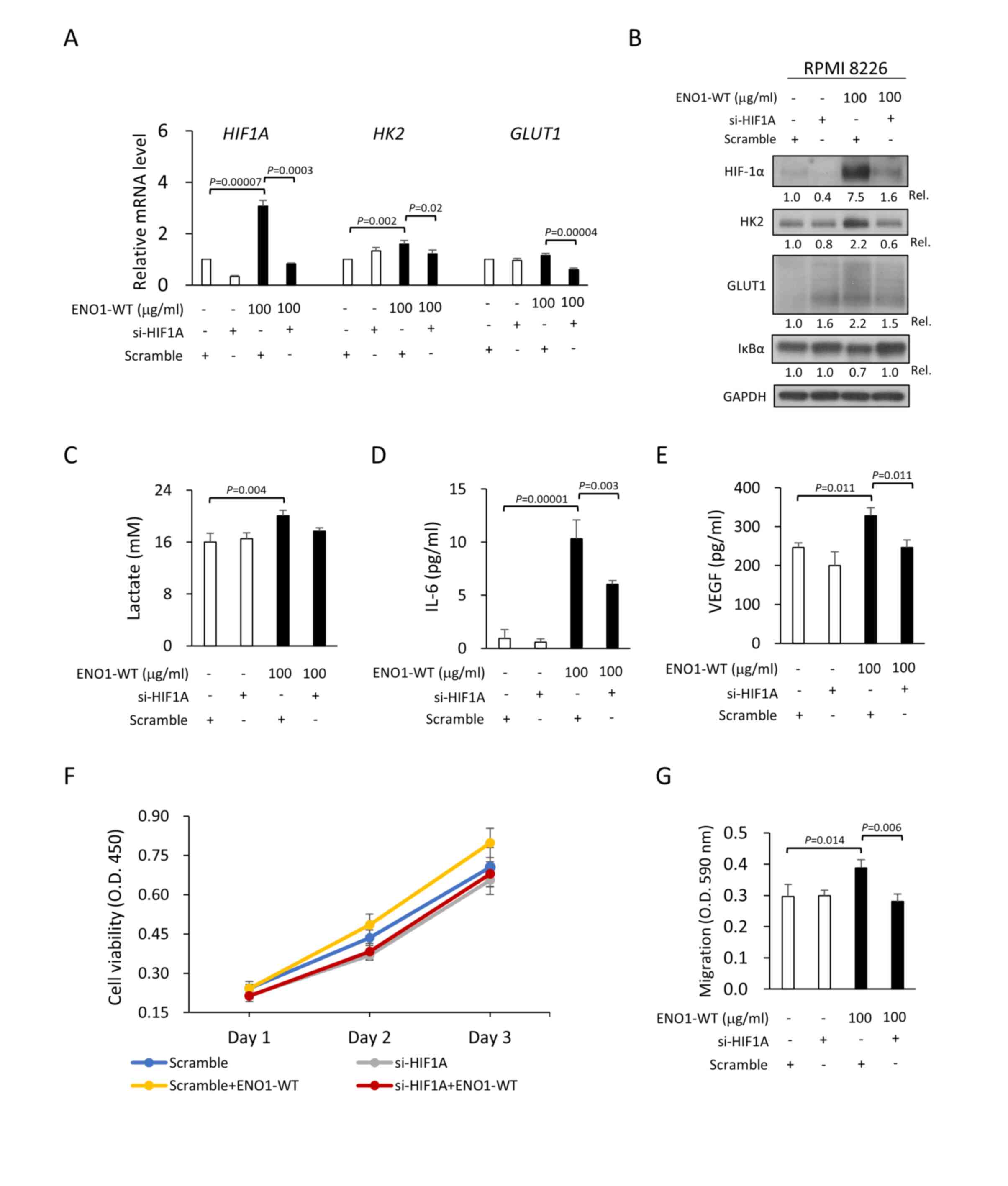 | Figure 4.Extracellular ENO1 enhances
glycolysis and pro-cancer activities through HIF-1α. RPMI-8226
cells were transfected with HIF-1α-targeting siRNA (si-HIF1A) or
control siRNA (scramble sequence) for 96 h. (A) The HIF1A,
HK2 and GLUT1 mRNA levels were quantified by reverse
transcription-quantitative PCR after 6 h of treatment with or
without 100 µg/ml ENO1-WT. (B) The HIF-1α, HK2, GLUT1 and IκBα
protein levels and the (C) lactate, (D) IL-6 and (E) VEGF
concentrations in the culture medium were measured 24 h after
treatment with ENO1-WT. The amounts of studied proteins were first
normalized with GAPDH, and then the Rel. was calculated by
comparing with the scramble siRNA-treated cells, of which the value
is set to 1.0. (F) Cell viability and (G) Transwell migration
assays of RPMI-8226 cells with or without ENO1-WT treatment were
performed following transfection with si-HIF1A or control siRNA.
All results are presented as the mean ± SD of three independent
experiments. P-values were calculated with one-way ANOVA (with
Tukey's post hoc test). ENO1, enolase-1; ENO1-WT, wild-type ENO1;
GLUT1, glucose transporter 1; HIF1A or HIF-1α, hypoxia-inducible
factor 1-α; HK2, hexokinase 2; IL-6, interleukin 6; LDH, lactate
dehydrogenase; Rel., relative ratio; siRNA, small interfering RNA;
VEGF, vascular endothelial growth factor. |
Since NF-κB and HIF-1α have been reported to play
key roles in the pathogenesis of MM (32), the involvement of NF-κB activation
in the extracellular ENO1/HIF-1α axis in MM was next explored. As
shown in Fig. 4B, degradation of
IκBα (an obligatory step in NF-κB activation) following
extracellular ENO1 administration was observed, which was reversed
in the HIF-1α-knockdown cells. This suggested that HIF-1α may be
involved in the extracellular ENO1-induced NF-κB activation.
However, it was demonstrated that extracellular ENO1 upregulated
HIF-1α protein levels, and this increase was not altered in the
presence of an NF-κB inhibitor (BAY 11-7085), indicating that
extracellular ENO1-induced HIF-1α upregulation was not regulated in
a NF-κB-dependent manner (Fig.
S3). Additionally, extracellular ENO1 enhanced the protein
expression of HIF-1α. Furthermore, HIF-prolyl hydroxylase (PHD) is
responsible for the degradation of HIF-1α under normal condition
(23), thus it was investigated if
PHD expression is regulated by extracellular ENO1. RPMI 8226 cells
were treated with extracellular ENO1 followed by measurement of
PHD2 protein (a main regulator of HIF-1). As demonstrated in
Fig. S4, the levels of PHD2
protein were not altered in the presence of ENO1.
