Introduction
Breast cancer (BC) is a common form of cancer in
women, and its incidence and mortality have been increasing
(1). BC is categorized into four
molecular subtypes based on the presence of the estrogen receptor
(ER), progesterone receptor (PR), Ki67 and human epidermal growth
factor receptor 2 (HER2): Luminal A (ER+,
PR+, HER2−, Ki67−), luminal B
(ER+, PR+, HER2−/+,
Ki67+), HER2 overexpressing (ER−,
PR−, HER2+) and triple-negative
(ER−, PR−, HER2−) (2). Among these subtypes, triple-negative
BC (TNBC) accounts for 12–17% of all BCs and shows features, such
as aggressive disease with dismal outcomes, absence of a
therapeutic target for decades, heterogeneous disease and higher
incidence in the younger age group (3). Although conventional chemotherapy can
achieve a response in patients with TNBC, the risk of recurrence is
higher than that in other types of BC. Additionally, the adverse
effects of conventional chemotherapy can be difficult to overcome,
highlighting an urgent need to identify novel therapeutic options
for TNBC (4).
Numerous mammalian cell divisions are regulated by
cell-cycle checkpoint proteins. In cancer cells, these checkpoint
proteins are mostly overexpressed or mutated and could be potential
targets for cancer therapies (5).
The spindle assembly complex (SAC; also known as the mitotic
checkpoint) is a signaling pathway that plays a role in monitoring
daughter chromatid separation and arrests mitosis if the chromatid
is separated incorrectly (6). In
the SAC, monopolar spindle 1 kinase (Mps1) is the core protein that
recruits SAC-related proteins and is activated in unattached
kinetochores. Inhibition of Mps1/TTK causes abrogation of the
mitotic checkpoint complex and anaphase-promoting complex, thereby
inducing immature mitosis in cancer cells. This results in massive
aneuploidy and cell death due to mitotic catastrophe (7,8).
Mps1/TTK is overexpressed in some cancer cells that have a poor
prognosis, especially in TNBC (9–12).
Thus, the inhibition of Mps1/TTK can be a promising target for TNBC
therapy. Several Mps1/TTK inhibitors are under investigation in
clinical trials to treat solid cancers, and efforts are underway to
overcome hematologic and gastrointestinal toxicities (13,14).
Various combination strategies were tried to enhance therapeutic
index of Mps1/TTK inhibitors by reducing their doses (15,16).
Based on the synergic preclinical efficacy, most of Mps1/TTK
inhibitors undergoing clinical trials have been studied in
combination with paclitaxel (17).
CFI-402257 combined with weekly paclitaxel was well tolerated
during a Phase 1b trial [Philippe Bedard, Mihaela Mates, John
Hilton, et al: Abstract P3-07-10: CCTG IND.236: A Phase 1b
trial of combined CFI-402257 and weekly paclitaxel in patients with
HER2-negative (HER2−) advanced BC. Cancer Res (2023) 83
(5_Supplement): P3-07-10]. However, considerable toxicity was
observed in the combination of BAY 1217389 with paclitaxel without
a therapeutic window (18).
Therefore, novel combination approach with alternative drugs rather
than paclitaxel with Mps1/TTK inhibitors would be meaningful.
Adenosine monophosphate (AMP)-activated protein
kinase (AMPK) is a cellular energy stress sensor that is activated
by the AMP: Adenosine triphosphate (ATP) ratio in the cytoplasm
(19). Aneuploid cells are
sensitive to energy- and proteotoxic stress-inducing compounds,
including AMPK activators. Aneuploidy plays a dual role in
tumorigenesis and is a potential therapeutic vulnerability in
cancer. The antineoplastic activity of AMPK activators is more
effective in high-grade aneuploidy than in low-grade aneuploidy
(20,21). Moreover, the p-AMPK level in
patients with TNBC is ~90% lower than that in healthy patients and
related to a higher grade of cancer (22,23).
Therefore, the anticancer effects of AMPK agonists on the excessive
aneuploidy, induced by MPS1/TTK inhibitors in TNBC, were
investigated.
In the present study, it was sought to evaluate the
synergistic effect of the combination strategy of Mps1/TTK
inhibitors with AMPK agonist in order to increase therapeutic range
through dose reduction. Based on understanding the mechanisms
underlying the effects of the combination of CFI-402257 and AICAR,
combining Mps1/TTK inhibitors with AMPK agonists appears to be a
rational option for BC.
Materials and methods
Ethics statement
All applicable national and institutional guidelines
for the care and use of animals were followed. The protocols for
animal experiments were approved (approval no. 2021-7F-08-02;
approval date: 2022-08-27) by the Animal Ethics Committee of the
Korea Research Institute of Chemical Technology (Daejeon, Korea).
The study was conducted in compliance with the ARRIVE
guidelines.
Chemicals and reagents
CFI-402257, empesertib and BOS-175722 were purchased
from MedChemExpress. AICAR was purchased from Cayman Chemical
Company. A-23187, metformin and nocodazole were provided from
Sigma-Aldrich; Merck KGaA. The primary antibodies for
phosphorylated (p-)TTK (cat. no. 44-1325G; RRID: AB_2533594; Thermo
Fisher Scientific, Inc.), Cyclin B1 (cat. no. sc-7393; RRID:
AB_627336), Cyclin D1 (cat. no. sc-8396; RRID: AB_627344), Cyclin E
(cat. no. sc-248; RRID: AB_627362) and p27 (cat. no. sc-1641; RRID:
AB_628074; all from Santa Cruz Biotechnology, Inc.) were used. The
following primary antibodies were obtained from Cell Signaling
Technology, Inc.: TTK (cat. no. 5469; RRID: AB_10.692670), p-AMPK
(cat. no. 2535; RRID: AB_331250), AMPK (cat. no. 2532; RRID:
AB_33033198), p-cdc2 (cat. no. 9111; RRID: AB_331460), CDK2 (cat.
no. 2546; RRID: AB_2276129), cleaved caspase-3 (cat. no. 9661;
RRID: AB_234188), cleaved PARP (cat. no. 5625; RRID: AB_10699459),
LC3B (cat. no. 2775; RRID: AB_915950), beclin-1 (cat. no. 3738;
RRID: AB_490837), Atg3 (cat. no. 3415; RRID: AB_2059244), Atg7
(cat. no. 8558; RRID: AB_10831194), p-PI3K (cat. no. 4228; RRID:
AB_659940), PI3K (cat. no. 4292; RRID: AB_329869), p-Akt (cat. no.
9271; RRID: AB_329825), p-mTOR (cat. no. 2971; RRID: AB_330970),
mTOR (cat. no. 2983; RRID: AB_2105622), ERK (cat. no. 9102; RRID:
AB_330744), p-p38 (cat. no. 9211; RRID: AB_331641) and p38 (cat.
no. 9212; RRID: AB_330713). The secondary antibodies were purchased
from Novus Biologicals, LLC (cat. no. NB7561) and GeneTex (cat. no.
GTX213110-01) respectively. Phenol Red-free Matrigel was purchased
from Corning, Inc. (cat. no. 356237).
Cell culture
Human mammary epithelial cells (HMEC; cat. no.
CC-2551) were obtained from Lonza Group, Ltd. and incubated in
MEBM™ basal medium (cat. no. CC-3151) and MEGM™ SingleQuits™
supplements (cat. no. CC-4136). MCF-7 (cat. no. HTB-22) and
MDA-MB-231 (cat. no. CRM-HTB-26) were obtained from the American
Type Culture Collection and incubated in RPMI-1640 medium (cat. no.
SH30027.01; Hyclone; Cytiva) containing 10% fetal bovine serum
(cat. no. SH030919.03; Hyclone; Cytiva) and 1%
penicillin/streptomycin at 37°C in a 5% CO2 incubator.
All cell lines were tested negative for mycoplasma contamination
using a MycoAlert® Mycoplasma Detection Kit (Lonza
Group, Ltd.).
Cytotoxicity assay
Cell viability was measured using Cyto X reagent
(cat. no. CYT1000; LPS Solution; http://lpss.co.kr). Cells were seeded at a density of
2×103 cells per well (MCF-7 and MDA-MB-231) and
4×103 cells per well (HMECs) in a 96-well plate. After
24 h, treatment with 0–1,000 nM CFI-402257, Empesertib and
BOS-172722, 0–1,200 µM AICAR, 0–10 µM A-23187 and 100 mM metformin
was conducted according to the experimental conditions. After 72
and 120 h of incubation, 10-µl Cyto X reagent (LPS solution) was
added and incubated at 37°C in a 5% CO2 incubator for an
additional 2 h. Thereafter, the absorbance was detected on a
spectrophotometer (BioTek; Agilent Technologies, Inc.), and the
IC50 values for each compound were determined using
GraphPad Prism 8 (Dotmatics), including logarithmic transformation,
normalization and non-linear regression (RRID: SCR_002798).
Cell-cycle distribution analysis
MDA-MB-231 cells were seeded at a density of
1×105 in 6-well plates. After 24 h of incubation, the
cells were treated with CFI-402257 (100 nM), AICAR (300 µM), or a
combination of both for 72 h. The total number of cells
(1×106 cells) were harvested and washed. Subsequently,
the cells were fixed in 70% ice-cold ethanol at −20°C for at least
3 h prior to staining. Cells were stained with 200 µl of
Muse® Cell Cycle Reagent (cat. no. 4700-1495; Merck
KGaA) and incubated for 30 min at 20–25°C in the dark. Data were
analyzed using a Muse® Cell Analyzer (software version
1.5; Merck KGaA). All procedures were conducted according to the
manufacturer's instructions.
