Introduction
As of 2020, lung cancer was responsible for the
deaths of ~1.8 million individuals worldwide, making it the leading
cause of cancer mortality. Non-small cell lung cancer (NSCLC)
constitutes the majority of these cases, accounting for ~85%
(1,2). The surgical resection, despite its
inherent risks, has unequivocally exhibited a remarkable decline in
mortality rates among patients afflicted with operable early-stage
and locally advanced NSCLC; 25–70% of surgical patients eventually
relapse, yielding a worse prognosis (3). The current landscape of advanced lung
cancer treatment has witnessed the ascendancy of immunotherapy as
the foremost therapeutic modality (4). Among them, the application of immune
checkpoint blockade has demonstrated remarkable efficacy in
improving survival outcomes; however, no response cases still
prevail among a substantial proportion of patients undergoing
treatment (5,6). The mechanisms by which the body
resists immunotherapy and the development of strategies to make
advanced and metastatic disease manageable are urgently needed to
be explored.
Blockers targeting programmed cell death protein 1
(PD-1) and its ligand PD-L1 have been granted approval for the
treatment of patients with NSCLC, owing to their remarkable
efficacy in improving overall survival (7). The administration of PD-1 or PD-L1
antibodies rescues T cells from a state of exhaustion,
reinvigorating the immune response against cancer cells (8). However, some patients could not
significantly improve their survival rate after treatment with PD-1
blockers, and radiotherapy also did not increase responses to
combination with PD-1/PD-L1 therapy (9). The risk of mortality was significantly
diminished in both PD-L1 positive and PD-L1 negative patients when
subjected to PD-1/PD-L1 blockade as compared with conventional
therapy in clinical trials (10).
Notably, the combination therapy involving PD-1/PD-L1 inhibitors
has exhibited a remarkable augmentation in clinical outcomes for
patients with NSCLC and brain metastases (11). Therefore, combining PD-1/PD-L1
inhibitors with co-stimulatory molecule agonists can
synergistically enhance multiple processes in the cancer-immunity
cycle and improve the tumor microenvironment by reducing
immunosuppression (8).
The non-transmembrane enzyme, protein tyrosine
phosphatase 1B (PTP1B), is widely acknowledged as a negative
regulator of metabolic signaling pathways (12). Recently, a survival analysis
revealed a significant upregulation of PTP1B in the majority of
tumor tissues, which exhibited a strong association with an
unfavorable prognosis (13). The
PTP1B inhibitor MSI-1436 demonstrated remarkable drug sensitivity
by significantly suppressing tumor cell viability, thereby
suggesting its potential to augment the efficacy of PD-1 inhibitors
across diverse cancer types (13).
Moreover, the PTPN1 gene, encoding for PTP1B, serves as a negative
regulator in various cytokine signaling pathways and T cell
receptor signaling (14). PTP1B was
identified a novel intracellular checkpoint that exert the function
of modulating immune cell-tumor cell interaction as a tumor
suppressing factor (15). The
inhibition of PTP1B can potentiate the expansion and cytotoxicity
of antigen-induced CD8+ T cells, enhance the efficacy of
PD-1 blockade and impede tumor progression (15,16).
The findings suggest that PTP1B plays a crucial role in the cancer
tumor microenvironment, but the exceptional treatment strategy and
modulation mechanism in NSCLC immunotherapy remain unclear. A
previous research by the authors has revealed a remarkable surge in
the infiltration of cytotoxic T lymphocytes (CTLs) within both the
tumor microenvironment and spleen subsequent to anti-TNFR2
immunotherapy (17). The
simultaneous induction of CTLs and modulation of the tumor's
immunosuppressive microenvironment synergistically culminatea in a
curative efficacy against multiple malignancies (18). Therefore, the modulation of PTP1B
and TNFR2 has the potential to augment the sensitivity of PD-1
blockade, amplify its antitumor efficacy, and protract patient
survival by orchestrating dynamic alterations within the tumor
microenvironment. Notably, PTP1B emerges as a pivotal target in
relation to tumor immunotherapy.
In the present study, the objective was to
investigate a more effective combination immunotherapy approach
involving anti-PTP1B, anti-PD-1 and anti-TNFR2 for NSCLC while
concurrently elucidating the contributions of multiple immune
cells, thereby providing innovative targeted strategies for
identifying patients with NSCLC.