The aforementioned results demonstrated that
glycolysis and pro-cancer activities were suppressed by ENO1
knockdown and promoted by extracellular ENO1 treatment. Thus, these
observations were further investigated by increasing the overall
expression of ENO1 in MM cells. The overexpressed ENO1
(Myc/DDK-tagged) protein level was observed in the KMS-11 MM cell
line (oeENO1) compared with that of mock cells (Fig. S5A). Overexpression of ENO1 in the
KMS-11 cells slightly increased the HIF-1α protein expression level
compared with the mock cells, but an increase in GLUT1 and HK2
protein levels was not observed (Fig.
S5A). However, a slight increase in lactate production
(P=0.080) following ENO1 overexpression was observed (Fig. S5B). Interestingly, overexpression
of ENO1 promoted certain pro-cancer activities, including cell
viability (Fig. S5C) and migration
(Fig. S5F), but had no impact on
IL-6 (Fig. S5D) and VEGF (Fig. S5E) secretion levels.
ENO1 mAb reduces glycolytic
activities
Following the results of the extracellular ENO1
studies (Figs. 3 and 4), it was of further interest to determine
whether surface (or membrane bound) ENO1 could mediate glycolysis,
using the proprietary HuniLife ENO1 mAb as an investigatory tool.
Without the addition of ENO1 protein, it was demonstrated that ENO1
mAb treatment reduced the levels of secreted lactate in two MM cell
lines (RPMI-8226 and U266) in a dose-dependent manner (Fig. 5A). To delineate the underlying
mechanism, it was first revealed that ENO1 mAb did not directly
affect total cellular enolase activity (Fig. 5B). However, it was demonstrated that
ENO1 mAb decreased glucose uptake in RPMI-8226 and U266 cells in a
dose-dependent manner, compared with the hIgG1 control (Fig. 5C). Moreover, the ENO1 mAb reduced
the protein expression levels of HIF-1α, HK2 and PFKFB3 in
RPMI-8226 cells, but did not affect the expression levels of GLUT1
and ENO1 (Fig. 5D). Similarly, ENO1
mAb significantly reduced lactate production in PC-3 cells
following stimulation with TNF-α, which mimicked the inflammatory
environment of advanced PCa tumors (Fig. S2F). Collectively, these results led
to the hypothesis that cell surface ENO1 may be involved in
controlling glycolytic flux in cancer cells.
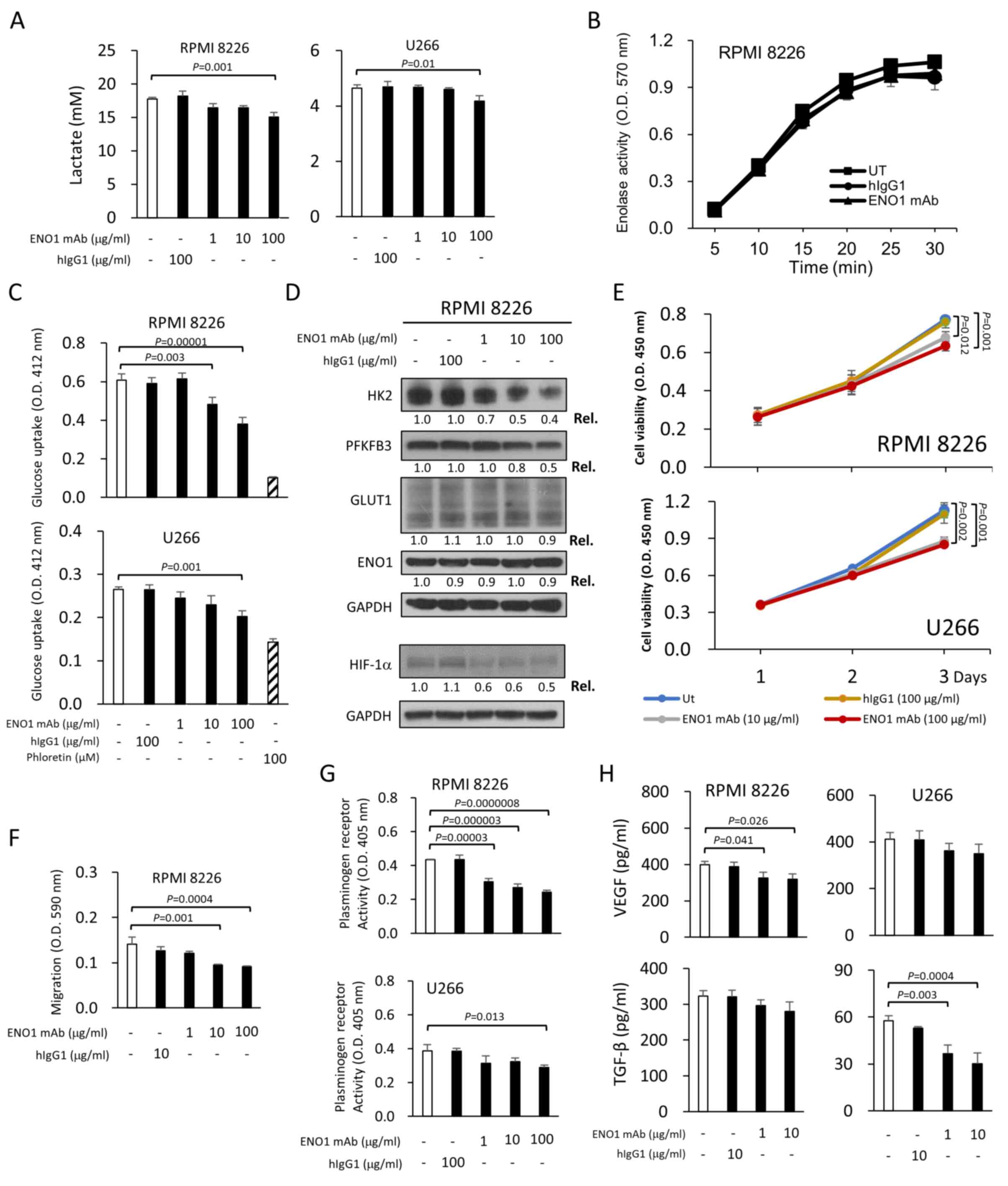 | Figure 5.ENO1 mAb reduces glycolytic and
pro-cancer activities. (A) The lactate concentrations in the
culture medium, collected from ENO1 mAb-treated RPMI-8226 (left
panel) and U266 (right panel) cells, were measured after 48 h of
ENO1 mAb treatment. In all studies, hIgG1 at the indicated
concentrations was included as a specificity control for ENO1 mAb.