Annexin V apoptosis assay
A total of 1×105 of MDA-MB-231 cells in
6-well plates were cultured with CFI-402257 (100 nM), AICAR (300
µM), or a combination of both. After 72 h, the suspended and
adherent cells were harvested and washed with Dulbecco's Phosphate
Buffered Saline (DPBS). After washing, each tube was stained with
100 µl of the Muse® Annexin V & Dead Cell reagent
(cat. no. MCH100105; Merck KGaA) and incubated for 20 min at room
temperature in the dark. Samples were prepared and stained
according to the manufacturer's protocol and analyzed using a
Muse® Cell Analyzer (software version 1.5; Merck
KGaA).
Acridine orange (AO) staining
AO stain, a cell-permeable reagent, conjugates with
nuclear acid. When it combines with DNA, the green signal appears;
however, a red signal appears in an acidic environment, such as an
autophagosome. MDA-MB-231 cells were seeded in 35-mm confocal
dishes. After 24 h, the cells were treated with CFI-402257 (100
nM), AICAR (300 µM), or a combination of the two for 72 h.
Thereafter, the cells were stained with 1 µg/ml of AO (cat. no.
A8097; Sigma-Aldrich; Merck KGaA) for 20 min at room temperature in
the dark. Images were obtained using an inverted fluorescence
phase-contrast microscope (K1-Fluo; Nanoscope Systems, Inc.).
Western blot analysis
The cells were trypsinized and rinsed with cold
DPBS. Cells were then lysed on ice using RIPA protein extract
solution (cat. no. EBA-1147; Elpis Bio-tech, Inc.), in which
Pierce™ Protease inhibitor mini tablets (cat. no. A32955) and
Pierce™ Phosphatase inhibitor mini tablets (cat. no. A32957; both
from Thermo Fisher Scientific, Inc.) were added. A total of 20
micrograms of total protein were quantitated using bicinchoninic
acid (BCA) and loaded onto 5–20% E-pagel (cat. no. E-T520L; ATTO
Corporation) filled with 1X Tris-Glycine SDS Buffer (cat. no.
CBT015; LPS solution). The gels were transferred to a Q-blot kit
(cat. no. WSE-4055; ATTO Corporation). After transfer, the PVDF
membranes were blocked with 1X TBS buffer (cat. no. CBT005; LPS
solution) containing 5% BSA (cat. no. BSAS0.1; Bovogen Biologicals)
for 30 min at room temperature and incubated with indicated primary
antibodies (1:1,000) overnight at 4°C. Thereafter, the membranes
were incubated with anti-mouse IgG (1:10,000) or anti-rabbit IgG
horseradish peroxidase (HRP)-conjugated secondary antibodies
(10,000) for 1 h at room temperature. Finally, the blots were
developed by using an Immobilon® Western
Chemiluminescent HRP substrate (cat. no. WBKLS0500;
MilliporeSigma). Densitometric analysis was performed using
Luminograph (cat. no. WSE-6100; software version ImageSaver 6; ATTO
Corporation).
In vivo tumor xenograft model
Total 20 of Female CAnN.Cg-Foxn1nu/CrlOri (BALB/c
nude) mice (8 weeks-old; weight, 21±1 g) were obtained from Orient
Bio, Inc. The mice were acclimated for 1 week before experiment at
a temperature of 24±1°C with 45±5% humidity, and a 12/12-h
light/dark cycle and were provided food and water ad
libitum. The animals were injected subcutaneously with
MDA-MB-231 cells (5×106 cells/mouse) in a mixture of
phenol-red free RPMI and phenol-red free Matrigel (total volume of
100 µl at a 1:1 ratio). When the tumor size reached 50–60
mm3, the animals were randomly classified into vehicle,
CFI-402257, AICAR and combination groups (n=5 per group).
CFI-402257 and the vehicle (10% NMP:40% PEG400:50% D.W.) were
administered via oral gavage (P.O.), whereas AICAR (diluted in PBS)
was administered via intraperitoneal (I.P.) injection (QD, 5
days/week). Tumor sizes and animal weight were measured. Animals
was euthanized using a 50% per min displacement of chamber air with
compressed CO2. Tumor tissue sample was collected,
images were captured and the weight was measured. Tumor volume was
calculated using the standard formula: 0.52 × length ×
width2. The tumor growth inhibition (TGI) rate was
calculated using the following equation: [1-(Vf, drug
treated-Vi, drug treated)/(Vf,
vehicle-Vi vehicle)] ×100, where Vf was
the average volume of tumor on the final day of the study, and
Vi was the average volume of tumor on the first day of
the study (24).
Statistical analysis
The experimental data were performed at least three
times, and are expressed as the mean±SEM or the mean ± SD. For the
determination of statistically significance, a one-way analysis of
variance (ANOVA) was used and then compared it with Bonferroni's
multiple comparison tests. *P<0.05 was considered to indicate
statistically significant differences between the control and the
treatment groups. All of the statistical comparisons were performed
using GraphPad Prism Software Version 8.0 (Dotmatics).
Results
Anticancer activity of CFI-402257,
AICAR and their combination in MDA-MB-231 cells
Cytotoxicity of three Mps1/TTK inhibitors
(CFI-402257, empesertib and BOS-172722) and three AMPK activators
(AICAR, A-23187 and metformin) was tested either alone or in
combination in MDA-MB-231 cells. AICAR and CFI-402257 were
investigated alone or in combination to evaluate the enhanced
anticancer effect of a combination of drugs. The viability of
MDA-MB-231 cells treated with the combination of AICAR or metformin
with CFI-402257 was reduced significantly compared with that of
cells treated with all drugs alone (Figs. 1B, S1 and S2).
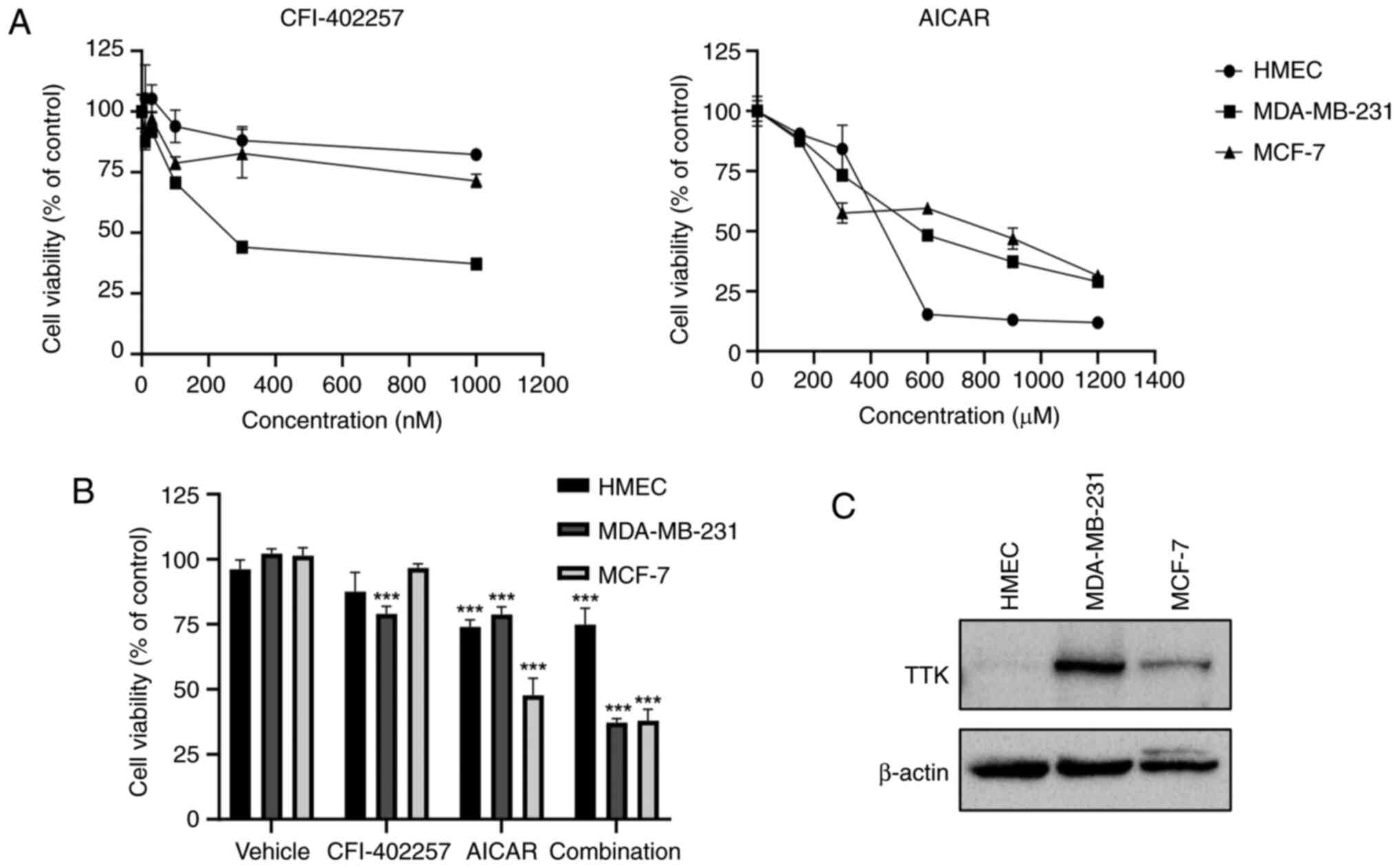
The inhibition of cell cytotoxicity after treatment
with CFI-402257 and AICAR was examined in a normal breast cell line
(HMEC) and a luminal BC cell line (MCF-7). As demonstrated in
Fig. 1A, CFI-402257 selectively
induced MDA-MB-231 TNBC cell cytotoxicity with high Mps1/TTK
expression (Fig. 1C), whereas AICAR
showed low selectivity in three breast cell lines. The combination
therapy of CFI-402257 and AICAR was effective on both of MDA-MB-231
and MCF-7 cells. However, the combined effects were similar to that
of AICAR alone in the MCF-7 cell line (Fig. 1B).