Materials and methods
Animals and tumor models
Male C57BL/6 wild-type mice (aged 6–8 weeks) were
procured from Spaefer Biotechnology Co, Ltd. The animal study was
ethically approved (approval no. 2305196) by the Animal Research
Ethics Committee at Guizhou University [Laboratory Animal License
Number: SCXK (Guizhou) 2023–0002; Guiyang, China]. The mice were
housed in SPF under standard conditions (22±2°C, 50–70% humidity)
and maintained on a 12-h light/dark cycle (light on 7:00 a.m.) with
food and water available ad libitum. The mice were subcutaneously
injected with 0.1 ml of PBS containing Lewis lung cancer cells
(LLC, 2×106/ml) in the unilateral flank (right side) to
establish tumor models. Drug or administration interventions were
initiated once the tumor diameter reached a pre-determined range of
6–8 mm (Fig. 1A-C). Each
experimental group consisted of 5 mice. Additionally, to evaluate
sustained immunocompetence induced by these immunotherapies in
mice, an identical treatment regimen on a separate batch of mice
was employed with the same number to assess their survival curve,
as shown as Fig. 1D.
Combined immunotherapy
To evaluate the effect of immunotherapy in the lung
cancer-bearing mice, the mice were randomized into different
groups: (i) PBS group: intraperitoneal (i.p.) injection of PBS (Bio
X Cell, 200 µl/mouse); (ii) i.p. injection of anti-PD-1 (cat. no.
HY-P99144; MedChemExpress, 200 µg/200 µl/mouse); (iii) i.p.
injection of anti-PD-1 (200 µg/100 µl/mouse) and i.p. injection of
anti-TNFR2 (cat. no. HY-12219A, Bio X Cell, 100 µg/mouse); (iv)
i.p. injection of anti-PD-1 (200 µg/100 µl/mouse) and injection of
anti-PTP1B (MSI-1436; cat. no. HY-12219A; MedChemExpress; 100
µg/100 µl/mouse); and (v) i.p. injection of anti-PD-1 (200 µg/100
µl/mouse), anti-PTP1B (100 µg/100 µl/mouse) and anti-TNFR2 (100
µg/100 µl/mouse) (triple therapy). Intraperitoneal injections of
anti-PD-1 and anti-TNFR2 were administered every 4 days for 2
weeks, while anti-PTP1B was injected once every 3 days for 2 weeks.
Tumor size and body weight of mice were monitored every 3 days,
with humane endpoints set at a tumor diameter >2,000 mm. Due to
the weight loss often induced by immunotherapy mouse weights were
rigorously monitored to ensure that any decrease did not exceed
20%, thereby mitigating potential suffering. The mice were
euthanized through an overdose of sodium pentobarbital anesthesia
(100 mg/kg, i.p.). The experiments adhered to institutional
guidelines and ethical standards in relation to euthanasia and
death verification. Upon observing indicators such as the absence
of respiration, lack of cardiac activity, unresponsiveness to
stimuli, muscular rigidity, and cyanosis without any signs of life,
the occurrence of death was confirmed. These conditions were
observed initially 5–10 min after injection and then confirmed
again 30 min later.
Cell culture
The lung adenocarcinoma LLC cells (obtained from the
Zhongqiao Xinzhou Biotechnology Co., Ltd. (cat. no. ZQ0203) were
utilized for the experiments. The cells were cultured in complete
DMEM medium (Gibco; Thermo Fisher Scientific, Inc.), supplemented
with 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.). The LLC cells
were maintained at a temperature of 37°C in a humidified incubator
with a gas mixture consisting of 5% CO2 and 95% air.
Flow cytometry
To discern the infiltration of distinct leukocyte
subsets in tumor tissues, spleen and lymph nodes, the antibodies
used for flow staining diluted at 1:100 in single-cell suspensions;
all the single-cell suspensions were adorned with rat anti-mouse
CD45 conjugated to BV510 (BD Biosciences; clone 30-F11), hamster
anti-mouse CD3 conjugated to BV605 (BD Biosciences; clone
145-2C11), rat anti-mouse CD4 conjugated to FITC (BD Biosciences;
clone GK1.5), rat anti-mouse CD8 conjugated to Percp cy5.5 (BD
Biosciences; clone 53–6.7) and rat anti-mouse CD25 conjugated to
APC (BD Biosciences; clone PC61), rat anti-mouse IFN-gamma
conjugated to PE (BD Biosciences; clone XMG1.2), rat anti-mouse
Foxp3 conjugated to PE (BD Biosciences; clone R16-715), rat
anti-mouse CD16/32 (BD Biosciences; clone 93), AR700-anti-Dead/Live
(BD Biosciences; cat. no. 564997) at 4°C for 30 min. After
undergoing a meticulous triple washing process with FACS buffer,
the data obtained from the stained samples was acquired using the
state-of-the-art flow cytometer (BD FACSCelestaTM; BD
Biosciences). Cell debris was excluded based on forward and side
scatter gating, and viable cells were analyzed using live/dead
staining antibodies. The data analyses were conducted utilizing the
FlowJo software (v10.8.1; FlowJo LLC), which enabled precise
discrimination between cell populations.