RPMI-8226 and U266 cells were treated with 100 µg/ml ENO1 mAb or
100 µg/ml hIgG1 for 48 h. (B) Enolase activity and (C) glucose
uptake in lysates were further analyzed. Phloretin (a GLUT1
inhibitor) was added at 100 µM as a positive control to inhibit
glucose uptake. (D) RPMI-8226 cells were treated with the indicated
concentrations of ENO1 mAb or hIgG1 for 24 h. The HIF-1α, HK2,
PFKFB3, GLUT1 and ENO1 protein levels were analyzed by
immunoblotting. The amounts of studied proteins were first
normalized with GAPDH, and then the Rel. was calculated by
comparing with the untreated cells, of which the value was set to
1.0. (E) RPMI-8226 (upper panel) or U266 (lower panel) cells were
treated with the indicated concentrations of ENO1 mAb or hIgG1.
Cell viability was assessed 1, 2 or 3 days after treatment using
the Cell Counting Kit-8. The OD 450 nm value was positively
associated with the number of viable cells. (F) RPMI-8226 cells
were treated with the indicated concentrations of ENO1 mAb or
hIgG1. Cell migration was measured by Transwell assay after 18 h of
treatment. (G) RPMI-8226 (upper panel) and U266 (lower panel) cells
were treated with the indicated concentrations of ENO1 mAb or hIgG1
followed by measurement of the plasminogen receptor activity. (H)
RPMI-8226 (left panels) or U266 (right panels) cells were treated
with the indicated concentrations of ENO1 mAb or hIgG1 for 48 h.
Supernatant was collected for determination of human VEGF (upper
panel) and TGF-β (lower panel) levels by ELISA. All results are
presented as the mean ± SD of three independent experiments.
P-values were calculated with one-way ANOVA (with Tukey's post hoc
test). ENO1, enolase-1; GLUT1, glucose transporter 1; HIF-1α,
hypoxia-inducible factor 1-α; hIgG1, human IgG1; HK2, hexokinase 2;
mAb, monoclonal antibody; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6 biphosphatase 3; Rel.,
relative ratio; TGF, transforming growth factor; Ut, untreated;
VEGF, vascular endothelial growth factor. |
ENO1 mAb reduces cell viability,
migration and cytokine production
In addition to the inhibition of glycolysis, the
anticancer effects of the proprietary HuniLife ENO1 mAb on cell
viability, cell migration and the production of tumor-secreted
factors were further explored in vitro. It was demonstrated
that the ENO1 mAb inhibited the viability of RPMI-8226 and U266
cells by 18 and 25%, respectively (Figs. 5E and S6). The ENO1 mAb also inhibited the
migration of RPMI-8226 cells in a dose-dependent manner (Fig. 5F). A similar inhibition of
TNF-α-stimulated migration of PC-3 cells by ENO1 mAb was also
observed (Fig. S2E). Cell surface
ENO1 has been reported to be a receptor for activating plasminogen
to stimulate migration of cells (6). Consistently, a reduction in
plasminogen receptor activity was observed in RPMI-8226 and U266
cells following treatment with ENO1 mAb (Fig. 5G). The secretion levels of VEGF and
TGF-β (a key modulator of the TME) in these two MM cell lines were
also reduced by administration of the ENO1 mAb (Fig. 5H). Although several ENO1-specific
antibodies have also been reported to inhibit cancer invasiveness,
metastasis and stemness (8,12,17–20),
to the best of our knowledge, none have been investigated for their
effects on glycolysis (the Warburg effect). In summary, the
proprietary HuniLife ENO1 mAb demonstrated both an antiglycolytic
and antitumor potential in MM and PCa cells (Figs. 5 and S2).
In addition, the alternative spliced nuclear isoform
of ENO1, named c-myc promoter binding protein 1 (MBP-1), acts as a
tumor suppressor (1). It was
further determined whether ENO1 mAb is involved in the regulation
of MBP-1 expression. RPMI-8226 cells were treated with ENO1 mAb
followed by measuring MBP-1 protein levels. As revealed in Fig. S7A, the MBP-1 protein levels were
not significantly altered in the presence of ENO1 mAb. Moreover, it
has been reported MBP1 bound to the c-Myc promoter acts as a
transcriptional suppressor (1). The
c-MYC mRNA levels in RPMI-8226 cells upon the treatment of
ENO1 mAb were therefore analyzed. The results indicated that ENO1
mAb had no significant impact on regulation of c-Myc
transcription (Fig. S7B).
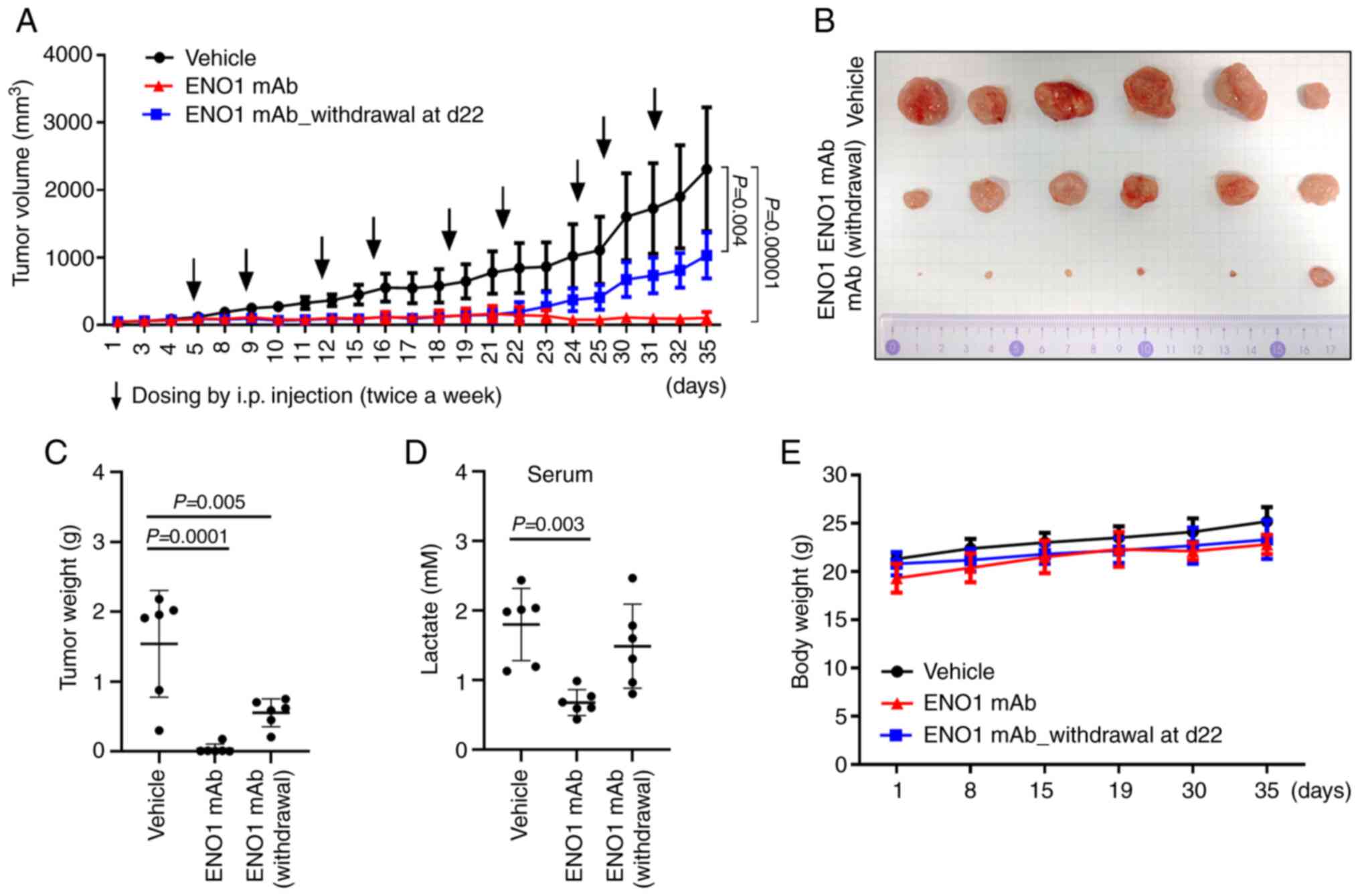
ENO1 mAb reduces tumor growth and
glycolysis in a RPMI-8226 subcutaneous xenograft mouse model
It has previously been reported that the ENO1 mAb
reduced tumor growth in PC-3 subcutaneous xenograft and
intra-tibial implantation mouse models (12). Therefore, it was further
investigated whether the ENO1 mAb could exert anticancer effects on
MM cells in vivo. Nude mice were injected with RPMI-8226
cells, and the subcutaneous tumors were treated with 30 mg/kg ENO1