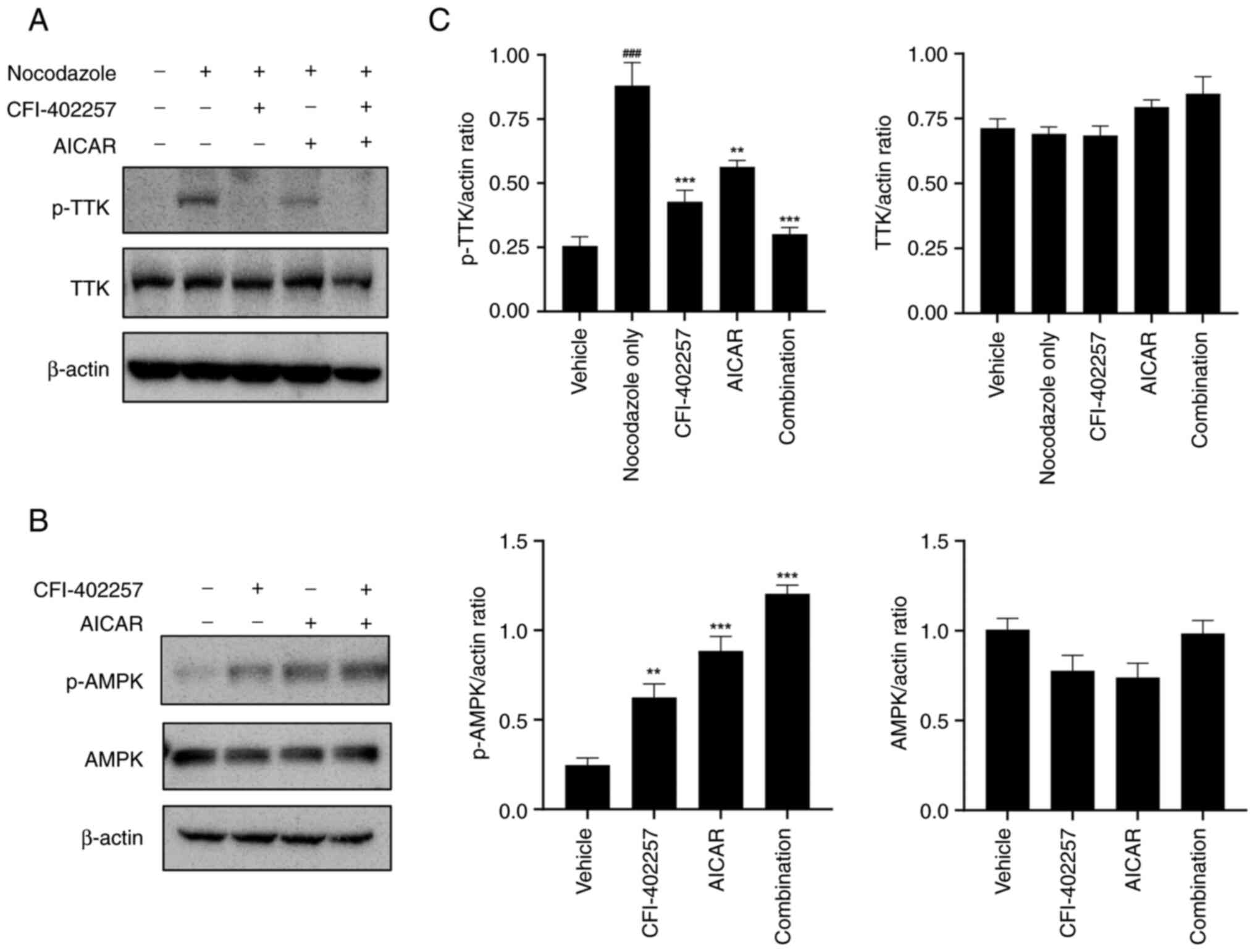
To validate the effects of CFI-402257 and AICAR on
their respective targets, the expression levels of p-TTK, TTK,
p-AMPK and AMPK were determined using western blot analysis. The
robust SAC was activated by a spindle poison, nocodazole (50
ng/ml), to determine SAC modulation by Mps1/TTK inhibitors
(25). Mps1/TTK dephosphorylation
by Mps1/TTK inhibitors was evaluated (15,24).
Combination therapy with CFI-402257 and AICAR showed synergistic
effects by decreasing p-TTK and increasing p-AMPK protein levels
(Figs. 2 and S3).
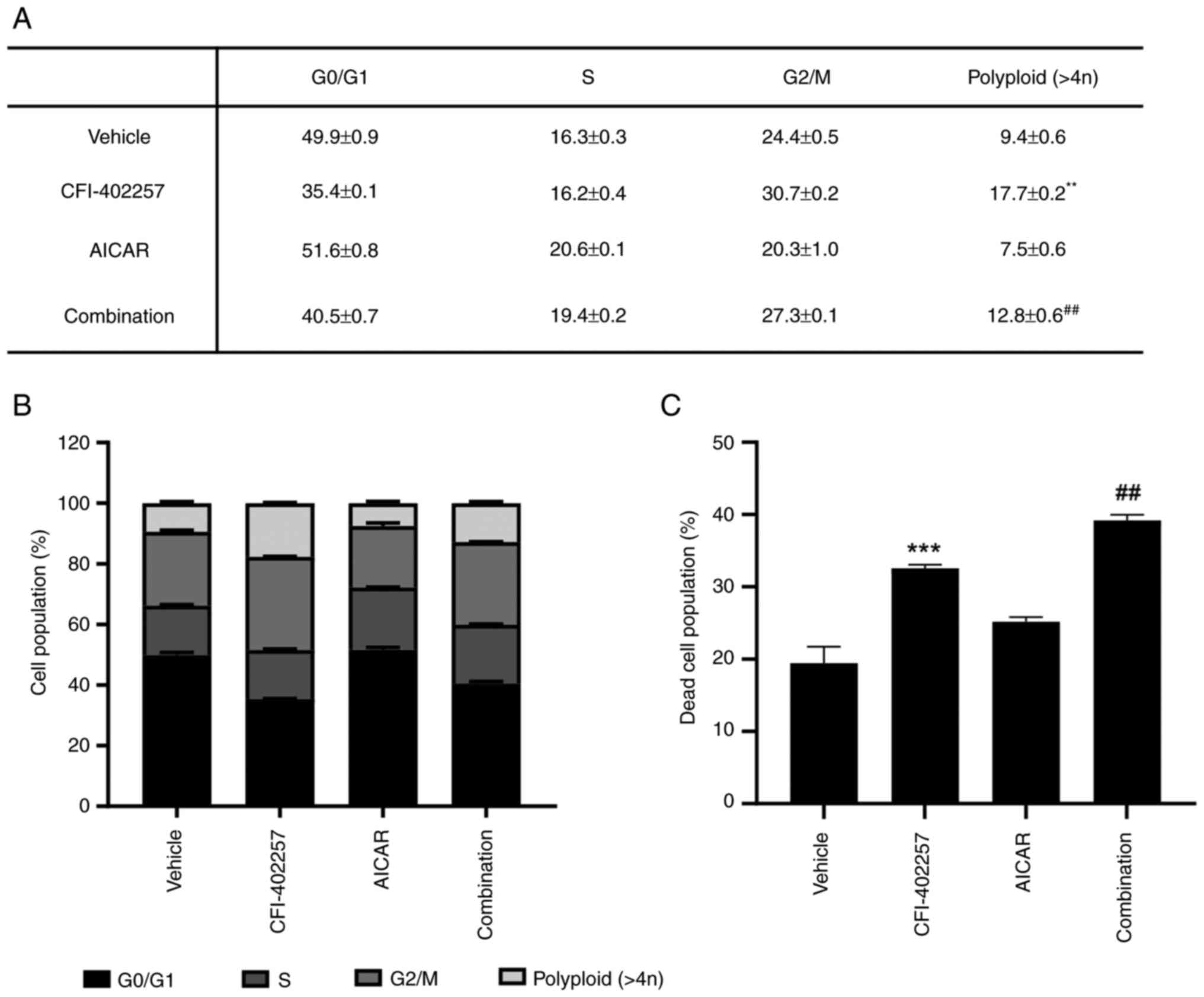
Cell-cycle distribution after
combination therapy with CFI-402257 and AICAR
CFI-402257 increased the proportion of G2/M phase
(24.4–30.7%) and polyploid cells (9.4–17.7%), while AICAR slightly
increased the proportion of cells in the S phase (16.3–20.6%).