Immunohistochemistry
The mouse tumor tissues were fixed in formalin,
embedded in paraffin, cut into 4-µm sections, dewaxed with xylene,
and rehydrated using a gradient of alcohol. After washing with
distilled water, heat-induced antigen retrieval was performed using
the decloaking chamber with low pH (pH 6.0) citrate buffer (10%,
Thermo Fisher Scientific, Inc., cat. no. J61249) at 95°C for 20
min, followed by cooling to room temperature for 15 min. The slides
were then rinsed three times in PBS (pH 7.4) on a decolorizing
shaker for a duration of 5 min per rinse before being blocked with
a solution of 5% BSA (Sigma-Aldrich; Merck KGaA; cat. no.
9048-46-8) at room temperature for 1 h. After undergoing natural
cooling, the slides were meticulously rinsed thrice in PBS (pH 7.4)
on a decolorizing shaker for a duration of 5 min per rinse.
Subsequently, the sections were treated with a 3% hydrogen peroxide
solution and subsequently underwent three rounds of washing in PBS
(pH 7.4) on a decolorizing shaking table for 5 min each time. Then,
at room temperature, the tissue was covered with 3% BSA for 30 min.
Primary antibodies including rabbit anti-PD-L1 (Absin; cat. no.
abs136046; 1:200), anti-TNFR2 (Affinity Biosciences; cat. no.
AF0364; 1:200) and anti-PTP1B (Absin; cat. no. abs131747; 1:200)
were applied to the section in PBS at a specific ratio and
incubated overnight at 4°C in a humidified chamber. After being
washed again in PBS on a shaking platform three times for 5 min
each time, the sections were incubated with Goat anti-Rabbit
HRP-labeled antibodies (Thermo Fisher Scientific, Inc.; cat. no.
31460; 1:1,000) at room temperature for 50 min. After undergoing an
additional round of gentle agitation in PBS for three consecutive
intervals of 5 min each, a freshly prepared peroxidase substrate
solution DAB (Thermo Fisher Scientific, Inc.; cat. no. 34065)
color-developing agent was introduced to facilitate the
visualization of positive staining by a light microscope (BX53;
Olympus Corporation).
Statistical analysis
Unless otherwise specified, all experiments were
performed at least three times. The statistical analysis was
conducted employing the one-way ANOVA test for multiple groups and
the Scheffe post hoc test. The assessment of survival advantage was
performed using the log-rank test. Statistical analyses were