mAb twice a week. After nine doses in 35 days, both the tumor
volume (Fig. 6A and B) and tumor
weight (Fig. 6C) were reduced by
ENO1 mAb treatment, compared with the vehicle control group. This
reduction in tumor growth was attenuated when ENO1 mAb treatment
was withdrawn from day 22. High serum lactate levels have been
reported to be associated with poor prognosis and a reduced overall
survival in patients with various types of cancer (33,34).
Elevated lactate levels in mice were also associated with tumor
growth in the present xenograft study (Fig. 6D). Notably, ENO1 mAb treatment
reduced serum lactate levels in the tumor-bearing mice, and this
reduction was abrogated upon withdrawing ENO1 mAb treatment. In
addition, ENO1 mAb administration did not result in an apparent
difference in body weight gain (Fig.
6E), which indicated minimal toxicity of ENO1 mAb treatment in
animals. Moreover, hIgG1 isotype was included as a control in the
present study, which did not have significant effects on the tumor
volume (Fig. S8A and B), tumor
weight (Fig. S8C), serum lactate
levels (Fig. S8D) and body weight
gain (Fig. S8E) in animals. A
previous study indicated that drugs need to achieve systemic
concentrations in the blood that permit adequate penetration and
target binding into the tumor tissue (35). Based on the ENO1 concentration
(1–100 µg/ml) studied in vitro, higher dose (30 mg/kg=625
µg/ml serum drug concentration) of ENO1 mAb than theoretically
required was used to ensure sufficient drug distribution into tumor
site in vivo. Moreover, it was determined that the half-life
of proprietary ENO1 mAb was ~10 days in mice. In the MM
subcutaneous xenograft studies, the frequency of ENO1 mAb
administration was twice a week, to ensure there was a stable level
of ENO1 mAb for the treatment duration in animals. Taken together,
these results suggested that the ENO1 mAb could serve as a novel
therapeutic by targeting glycolysis and associated pro-cancer
activities in MM.
Discussion
ENO1 is a multifunctional protein with a ‘main’
function as a glycolytic enzyme in the cytosol and a ‘moonlighting’
function as a plasminogen receptor on the cell surface (10). Both the main and moonlighting roles
of ENO1 have been implicated in cancer progression (24), but, to the best of our knowledge,
there has not yet been a study to address the role of extracellular
ENO1 in the context of glycolysis. Therefore, the present study was
the first to provide evidence that extracellular ENO1 could enhance
the intracellular glycolytic pathway. Studies of two cancer types,
MM (the present study) and PCa (12) have been conducted, where high
prevalence of ENO1 expression has been correlated to cancer
progression (10,11). It was therefore conceivable that
knockdown of ENO1 expression in both MM and PCa cells would lead to
a decrease in pro-cancer activities, as previously reported in
other cancer types (5,18). However, it was surprising that
extracellular ENO1 increased glycolysis (lactate production and LDH
activity), cell viability, cell migration and the production of
VEGF in the present study. In addition, the present study revealed
that the extracellular ENO1-induced glycolysis and pro-cancer
activities were mediated by the upregulated expression of HIF-1α
and its downstream glycolysis-related proteins, HK2 and GLUT1.
Notably, it was demonstrated in the present study
that enzymatic activity of administered extracellular ENO1 was not
required for the glycolysis, cell migration and VEGF production
induced by extracellular ENO1. It has also been previously reported
that ENO1 enzymatic activity was not essential for the enhanced
cell invasion and proliferation in lung cancer (18). These results therefore exclude the
possibility that extracellular ENO1 enters the cytosol to directly
enhance glycolysis with its enzymatic activity. However, the
authors were unable to address the issue that if free unbound ENO1
in the medium could have any effect on glycolysis in their
experiment settings. The use of an ENO1 mAb in the present study
further demonstrated that surface ENO1 of MM or PCa (following
stimulation with TNF-α) cells enhanced glycolysis, cell viability,
cell migration and the production of VEGF and TGF-β. The ENO1 mAb
suppressed glycolysis and pro-cancer activities by decreasing
protein expression of HIF-1α and GRGs, such as HK2 and PFKFB3.
Lastly, the results of the present study demonstrated that the ENO1
mAb inhibited MM tumor growth in nude mice and reduced the elevated
serum lactate. The elevated serum lactate levels observed in the MM
mice were associated with tumor growth, which was indicated by the
increased lactate levels and tumor weight upon withdrawing of ENO1
mAb treatment.