Interestingly, the combination therapy group revealed decreased
proportions of G2/M phase and polyploid cells and induced a higher
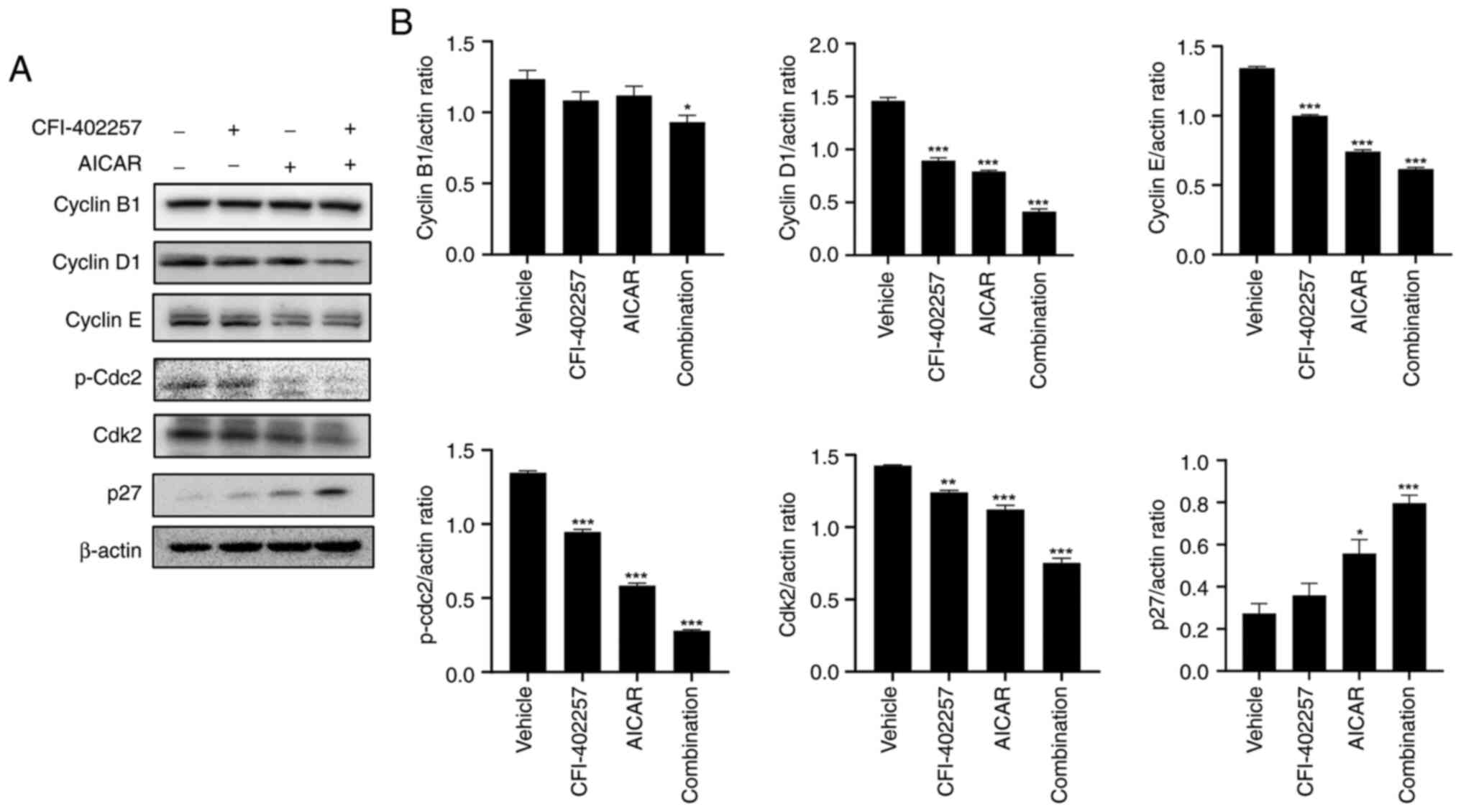
cell death rate than the CFI-402257 monotherapy group (Fig. 3). The regulatory proteins involved
in cell-cycle distribution were also evaluated using western blot
analysis. Combination therapy reduced the levels of cyclin D1,
cyclin E, p-cdc2 and cyclin-dependent kinase 2 (cdk2) in MDA-MB-231
cells. The protein level of cyclin B1 decreased slightly. Lastly,
combination therapy significantly increased the protein levels of
p27 (Figs. 4 and S3).
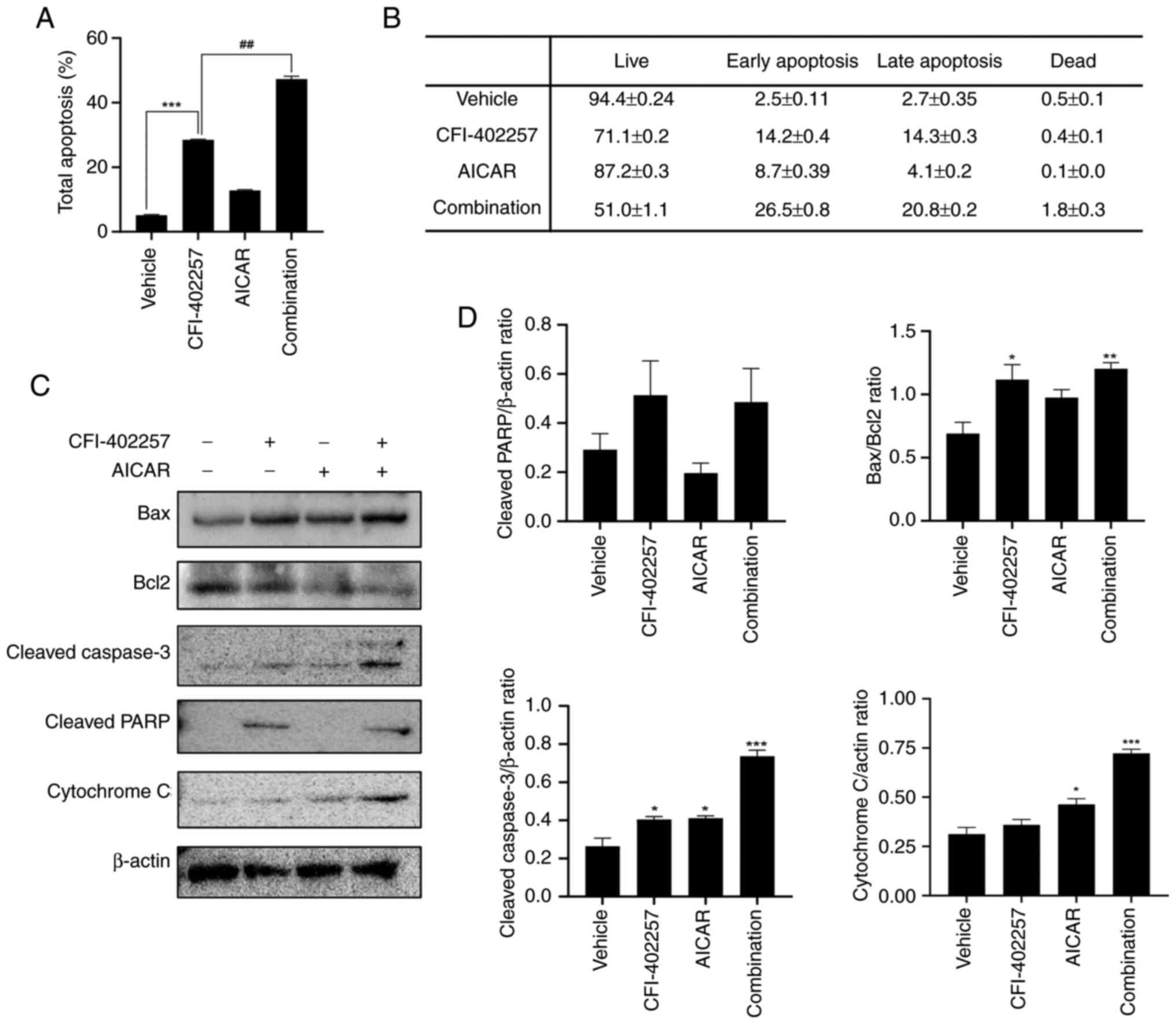
Apoptosis induced by combination
therapy with CFI-402257 and AICAR
Annexin V-7AAD double-staining was performed to
evaluate cell death induced by the combination of CFI-402257 and
AICAR. Live (annexin V−, 7-AAD−), early
apoptotic (annexin V+, 7-AAD+), late
apoptotic (annexin V+, 7-AAD+), and dead
(annexin V−, 7-AAD+) cells were identified
using a Muse® Cell Analyzer. The total number of
apoptotic cells was calculated by adding the early and late
apoptotic cells. As demonstrated in Figs. 5A, B and S4, the combination therapy significantly
increased the rate of total apoptosis in MDA-MB-231 cells in
comparison with the CFI-402257 monotherapy group (28.5 vs. 47.3%).
In addition, the expression levels of apoptosis-related proteins
were analyzed using western blotting. Combination therapy increased
the levels of cleaved poly (ADP-ribose) polymerase, cleaved
caspase-3 and Bax in MDA-MB-231 cells. The cytoplasmic release of
cytochrome C also increased, and the Bcl2 level was decreased.