executed using GraphPad Prism 10 software (Dotmatics). P<0.05
was considered to indicate a statistically significant
difference.
Results
Combination immunotherapy suppresses
NSCLC tumor growth in mice
To evaluate the potential synergistic impact of
combining anti-PD-1 or anti-TNFR2 with anti-PTP1B on enhancing the
antitumor immune response mediated in NSCLC, murine models
(C57BL/6) were utilized to subcutaneously implant LLC lung cancer
cells and the effect on tumor volume was assessed based on group
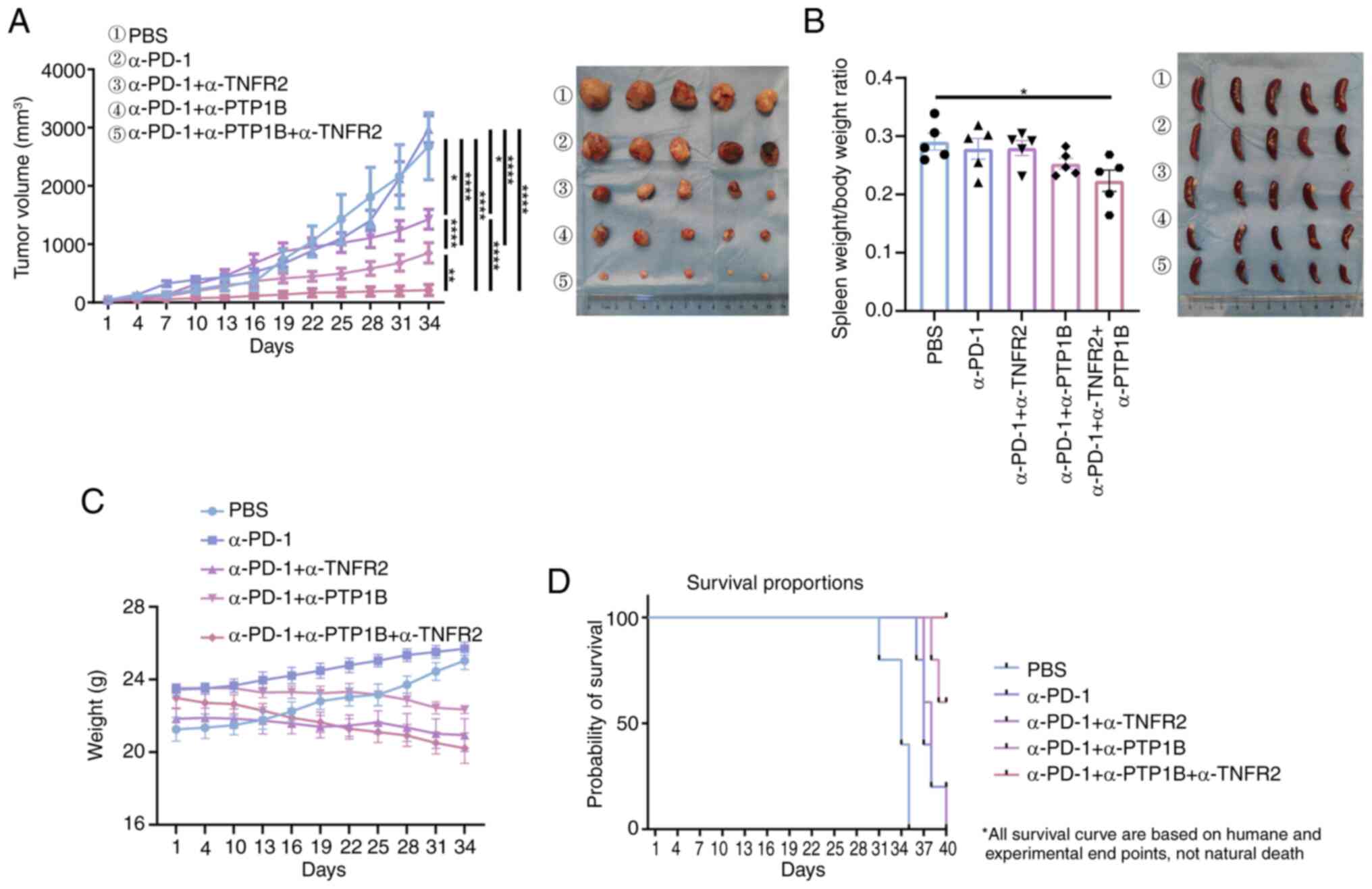
allocation and treatment regimen. The results demonstrated that
monotherapy with anti-PD-1 had minimal impact on tumor volume in
murine models of lung cancer. Treatment with a combination of
anti-PD-1 and either anti-PTP1B or anti-TNFR2 in mice resulted in a
significant reduction in tumor volume. Moreover, the combination of
anti-PD-1 and anti-PTP1B exhibited superior efficacy compared with
the combination of anti-PD-1 with only anti-PTP1B. Notably, the
triple therapy consisting of anti-PD-1, anti-PTP1B and anti-TNFR2
demonstrated significantly smaller tumor volumes compared with the
PBS control group or any single or double therapy groups, as
revealed in Fig. 1A. The ratio of
spleen to body weight had a decrease in the triple therapy compared
with PBS group mice (Fig. 1B). This
suggests that the triple therapy may significantly enhance the
systemic immune response, potentially boosting the immune activity
against tumors in spleen, while the PBS group resulted in
splenomegaly or inflammation. No significant weight loss was
observed with immunotherapy in all groups (Fig. 1C). Thus, the triple therapy
demonstrated superior efficacy compared with all other treatments.
Meanwhile, humane euthanasia was performed as follows: all mice in
the PBS group, PD-1 group and PD-1 + TNFR2 group were euthanized
(n=5 per group; 100% mortality rate). In the PD-1 + PTP1B group,
two out of five mice were euthanized (40% mortality rate within
this group). Notably, triple therapy demonstrated remarkable
efficacy with a 100% survival rate at the end of a 40-day treatment
period (Fig. 1D). The findings
indicated that the triple therapy significantly inhibits tumor
growth and prolongs survival in mice with NSCLC tumor, while
exhibiting negligible toxic side effects, rendering it a safe and
efficacious immunotherapeutic approach for this malignancy.