Normal cells typically catabolize glucose by
oxidative phosphorylation in the mitochondria, whereas tumor cells
tend to convert glucose into lactate even in conditions of
sufficient oxygen (36). This
phenomenon was first studied by Otto Warburg and termed aerobic
glycolysis or the Warburg effect (37). This reprogramming of energy
metabolism has emerged as another hallmark of cancer (38). The upregulation of HIF-1α, numerous
glycolytic enzymes (including LDH, PFK, HK2 and ENO1) or molecules
associated with glucose uptake (such as GLUT1) have been reported
in numerous types of cancer (39).
HIF-1α regulation involves PHD, which induces HIF-1α degradation in
normoxia, but allows stabilization of HIF-1α in hypoxia. However,
in certain circumstances, HIF-1α regulation involves
PHD-independent mechanism (40).
Interestingly, it was identified that extracellular ENO1 had no
significant impact on the regulation of PHD protein expression, and
thus it was suggested by the authors that extracellular ENO1 could
promote the HIF-1α protein expression through a PHD-independent
mechanism. Moreover, it was investigated if HIF-1α expression could
be regulated by ENO1 mAb under low oxygen or
CoCl2-mimicked condition. However, the authors' studies
could not lead to consistent results so far from these two
experimental systems. Efforts have been made to develop inhibitors
to the aforementioned molecules (41), including intracellular ENO1
(42). However, no satisfactory
clinical development to treat cancer by targeting the Warburg
effect has been achieved, due to dose-limiting toxicities, low
specificity and low potency of drugs (43). Although glycolysis is an ideal
target pathway for cancer therapy, avoiding the risk of shutting
down glycolysis in all cells remains challenging. In the present
study, it was demonstrated that surface ENO1 enhanced the
glycolytic pathway in cancer cells, which makes the ENO1 antibody
an ideal modality for targeting glycolysis in cancer cells
expressing high levels of surface ENO1, such as in MM (11).
Lactate, the metabolic intermediate generated during
aerobic glycolysis, is not only utilized as a fuel for growth but
also provides acidity to the TME, which promotes the invasion and
metastasis of cancer cells (44).
Elevated lactate levels can inhibit the function of a variety of
immune cells, such as T cells and natural killer cells (45). Certain strategies have been
developed to target lactate production, such as inhibition of
monocarboxylate transporters (46)
and pyruvate dehydrogenase kinase-1 (47). It has also been reported that
knockdown of the ENO1 gene could reduce the production of
lactate (48). The present study
demonstrated that the HuniLife proprietary ENO1 mAb reduced lactate
production and LDH activity in MM cells, which suggested a novel
and feasible approach to regulate the TME.
It has previously been reported that aerobic
glycolysis signatures were correlated with drug resistance in MM
(49), and LDH A and HIF-1α were
identified as valid targets to prevent MM drug resistance and
progression in vivo (49).
Moreover, HK2 was found to be upregulated in MM, which was
significantly correlated with poor prognosis (50). Surface ENO1 was significantly
upregulated in MM cells from patients, particularly after
stimulation by pDCs in the TME (bone marrow) (11). The results of the present study
indicated that the ENO1 mAb downregulated HIF-1α and HK2 protein
expression, therefore the therapeutic potential of the ENO1 mAb in
a clinical setting can be anticipated. In line with the
aforementioned observations, in the present study, administration
of extracellular ENO1 to MM cell culture increased HIF-1α and HK2
expression, and ENO1 mAb administration inhibited HIF-1α and HK2
expression in the absence of an additional source of ENO1. However,
it remains difficult to reconcile the observations regarding the
regulation of other glycolytic enzymes with these experimental
results. In the present study, GLUT1 expression increased upon the
addition of extracellular ENO1, while ENO1 mAb treatment of MM
cells reduced the expression of PFKFB3 but not GLUT1. An improved
understanding of the underlying mechanism of extracellular
ENO1-regulated glycolysis is required to improve interpretation of
these results.
The HuniLife proprietary ENO1 mAb was originally
selected for its inhibitory activity on plasmin activation (close
to 100%) in LPS-stimulated monocyte cells (U937), and it also
exhibited an inhibitory activity (about 65.5±0.3%) against the
invasion capabilities of ENO1-expressing lung cancer cells (CL1-5)
(Patent: PCT/US2013/076877). The present study further demonstrated
that the ENO1 mAb inhibited glycolysis via HIF-1α and
glycolysis-related proteins in MM cells, but the detailed
mechanisms by which ENO1 mAb regulates HIF-1α remain to be
elucidated. The involvement of surface ENO1 in the uPAR/plasmin
pathway added further complexity to the mechanism of study
(51). It was hypothesized that the
mechanism may involve ENO1-associated proteins on the cell surface,
such as the hepatocytes growth factor receptor (HGFR) (18), B7-H3 (52) or fibroblast activation protein α
(FAP) (9). Notably, B7-H3 promotes
aerobic glycolysis by regulating HK2 (53), which coincides with the observation
in the present study that ENO1 mAb downregulates HK2 expression.
Further investigation of the regulation of HIF-1α expression by
surface ENO1 in other ENO1-related signaling pathways, including
the HGFR-mediated PI3K/AKT and Wnt/β-catenin pathways or
FAP-mediated NF-kB, is required.
A previous study demonstrated that the ENO1 mAb
targeted multiple TME niches involved in PCa progression and bone
metastasis (12). In the present
study, the ENO1 mAb significantly reduced tumor growth in a MM
subcutaneous xenograft mouse model. Indeed, there is a limitation
on subcutaneous xenograft mouse model for the efficacy studies.
However, the FDA-approved MM drugs, such as bortezomib (54) and daratumumab (55), all have demonstrated favorable
efficacy in shrinking tumor in MM xenograft mice, indicating that
such animal model still have clinical relevance to screening
potential new drugs. The ENO1 mAb also reduced the viability of MM
cells in vitro, but this reduction effect was modest even
with statistical significance. By contrast, the ENO1 mAb had a
marked inhibitory effect on tumor growth in vivo, suggesting
that the anticancer mechanism of the ENO1 mAb may involve not only
cancer cells but also TME niches (12). Therefore, it was reasonably
suggested that the tumor inhibition activity of ENO1 mAb could
result from its effects on TME, owing to its cross reactivity to
mouse. MM cells grow in hypoxic niches inside the bone marrow with
a metabolic shift towards aerobic glycolysis (56). Inhibition of ENO1 activates pDCs in
the bone marrow microenvironment, leading to enhanced MM cell
cytotoxicity (11). In addition,
ENO1 is expressed in the cytosol and on the cell surface and can be
found in the secretome (57). In
the present study, it was shown that the addition of ENO1 (as a
mimic of secreted protein) enhanced glycolytic activity through
HIF-1α, and this enhancing effect was attenuated by the
administration of ENO1 mAb. These data therefore provide evidence
of a role of extracellular ENO1 (surface or secreted forms) in
glycolytic modulation, and also provide the preclinical rationale
for targeting extracellular ENO1-mediated metabolic reprogramming
in cancer cells and the TME.