Overall, the Bax/Bcl2 ratio, a marker of apoptosis, increased
(Figs. 5C and D and S3). These results indicated that
combination therapy with CFI-402257 and AICAR effectively induced
apoptosis.
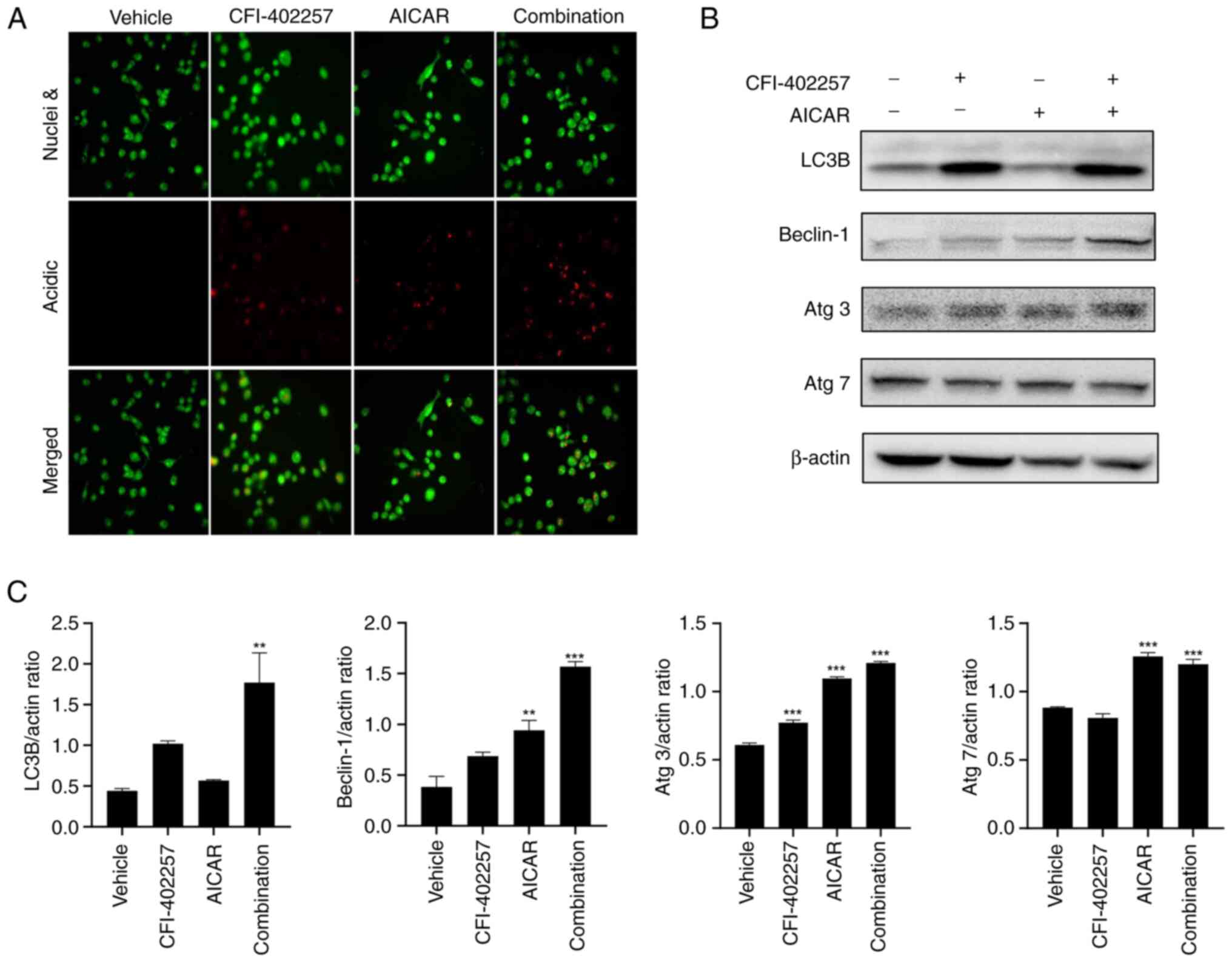
Autophagic cell death after
combination therapy with CFI-402257 and AICAR
To improve understanding of the mechanisms
underlying cell death, AO staining (which changes to bright red
under acidic conditions) was used to highlight autophagosomes
(26). The combination therapy with
CFI-402257 and AICAR significantly increased the number of acidic
vacuoles. Both monotherapies also induced the formation of acidic
vacuoles (Fig. 6A). Another marker
of autophagosomes, LC3B-II (membrane-bound form, lower band), was
also significantly upregulated in the combination group. The
expression levels of other autophagy-related proteins, namely,
Beclin-1, Atg3 and Atg7, also increased in the combination therapy
group (Figs. 6B, C and S3). Thus, combination therapy with
CFI-402257 and AICAR mediated autophagic cell death in MDA-MB-231
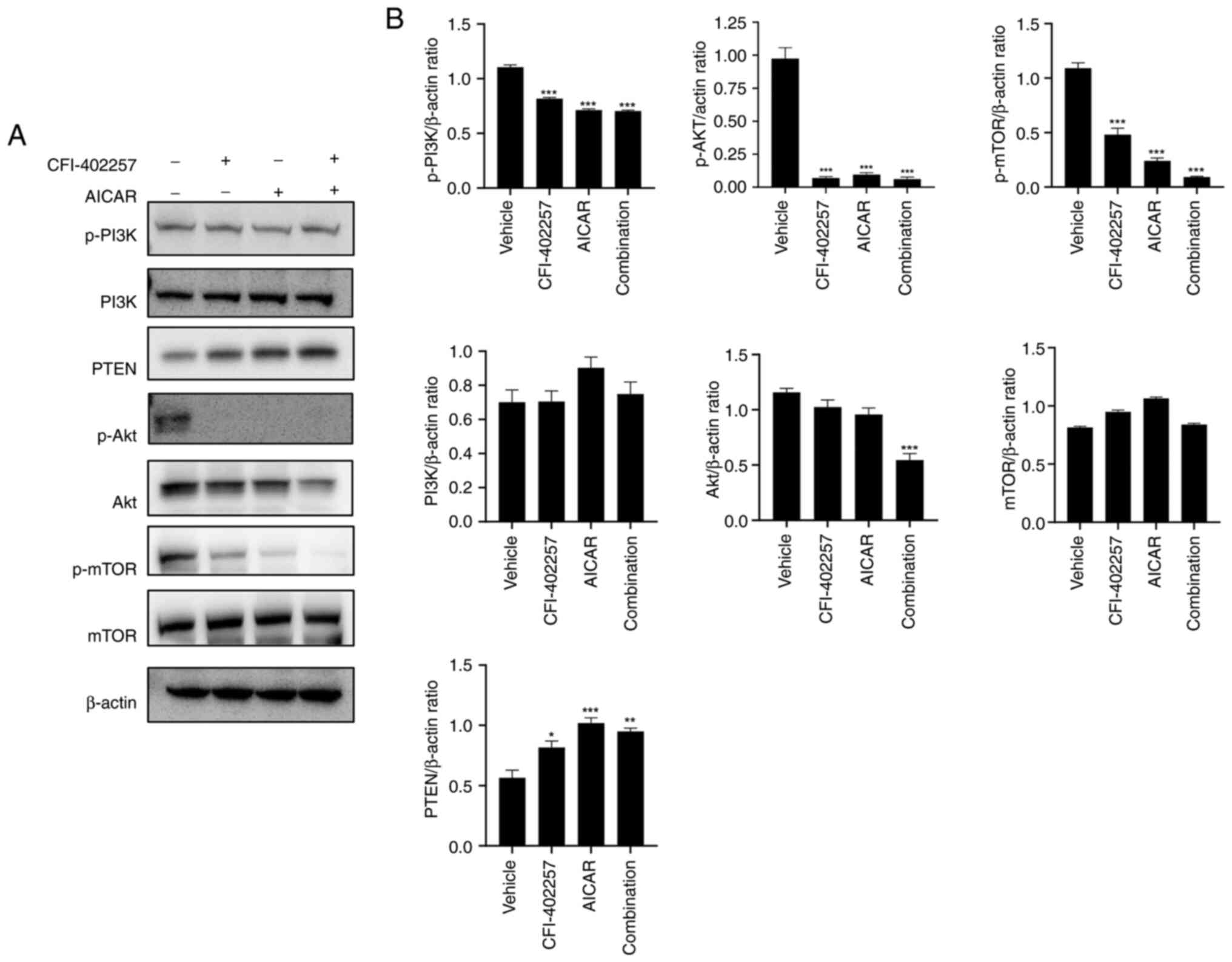
cells. Western blot analysis was used to determine the effect of
combination therapy on the PI3K/Akt/mTOR and MAPK pathways; it was
revealed that the expression levels of p-PI3K, p-Akt, p-mTOR and
Akt were downregulated, while the expression of the tumor
suppressor regulator PTEN, which inhibits the Akt/mTOR signaling
pathway (27), increased in the
combination therapy group (Figs. 7
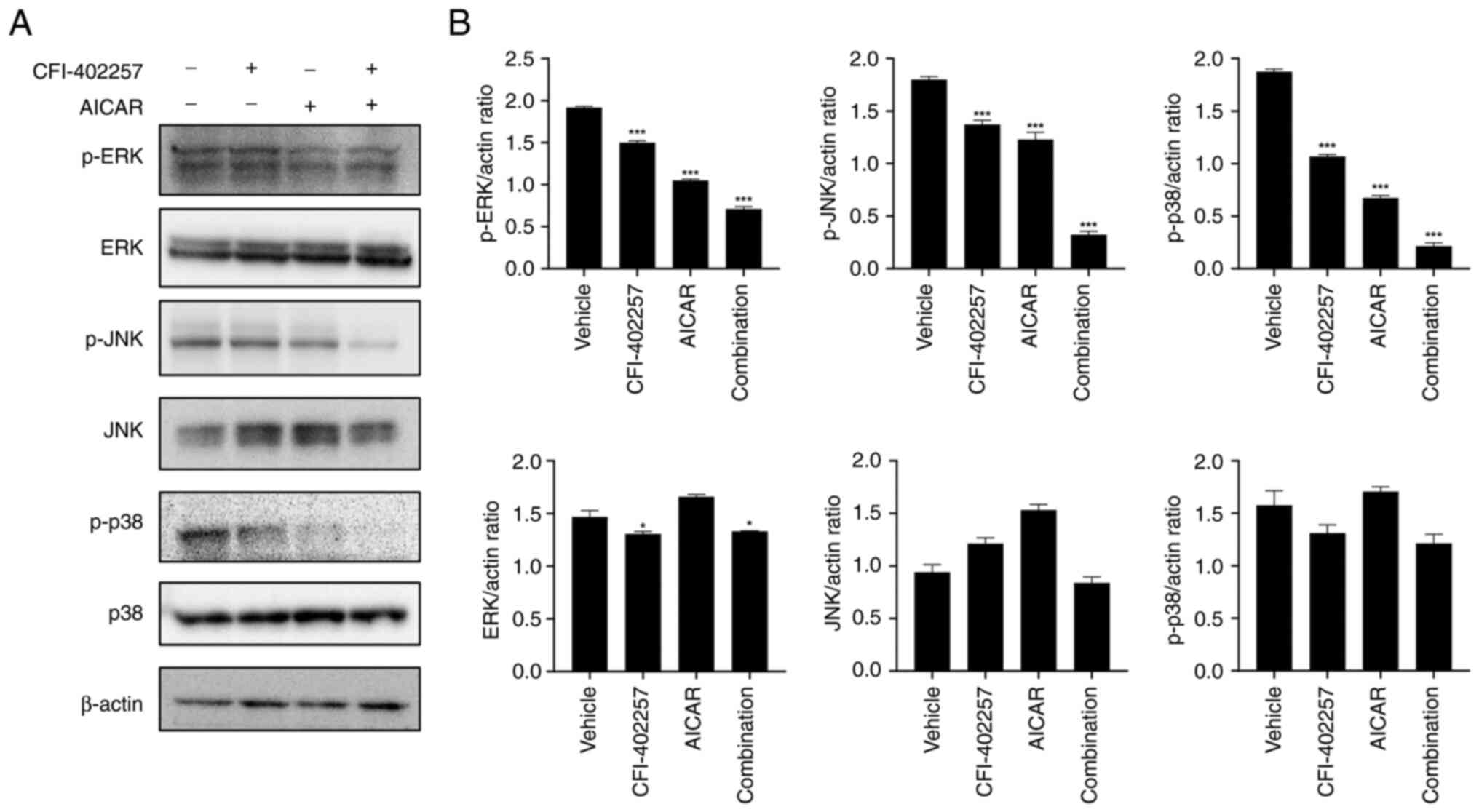
and S3). The expression levels of
p-ERK, p-JNK and p-p38 were also downregulated in the combination
therapy group (Figs. 8 and S3).
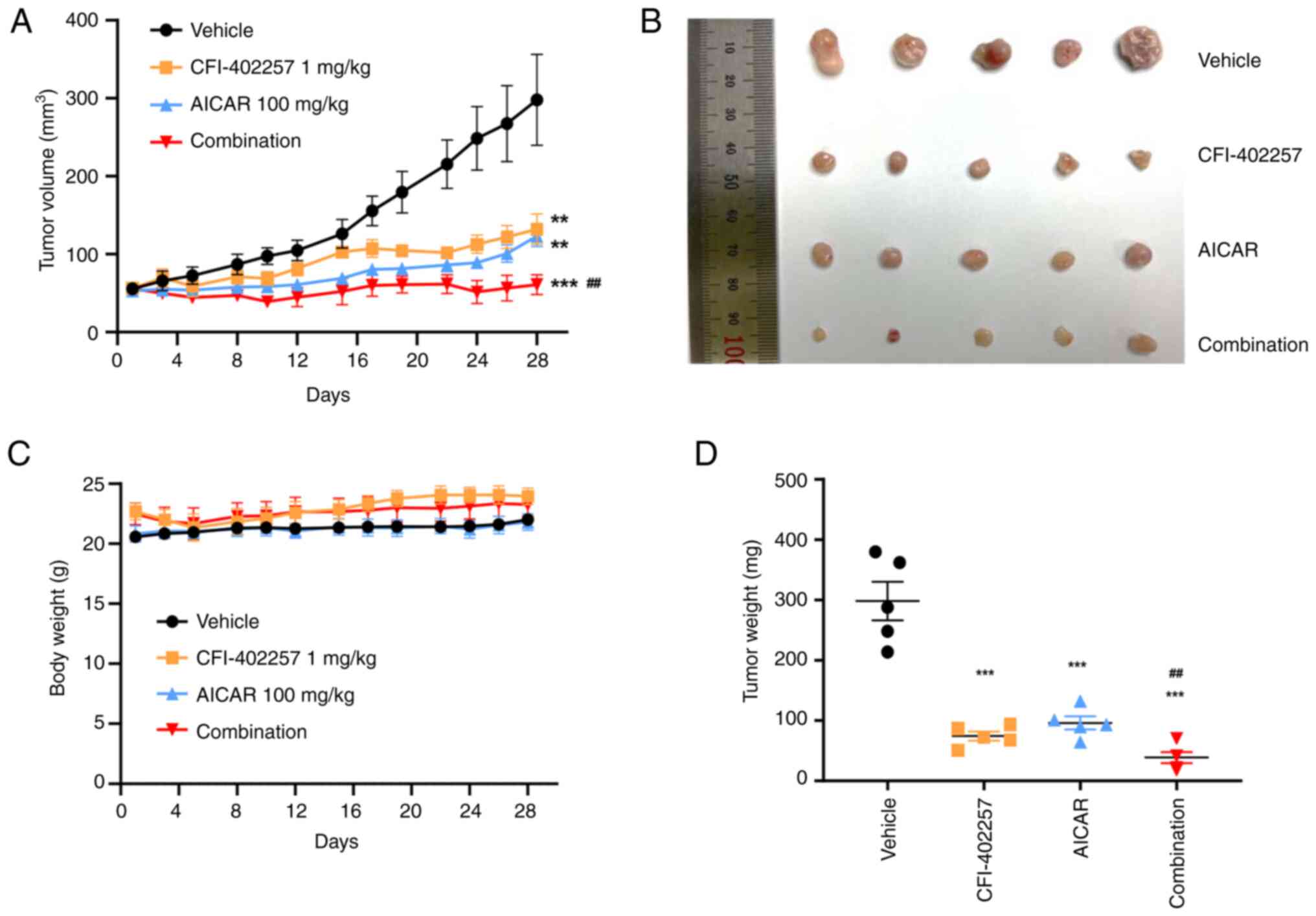
The combination of CFI-402257 and
AICAR effectively inhibits TNBC growth in the tumor xenograft
model
The in vivo antitumor activity of the
compounds was examined in mice bearing MDA-MB-231 tumor xenografts.
After the treatment, the longest tumor lengths observed were 14.33,
7.36, 7.92 and 5.85 mm in the vehicle group, CFI-402257
monotherapy, AICAR monotherapy and the combination therapy group,
respectively. Correspondingly, the final mean tumor volumes were
calculated as 297.9 mm3 (range, 133.4~485.28
mm3), 132.04 (range, 64~179.06 mm3), 123.12
(range, 83.7~153.32 mm3) and 60.96 mm3
(range, 30.61~101.63 mm3) for the vehicle treatment,
CFI-402257 alone, AICAR alone, and the combination treatment,
respectively. Complete inhibition of tumor growth was observed in
the combination group, with a TGI rate of 98%, whereas the TGI
rates for monotherapy with CFI-402257 and AICAR were 69 and 71%,
respectively (Fig. 9). No
significant adverse effects, such as weight loss (>10% of total
weight) or strange behaviors, were observed in the studies.
Discussion
Mps1/TTK, which is the core protein of the SAC
complex, is overexpressed in TNBC in comparison with other BC cells
(9–12). Mps1/TTK inhibition triggers genomic
instability and results in cancer cell apoptosis. Aneuploid cancer
cells show resistance to multiple chemotherapeutic regimens, induce
tumor heterogeneity, and improve the tumors' adaptation to harsh
conditions (28,29). In particular, highly aneuploid
cancer cells are more sensitive to the energy and proteotoxic
stress induced by AMPK agonists than normal cells (20,30,31).
Therefore, the synergistic effect of the MPS1/TTK inhibitor (which
increases aneuploidy) with proteotoxic compounds that perturb
energy metabolism was investigated in the present study.