There are several notable observations regarding
cytosolic or extracellular ENO1. Firstly, cytosolic ENO1 is highly
expressed and utilized for glycolysis in cancer cells (48). Secondly, ENO1 is transported onto
the cell surface of cancer cells (8) or activated immune cells (7), which activates plasmin to facilitate
cell motility. Thirdly, the ENO1-induced biological activities are
all related to cell survival, such as viability, motility,
angiogenesis, cytokine and chemokine production, and energy
programing (10). Based on the
observation that extracellular ENO1 can promote glycolysis
reprograming, it is conceivable that ENO1 may play a pivotal role
in responding to stressed or hypoxic conditions, such as overgrown
tumor cells in inflammatory and necrotic milieu.
In conclusion, the present study revealed an
unexpected role of extracellular ENO1 in the crosstalk with the
intracellular glycolytic pathway via HIF-1α (Fig. 7). However, the coordination and
synchronicity of extracellular/intracellular ENO1 in regulating
energy programs and ensuing biological activities in response to
the ever-changing cellular environment remains to be elucidated. In
summary, in the present study, the HuniLife ENO1-targeting antibody
demonstrated multiple anticancer mechanisms via inhibition of
glycolysis, lactate production, cytokine secretion, cell viability
and migration. This ENO1 antibody is currently in a phase I
clinical trial for safety assessment, and future clinical studies
are required to validate its anticancer potential in treating
patients.
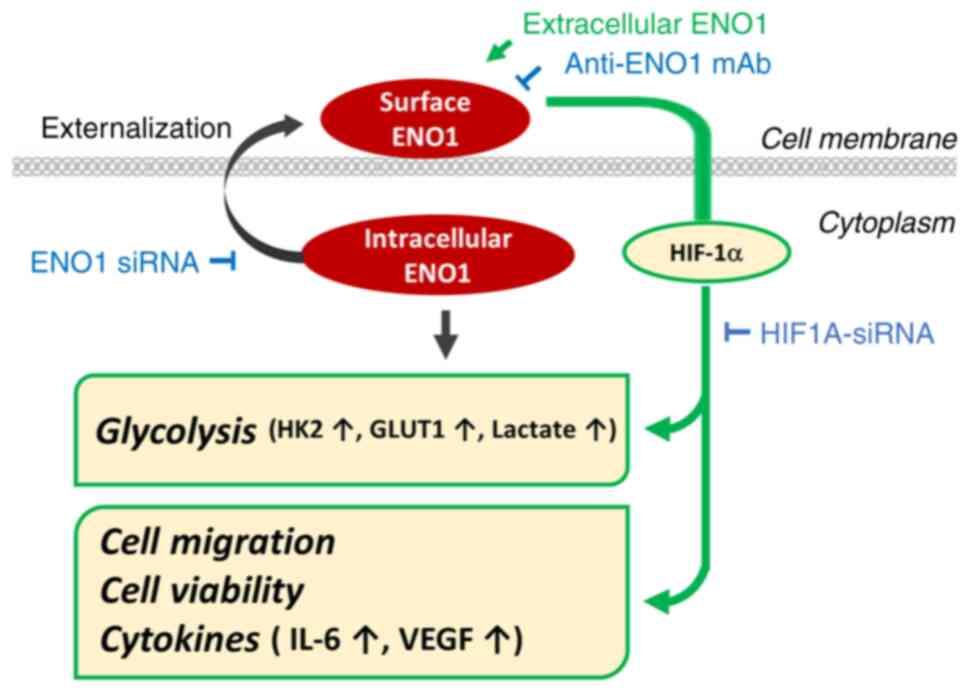 | Figure 7.Schematic diagram summarizing the
effects and possible mechanisms of extracellular ENO1 in regulating
glycolysis and pro-cancer activities. Extracellular ENO1 may
enhance glycolytic activity and glycolysis-related genes, including
HK2 and GLUT1, through HIF-1α. Moreover, extracellular ENO1 also
promoted HIF-1α-mediated pro-cancer activities, such as cell
migration, cell viability and production of tumor-promoting
cytokines. Consistently, these effects on cancer progression could
be attenuated by the ENO1 antibody or ENO1 siRNA. ENO1, enolase-1;
GLUT1, glucose transporter 1; HIF-1α, hypoxia-inducible factor 1-α;
HK2, hexokinase 2; siRNA, small interfering RNA. |
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank TFBS Bioscience for
their assistance to the in vivo studies during the
development of ENO1 mAb and Dr I-Shan Hsieh (former employee of
HuniLife Biotechnology, Inc.) for her contribution to Figs. 3F and 6D by measuring enolase activity and
lactate concentration, respectively.
Funding
The present study was supported by HuniLife Biotechnology,
Inc.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
ICC, WCH and TTY conceived the study, wrote the
original draft, reviewed and edited the manuscript. ICC, WCH, YTH,
MLC, AWT and PYW collected, analyzed and interpreted the data. TTY
supervised the study and acquired funding. MLC and AWT confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All animal studies were reviewed and approved by
The Ethics Committee of TFBS Bioscience (Taipei, Taiwan). All
animal procedures were performed according to approved protocols
from the Institutional Animal Care and Use Committee (IACUC) of
TFBS Bioscience (IACUC protocol no. TFBS2021-013).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BCRC
|
Bioresource Collection and Research
Center
|
|
ENO1
|
enolase-1
|
|
FAP
|
fibroblast activation protein α
|
|
FBS
|
fetal bovine serum
|
|
GLUT1
|
glucose transporter 1
|
|
GRGs
|
glycolysis-related genes
|
|
HGFR
|
hepatocytes growth factor
receptor
|
|
HIF-1α
|
hypoxia-inducible factor 1-α
|
|
HK2
|
hexokinase 2
|
|
hIgG1
|
human IgG1
|
|
IACUC
|
Institutional Animal Care and Use
Committee
|
|
IL-6
|
interleukin 6
|
|
LDH
|
lactate dehydrogenase
|
|
mAb
|
monoclonal antibody
|
|
MBP-1
|
c-myc promoter binding protein 1
|
|
MM
|
multiple myeloma
|
|
NF-κB
|
nuclear factor-κB
|
|
PCa
|
prostate cancer
|
|
PFK
|
phosphofructokinase
|
|
PFKFB3
|
6-phosphofructo-2-kinase/fructose-2,6
biphosphatase 3
|
|
PHD
|
HIF-prolyl hydroxylase
|
|
RNAi
|
RNA interference
|
|
TGF
|
transforming growth factor
|
|
TME
|
tumor microenvironment
|
|
VEGF
|
vascular endothelial growth
factor
|
References
|
1
|
Didiasova M, Schaefer L and Wygrecka M:
When place matters: Shuttling of enolase-1 across cellular
compartments. Front Cell Dev Biol. 7:612019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wold F and Ballou CE: Studies on the
enzyme enolase. II. Kinetic studies. J Biol Chem. 227:313–328.