To evaluate the anticancer effects of the MPS1/TTK
inhibitor in combination with a proteotoxic compound, cancer cell
cytotoxicity was evaluated after exposure to combinations of three
Mps1/TTK inhibitors (CFI-402257, Empesertib and BOS-172722) and
three AMPK activators (AICAR, A-23187 and Metformin). As
demonstrated in Fig. S2, the three
Mps1/TTK inhibitors showed similar cytotoxic potency with each AMPK
activator. However, A-23187 exhibited less synergism than the other
AMPK activators. Metformin is an indirect AMPK activator that
inhibits complex 1 in the mitochondrial respiratory chain and
eventually increases the AMP:ATP ratio by inhibiting oxidative
phosphorylation (OXPHOS). However, numerous resistant cancer cells
produce ATP via glycolysis rather than OXPHOS even in the presence
of oxygen, a phenomenon called the Warburg effect. Thus, inhibition
of OXPHOS may cause activation of glycolysis, which could result in
resistance. By contrast, AICAR is an allosteric AMPK activator that
mimics the structure of AMP, potentially allowing it to act on AMPK
while causing fewer changes in OXPHOS and glycolysis than metformin
(19,32,33).
Previous studies have also reported that AICAR is effective against
aneuploid cancer cells, but metformin is not (20,34).
Therefore, the subsequent studies were conducted using CFI-402257,
an MPS1/TTK inhibitor, and AICAR, an AMPK activator.
The in vitro anticancer effects of
CFI-402257, AICAR and their combination were tested in three breast
cell lines: HMECs, MCF-7 and MDA-MB-231. The MDA-MB-231 cell line
is commonly used as late-stage BC model. This cell line is
ER−, PR− and HER2-negative and expresses
mutated p53. Therefore, MDA-MB-231 represents a favorable model of
TNBC. CFI-402257 exhibited the most potent effect on MDA-MB-231
cells. HMECs may be more vulnerable to the energy stress induced by
AICAR than cancer cells. AICAR monotherapy exhibited cytotoxicity
in the MCF-7 cell line. The cytotoxicity of the drug combination
was compared with that of each drug treated alone to evaluate the
beneficial effects of combination therapy. The combination of AICAR
with CFI-402257 resulted in a notable decrease in the viability of
MDA-MB-231 cells compared with when each drug treated alone. In
MCF-7 cells, the combination of CFI-402257 and AICAR exhibited
cytotoxicity, while the combined effects were similar to that of
AICAR alone. Based on the results of combination therapy, expanding
the indication to include ER+ BC patients with high
Mps1/TTK expression could be considered. Phosphorylation of
Mps1/TTK is activated only in the tumor microenvironment;
nocodazole, which interferes with microtubule polymerization, was
treated with other test compounds to evaluate changes in p-Mps1/TTK
level (7,24,35).
In the present study, CFI-402257 and AICAR synergistically
inhibited cell proliferation and decreased p-Mps1/TTK and p-AMPK
protein levels in MDA-MB-231 cells.
The SAC inhibition and massive chromosome
segregation caused by CFI-402257 affect cell-cycle distribution
(24). In the CFI-402257 treatment
group, the levels of G2/M phase and polyploid cell counts were
substantially greater than those in the vehicle group. By contrast,
in the AICAR treatment group, the cell counts in the G0/G1 and S
phases were slightly greater than those in the vehicle group.
Notably, the annexin V apoptosis assay showed that the apoptotic
rate increased significantly with combined treatment with
CFI-402257 and AICAR. The potential benefits of utilizing an AMPK
agonist against various high-grade aneuploid human tumors was
proposed (20). In the present
study, the heightened apoptosis induced by AICAR was observed in
states of increased aneuploidy, facilitated by the Mps1/TTK
inhibitor (36). Future studies to
investigate the anticancer effects on sorted polyploidy cells are
required to understand the aneuploidy sensitivity of AMPK
agonist.
Apoptosis via the intrinsic pathway by Mps1/TTK
inhibitors has been widely reported, whereas autophagic cell death
has rarely been studied. To understand the precise molecular
mechanisms underlying this form of cell death, changes in autophagy
biomarkers were investigated, and the combination of CFI-402257 and
AICAR revealed dual inhibition of the PI3K/Akt/mTOR and MAPK
pathways (Figs. 7 and 8). The PI3K/Akt/mTOR-PTEN axis is
considered the predominant oncogenic pathway that is mutated in BC.
In particular, frequent alterations have been observed in PTEN,
which plays a crucial role in TNBC. Therefore, numerous clinical
attempts using PI3K/mTOR inhibitors have been made for TNBC
treatment. The MAPK signaling pathway consists of three main
groups: Extracellular signal-regulated protein kinases (ERK1/2),
p38 MAP kinases and c-Jun NH2-terminal kinases (JNK1/2/3), which
are associated with various malignancies, including breast, lung
and thyroid carcinomas as well as neck squamous cell carcinomas.
The MAPK pathway is frequently hyperactive in TNBC. One reason for
this is that TNBC shows aggressive division along with an increased
number of genomic copies in comparison with other BC cells. This
leads to genomic instability in these cells. Second, the loss of
crosstalk between other signaling pathways, such as the
PI3K/Akt/mTOR pathway, may cause uncontrolled MAPK pathway
activation. Therefore, dual targeting of the PI3K/Akt/mTOR and MAPK
pathways is important to effectively inhibit the cytotoxicity of
TNBC (37–39). Thus, the dual inhibition of the
PI3K/Akt/mTOR and MAPK pathways induced by the combination of
CFI-402257 and AICAR could be a potential strategy for treating
TNBC.
Interestingly, protein expression level of p27 was
increased (Fig. 4). The critical
role of p27 in regulating G1-to-S phase progression and its
prognostic significance have been reported in several BC studies
(40). p27 is eliminated by
ubiquitin-mediated proteolysis, and its degradation is inhibited by
decreased cyclin expression. Several investigators have reported
the effect of PI3K/Akt and Ras/Raf/Mek1 pathways on the abundance
and activity of p27. Considering the present study, the PI3K/Akt
pathway was inhibited by combination therapy. Inhibition PI3K/Akt
pathway suppresses the expression of oncogenes such as c-myc.
Therefore, the expression of p27, which are regulated by c-myc, was
increased (41).
The main limitation of the application of Mps1/TTK
inhibitors in vivo is their narrow therapeutic range due to
hematological and gastrointestinal toxicities. The maximum
tolerable dose of CFI-402257 in mice is 6.5 mg/kg (24). Considering the in vivo
profile results and the study designs of previous investigations on
CFI-402257 and AICAR (24,42,43),
the dosages for the combination therapy were chosen as 1 mg/kg for
CFI-402257 and 100 mg/kg for AICAR in the present study. This
combination inhibited tumor growth by 98% without any additional
adverse effects; in comparison, the inhibition rates with
CFI-402257 and AICAR monotherapy were 69 and 71%, respectively
(Fig. 9). The expression of markers
related to mechanisms such as the cell cycle, cell viability and
apoptosis in tumor tissues was not measured and it will be
considered in the further study. To the best of the author's
knowledge, the present study provides the first in vivo
evidence for the anticancer activity study of a combination of an
Mps1/TTK inhibitor and an AMPK activator.
In conclusion, to circumvent the excessive toxicity
of Mps1/TTK inhibitors, a combination of an Mps1/TTK inhibitor with
an AMPK agonist was explored in the present study. The combination
therapy showed enhanced anticancer effect mediated by dual
inhibition of the PI3K/Akt/mTOR and MAPK pathways, which regulate
cell cytotoxicity, apoptosis and autophagy. The combination of
CFI-402257 and AICAR significantly suppressed the growth of
MDA-MB-231-derived TNBC in immunodeficient mice. These findings
provide new insights into the potential for using CFI-402257 in
combination with an AMPK agonist to enhance its efficacy and
tolerability in TNBC therapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Korea Drug Development
Fund funded by the Ministry of Science and ICT, Ministry of Trade,
Industry, and Energy, and Ministry of Health and Welfare (grant no.
HN22C0694; Republic of Korea).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JSL, JS and SA designed the experiments. JSL
performed the experiments. SA, JSL and EK analyzed and interpreted
the data. SA, JSL, and EK wrote and revised the manuscript. JS and
SA supervised the study and confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All applicable national and institutional guidelines
for the care and use of animals were followed. The protocols for
animal experiments were approved (approval no. 2021-7F-08-02;
approval date: 2022-08-27) by the Animal Ethics Committee of the
Korea Research Institute of Chemical Technology with code number
(Daejeon, Korea). The study was carried out in compliance with the
ARRIVE guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AMP
|
adenosine monophosphate
|
|
AMPK
|
AMP-activated protein kinase
|
|
ATP
|
adenosine triphosphate
|
|
ER
|
estrogen receptor
|
|
HMEC
|
human mammary epithelial cells
|
|
Mps1
|
monopolar spindle 1 kinase
|
|
PR
|
progesterone receptor
|
|
SAC
|
spindle assembly complex
|
|
TNBC
|
triple-negative breast cancer
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sasmita AO and Wong YP: Organoids as
reliable breast cancer study models: An update. Int J Oncol Res.
1:0082018.
|
|
3
|
Schwentner L, Wolters R, Koretz K,
Wischnewsky MB, Kreienberg R, Rottscholl R and Wöckel A:
Triple-negative breast cancer: The impact of guideline-adherent
adjuvant treatment on survival-a retrospective multi-centre cohort
study. Breast Cancer Res Treat. 132:1073–1080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bai L, Zhou B, Yang CY, Ji J, McEachern D,
Przybranowski S, Jiang H, Hu J, Xu F, Zhao Y, et al: Targeted
degradation of BET proteins in triple-negative breast cancer.
Cancer Res. 77:2476–2487. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dominguez-Brauer C, Thu KL, Mason JM,
Blaser H, Bray MR and Mak TW: Targeting mitosis in cancer: Emerging
strategies. Mol Cell. 60:524–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lara-Gonzalez P, Westhorpe FG and Taylor
SS: The spindle assembly checkpoint. Curr Biol. 22:R966–R980. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Colombo R, Caldarelli M, Mennecozzi M,
Giorgini ML, Sola F, Cappella P, Perrera C, Depaolini SR, Rusconi
L, Cucchi U, et al: Targeting the mitotic checkpoint for cancer
therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res.
70:10255–10264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tardif KD, Rogers A, Cassiano J, Roth BL,
Cimbora DM, McKinnon R, Peterson A, Douce TB, Robinson R, Dorweiler
I, et al: Characterization of the cellular and antitumor effects of
MPI-0479605, a small-molecule inhibitor of the mitotic kinase Mps1.
Mol Cancer Ther. 10:2267–2275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maire V, Baldeyron C, Richardson M, Tesson
B, Vincent-Salomon A, Gravier E, Marty-Prouvost B, De Koning L,
Rigaill G, Dumont A, et al: TTK/hMPS1 is an attractive therapeutic
target for triple-negative breast cancer. PLoS One. 8:e637122013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salvatore G, Nappi TC, Salerno P, Jiang Y,
Garbi C, Ugolini C, Miccoli P, Basolo F, Castellone MD, Cirafici
AM, et al: A cell proliferation and chromosomal instability
signature in anaplastic thyroid carcinoma. Cancer Res.
67:10148–10158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Slee RB, Grimes BR, Bansal R, Gore J,
Blackburn C, Brown L, Gasaway R, Jeong J, Victorino J, March KL, et
al: Selective inhibition of pancreatic ductal adenocarcinoma cell
growth by the mitotic MPS1 kinase inhibitor NMS-P715. Mol Cancer
Ther. 13:307–315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuan B, Xu Y, Woo JH, Wang Y, Bae YK, Yoon
DS, Wersto RP, Tully E, Wilsbach K and Gabrielson E: Increased
expression of mitotic checkpoint genes in breast cancer cells with
chromosomal instability. Clin Cancer Res. 12:405–410. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fuentes-Antrás J, Bedard PL and Cescon DW:
Seize the engine: Emerging cell cycle targets in breast cancer.
Clin Transl Med. 14:e15442024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martinez R, Blasina A, Hallin JF, Hu W,
Rymer I, Fan J, Hoffman RL, Murphy S, Marx M, Yanochko G, et al:
Mitotic checkpoint kinase Mps1 has a role in normal physiology
which impacts clinical utility. PLoS One. 10:e01386162015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anderhub SJ, Mak GW, Gurden MD, Faisal A,
Drosopoulos K, Walsh K, Woodward HL, Innocenti P, Westwood IM, Naud
S, et al: High proliferation rate and a compromised spindle
assembly checkpoint confers sensitivity to the MPS1 inhibitor
BOS172722 in triple-negative breast cancers. Mol Cancer Ther.
18:1696–1707. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wengner AM, Siemeister G, Koppitz M,
Schulze V, Kosemund D, Klar U, Stoeckigt D, Neuhaus R, Lienau P,
Bader B, et al: Novel Mps1 kinase inhibitors with potent antitumor
activity. Mol Cancer Ther. 15:583–592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Novais P, Silva PMA, Amorim I and Bousbaa
H: Second-generation antimitotics in cancer clinical trials.
Pharmaceutics. 13:10112021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Atrafi F, Boix O, Subbiah V, Diamond JR,
Chawla SP, Tolcher AW, LoRusso PM, Eder JP, Gutierrez M, Sankhala
K, et al: A phase I study of an MPS1 inhibitor (BAY 1217389) in
combination with paclitaxel using a novel randomized continual
reassessment method for dose escalation. Clin Cancer Res.
27:6366–6375. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim J, Yang G, Kim Y, Kim J and Ha J: AMPK
activators: Mechanisms of action and physiological activities. Exp
Mol Med. 48:e2242016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang YC, Williams BR, Siegel JJ and Amon
A: Identification of aneuploidy-selective antiproliferation
compounds. Cell. 144:499–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vasudevan A, Schukken KM, Sausville EL,
Girish V, Adebambo OA and Sheltzer JM: Aneuploidy as a promoter and
suppressor of malignant growth. Nat Rev Cancer. 21:89–103. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao W, Li J, Hao Q, Vadgama JV and Wu Y:
AMP-activated protein kinase: A potential therapeutic target for
triple-negative breast cancer. Breast Cancer Res. 21:292019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hadad SM, Baker L, Quinlan PR, Robertson
KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG,
et al: Histological evaluation of AMPK signalling in primary breast
cancer. BMC Cancer. 9:3072009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mason JM, Wei X, Fletcher GC, Kiarash R,
Brokx R, Hodgson R, Beletskaya I, Bray MR, Mak TW, et al:
Functional characterization of CFI-402257, a potent and selective
Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc Natl
Acad Sci USA. 114:3127–3132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Ling Y, Guo Y, Bai Y, Shi X, Gong
F, Tan P, Zhang Y, Wei C, He X, et al: Mps1 kinase regulates tumor
cell viability via its novel role in mitochondria. Cell Death Dis.
7:e22922016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Millot C, Millot JM, Morjani H, Desplaces
A and Manfait M: Characterization of acidic vesicles in
multidrug-resistant and sensitive cancer cells by acridine orange
staining and confocal microspectrofluorometry. J Histochem
Cytochem. 45:1255–1264. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CY, Chen J, He L and Stiles BL: PTEN:
Tumor suppressor and metabolic regulator. Front Endocrinol
(Lausanne). 9:3382018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ippolito MR, Martis V, Hong C, Hong C,
Wardenaar R, Zerbib J, Spierings DCJ, Ben-David U, Foijer F and
Santaguida S: Aneuploidy-driven genome instability triggers
resistance to chemotherapy. Biorxiv. 2020.09.25.313924. 2020.
|
|
29
|
Vasan N, Baselga J and Hyman DM: A view on
drug resistance in cancer. Nature. 575:299–309. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo J, Solimini NL and Elledge SJ:
Principles of cancer therapy: Oncogene and non-oncogene addiction.
Cell. 136:823–837. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manchado E and Malumbres M: Targeting
aneuploidy for cancer therapy. Cell. 144:465–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song IS, Han J and Lee HK: Metformin as an
anticancer drug: A commentary on the metabolic determinants of
cancer cell sensitivity to glucose limitation and biguanides. J
Diabetes Investig. 6:516–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ly P, Kim SB, Kaisani AA, Marian G, Wright
WE and Shay JW: Aneuploid human colonic epithelial cells are
sensitive to AICAR-induced growth inhibition through EGFR
degradation. Oncogene. 32:3139–3146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang L, Li A, Lei Q and Zhang Y:
Tumor-intrinsic signaling pathways: Key roles in the regulation of
the immunosuppressive tumor microenvironment. J Hematol Oncol.
12:1252019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Herriage HC, Huang YT and Calvi BR: The
antagonistic relationship between apoptosis and polyploidy in
development and cancer. Semin Cell Dev Biol. 156:35–43. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Balko JM, Schwarz LJ, Bhola NE, Kurupi R,
Owens P, Miller TW, Gómez H, Cook RS and Arteaga CL: Activation of
MAPK pathways due to DUSP4 loss promotes cancer stem cell-like
phenotypes in basal-like breast cancer. Cancer Res. 73:6346–6358.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jeffrey KL, Camps M, Rommel C and Mackay
CR: Targeting dual-specificity phosphatases: Manipulating MAP
kinase signalling and immune responses. Nat Rev Drug Discov.
6:391–403. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuo WL, Duke CJ, Abe MK, Kaplan EL, Gomes
S and Rosner MR: ERK7 expression and kinase activity is regulated
by the ubiquitin-proteosome pathway. J Biol Chem. 279:23073–23081.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alkarain A, Jordan R and Slingerland J:
p27 deregulation in breast cancer: Prognostic significance and
implications for therapy. J Mammary Gland Biol Neoplasia. 9:67–80.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blain SW, Scher HI, Cordon-Cardo C and
Koff A: p27 as a target for cancer therapeutics. Cancer Cell.
3:111–115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jhaveri TZ, Woo J, Shang X, Park BH and
Gabrielson E: AMP-activated kinase (AMPK) regulates activity of
HER2 and EGFR in breast cancer. Oncotarget. 6:14754–14765. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Theodoropoulou S, Brodowska K, Kayama M,
Morizane Y, Miller JW, Gragoudas ES and Vavvas DG: Aminoimidazole
carboxamide ribonucleotide (AICAR) inhibits the growth of
retinoblastoma in vivo by decreasing angiogenesis and inducing
apoptosis. PLoS One. 8:e528522013. View Article : Google Scholar : PubMed/NCBI
|