1957. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feron O: Pyruvate into lactate and back:
from the Warburg effect to symbiotic energy fuel exchange in cancer
cells. Radiother Oncol. 92:329–333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Li H, Miao L and Ding J:
Silencing of ENO1 inhibits the proliferation, migration and
invasion of human breast cancer cells. J BUON. 25:696–701.
2020.PubMed/NCBI
|
|
6
|
Plow EF and Das R: Enolase-1 as a
plasminogen receptor. Blood. 113:5371–5372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wygrecka M, Marsh LM, Morty RE, Henneke I,
Guenther A, Lohmeyer J, Markart P and Preissner KT: Enolase-1
promotes plasminogen-mediated recruitment of monocytes to the
acutely inflamed lung. Blood. 113:5588–5598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Principe M, Ceruti P, Shih NY,
Chattaragada MS, Rolla S, Conti L, Bestagno M, Zentilin L, Yang SH,
Migliorini P, et al: Targeting of surface alpha-enolase inhibits
the invasiveness of pancreatic cancer cells. Oncotarget.
6:11098–11113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan Z, Hu H, Zhu Y, Zhang W, Fang Q, Qiao
T, Ma T, Wang M, Huang R, Tang Q, et al: Colorectal cancer cell
intrinsic fibroblast activation protein alpha binds to Enolase1 and
activates NF-κB pathway to promote metastasis. Cell Death Dis.
12:5432021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Almaguel FA, Sanchez TW, Ortiz-Hernandez
GL and Casiano CA: Alpha-enolase: Emerging tumor-associated
antigen, cancer biomarker, and oncotherapeutic target. Front Genet.
11:6147262020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ray A, Song Y, Du T, Chauhan D and
Anderson KC: Preclinical validation of alpha-enolase (ENO1) as a
novel immunometabolic target in multiple myeloma. Oncogene.
39:2786–2796. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen ML, Yuan TT, Chuang CF, Huang YT,
Chung IC and Huang WC: A novel enolase-1 antibody targets multiple
interacting players in the tumor microenvironment of advanced
prostate cancer. Mol Cancer Ther. 21:1337–1347. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kropff MH, Bisping G, Wenning D, Volpert
S, Tchinda J, Berdel WE and Kienast J: Bortezomib in combination
with dexamethasone for relapsed multiple myeloma. Leuk Res.
29:587–590. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar S and Rajkumar SV: Thalidomide and
lenalidomide in the treatment of multiple myeloma. Eur J Cancer.
42:1612–1622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lokhorst HM, Plesner T, Laubach JP, Nahi
H, Gimsing P, Hansson M, Minnema MC, Lassen U, Krejcik J, Palumbo
A, et al: Targeting CD38 with daratumumab monotherapy in multiple
myeloma. N Engl J Med. 373:1207–1219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gou Y, Li F, Huo X, Hao C, Yang X, Pei Y,
Li N, Liu H and Zhu B: ENO1 monoclonal antibody inhibits invasion,
proliferation and clone formation of cervical cancer cells. Am J
Cancer Res. 11:1946–1961. 2021.PubMed/NCBI
|
|
18
|
Li HJ, Ke FY, Lin CC, Lu MY, Kuo YH, Wang
YP, Liang KH, Lin SC, Chang YH, Chen HY, et al: ENO1 promotes lung
cancer metastasis via HGFR and WNT signaling-driven
epithelial-to-mesenchymal transition. Cancer Res. 81:4094–4109.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shu X, Cao KY, Liu HQ, Yu L, Sun LX, Yang
ZH, Wu CA and Ran YL: Alpha-enolase (ENO1), identified as an
antigen to monoclonal antibody 12C7, promotes the self-renewal and
malignant phenotype of lung cancer stem cells by AMPK/mTOR pathway.
Stem Cell Res Ther. 12:1192021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsiao KC, Shih NY, Fang HL, Huang TS, Kuo
CC, Chu PY, Hung YM, Chou SW, Yang YY, Chang GC and Liu KJ: Surface
α-enolase promotes extracellular matrix degradation and tumor
metastasis and represents a new therapeutic target. PLoS One.
8:e693542013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu L, Shi J, Cheng S, Zhu Y, Zhao X, Yang
K, Du X, Klocker H, Yang X and Zhang J: Estrogen promotes prostate
cancer cell migration via paracrine release of ENO1 from stromal
cells. Mol Endocrinol. 26:1521–1530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fujiwara S, Wada N, Kawano Y, Okuno Y,
Kikukawa Y, Endo S, Nishimura N, Ueno N, Mitsuya H and Hata H:
Lactate, a putative survival factor for myeloma cells, is
incorporated by myeloma cells through monocarboxylate transporters
1. Exp Hematol Oncol. 4:122015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Samec M, Liskova A, Koklesova L, Mersakova
S, Strnadel J, Kajo K, Pec M, Zhai K, Smejkal K, Mirzaei S, et al:
Flavonoids targeting HIF-1: Implications on cancer metabolism.
Cancers (Basel). 13:1302021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qiao G, Wu A, Chen X, Tian Y and Lin X:
Enolase 1, a moonlighting protein, as a potential target for cancer
treatment. Int J Biol Sci. 17:3981–3992. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zakrzewicz D, Didiasova M, Krüger M,
Giaimo BD, Borggrefe T, Mieth M, Hocke AC, Zakrzewicz A, Schaefer
L, Preissner KT and Wygrecka M: Protein arginine methyltransferase
5 mediates enolase-1 cell surface trafficking in human lung
adenocarcinoma cells. Biochim Biophys Acta Mol Basis Dis.
1864:1816–1827. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong
SW, Li RL, Zhao MY, Zhen Y, Yu XL, et al: Alpha-enolase promotes
cell glycolysis, growth, migration, and invasion in non-small cell
lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol.
8:222015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song Y, Luo Q, Long H, Hu Z, Que T, Zhang
X, Li Z, Wang G, Yi L, Liu Z, et al: Alpha-enolase as a potential
cancer prognostic marker promotes cell growth, migration, and
invasion in glioma. Mol Cancer. 13:652014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El Arfani C, De Veirman K, Maes K, De
Bruyne E and Menu E: Metabolic features of multiple myeloma. Int J
Mol Sci. 19:12002018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ahmad F, Cherukuri MK and Choyke PL:
Metabolic reprogramming in prostate cancer. Br J Cancer.
125:1185–1196. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamaguchi T, Iizuka N, Tsunedomi R,
Hamamoto Y, Miyamoto T, Iida M, Tokuhisa Y, Sakamoto K, Takashima
M, Tamesa T and Oka M: Glycolysis module activated by
hypoxia-inducible factor 1alpha is related to the aggressive
phenotype of hepatocellular carcinoma. Int J Oncol. 33:725–731.
2008.PubMed/NCBI
|
|
32
|
Cippitelli M, Stabile H, Kosta A, Petillo
S, Lucantonio L, Gismondi A, Santoni A and Fionda C: Role of NF-κB
signaling in the interplay between multiple myeloma and mesenchymal
stromal cells. Int J Mol Sci. 24:18232023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei Y, Xu H, Dai J, Peng J, Wang W, Xia L
and Zhou F: Prognostic significance of serum lactic acid, lactate
dehydrogenase, and albumin levels in patients with metastatic
colorectal cancer. Biomed Res Int. 2018:18040862018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vlachostergios PJ, Oikonomou KG, Gibilaro
E and Apergis G: Elevated lactic acid is a negative prognostic
factor in metastatic lung cancer. Cancer Biomark. 15:725–734. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Glassman PM and Balthasar JP: Mechanistic
considerations for the use of monoclonal antibodies for cancer
therapy. Cancer Biol Med. 11:20–33. 2014.PubMed/NCBI
|
|
36
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Warburg O, Wind F and Negelein E: The
Metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schwartz L, Supuran CT and Alfarouk KO:
The Warburg effect and the hallmarks of cancer. Anticancer Agents
Med Chem. 17:164–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu L, Chen X, Wang L and Chen S: The sweet
trap in tumors: Aerobic glycolysis and potential targets for
therapy. Oncotarget. 7:38908–38926. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iommarini L, Porcelli AM, Gasparre G and
Kurelac I: Non-canonical mechanisms regulating hypoxia-inducible
factor 1 alpha in cancer. Front Oncol. 7:2862017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen XS, Li LY, Guan YD, Yang JM and Cheng
Y: Anticancer strategies based on the metabolic profile of tumor
cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol
Sin. 37:1013–1019. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lung J, Chen KL, Hung CH, Chen CC, Hung
MS, Lin YC, Wu CY, Lee KD, Shih NY and Tsai YH: In silico-based
identification of human α-enolase inhibitors to block cancer cell
growth metabolically. Drug Des Devel Ther. 11:3281–3290. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Galluzzi L, Kepp O, Vander Heiden MG and
Kroemer G: Metabolic targets for cancer therapy. Nat Rev Drug
Discov. 12:829–846. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
de la Cruz-López KG, Castro-Muñoz LJ,
Reyes-Hernández DO, García-Carrancá A and Manzo-Merino J: Lactate
in the regulation of tumor microenvironment and therapeutic
approaches. Front Oncol. 9:11432019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brand A, Singer K, Koehl GE, Kolitzus M,
Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et
al: LDHA-associated lactic acid production blunts tumor
immunosurveillance by T and NK cells. Cell Metab. 24:657–671. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goodwin ML, Gladden LB, Nijsten MWN and
Jones KB: Lactate and cancer: Revisiting the warburg effect in an
era of lactate shuttling. Front Nutr. 1:272015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujiwara S, Kawano Y, Yuki H, Okuno Y,
Nosaka K, Mitsuya H and Hata H: PDK1 inhibition is a novel
therapeutic target in multiple myeloma. Br J Cancer. 108:170–178.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Capello M, Ferri-Borgogno S, Riganti C,
Chattaragada MS, Principe M, Roux C, Zhou W, Petricoin EF, Cappello
P and Novelli F: Targeting the Warburg effect in cancer cells
through ENO1 knockdown rescues oxidative phosphorylation and
induces growth arrest. Oncotarget. 7:5598–5612. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maiso P, Huynh D, Moschetta M, Sacco A,
Aljawai Y, Mishima Y, Asara JM, Roccaro AM, Kimmelman AC and
Ghobrial IM: Metabolic signature identifies novel targets for drug
resistance in multiple myeloma. Cancer Res. 75:2071–2082. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nakano A, Miki H, Nakamura S, Harada T,
Oda A, Amou H, Fujii S, Kagawa K, Takeuchi K, Ozaki S, et al:
Up-regulation of hexokinaseII in myeloma cells: Targeting myeloma
cells with 3-bromopyruvate. J Bioenerg Biomembr. 44:31–38. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gonias SL and Hu J: Urokinase receptor and
resistance to targeted anticancer agents. Front Pharmacol.
6:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zuo J, Wang B, Long M, Gao Z, Zhang Z,
Wang H, Wang X, Li R, Dong K and Zhang H: The type 1 transmembrane
glycoprotein B7-H3 interacts with the glycolytic enzyme ENO1 to
promote malignancy and glycolysis in HeLa cells. FEBS Lett.
592:2476–2488. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shi T, Ma Y, Cao L, Zhan S, Xu Y, Fu F,
Liu C, Zhang G, Wang Z, Wang R, et al: B7-H3 promotes aerobic
glycolysis and chemoresistance in colorectal cancer cells by
regulating HK2. Cell Death Dis. 10:3082019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishii T, Seike T, Nakashima T, Juliger S,
Maharaj L, Soga S, Akinaga S, Cavenagh J, Joel S and Shiotsu Y:
Anti-tumor activity against multiple myeloma by combination of
KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J.
2:e682012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sanchez L, Wang Y, Siegel DS and Wang ML:
Daratumumab: A first-in-class CD38 monoclonal antibody for the
treatment of multiple myeloma. J Hematol Oncol. 9:512016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Storti P, Bolzoni M, Donofrio G, Airoldi
I, Guasco D, Toscani D, Martella E, Lazzaretti M, Mancini C,
Agnelli L, et al: Hypoxia-inducible factor (HIF)-1α suppression in
myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis
and bone destruction. Leukemia. 27:1697–1706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
de Oliveira G, Freire PP, Cury SS, de
Moraes D, Oliveira JS, Dal-Pai-Silva M, Reis PP and Carvalho RF: An
integrated meta-analysis of secretome and proteome identify
potential biomarkers of pancreatic ductal adenocarcinoma. Cancers
(Basel). 12:7162020. View Article : Google Scholar : PubMed/NCBI
|