Triple therapy significantly induces
the alterations of the immune cells' population distribution
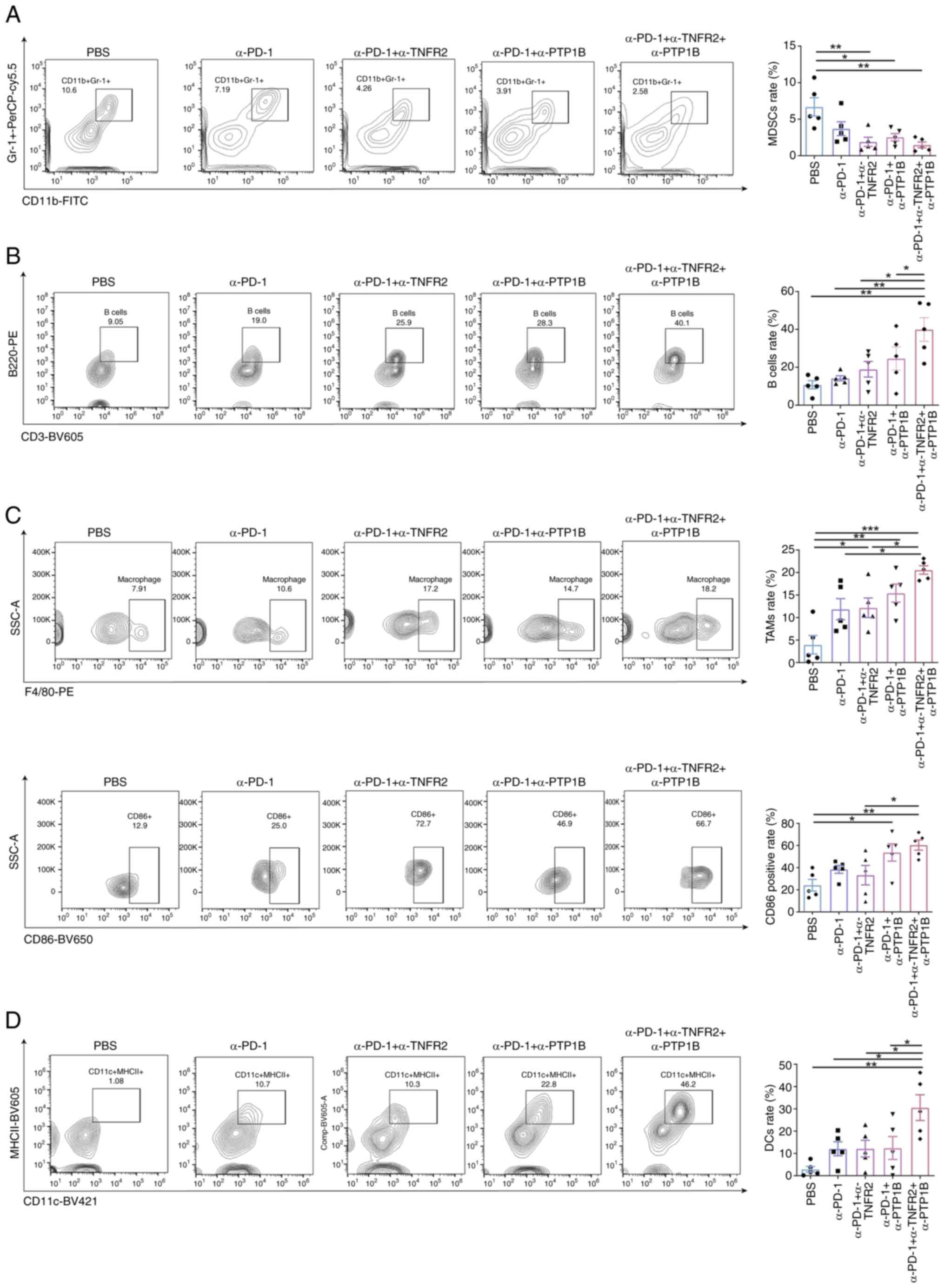
To explore whether combination therapies can elicit
multiple immune cells effects on the tumor growth, the expression
of immune cells NSCLC tumor in mice was initially assessed. It was
observed that the repression of tumor growth was accompanied by
alterations in the abundance of the tumor immune microenvironment
(TIME). Compared with the PBS group, both the α-PD-1 + α-TNFR2
group and α-PD-1 + α-PTP1B group exhibited a decrease in monocytic
myeloid-derived suppressor cells (MDSCs), which was
immunosuppressive, the α-PD-1 group did not exhibit any
statistically significant differences. As demonstrated in Fig. 2A, the triple therapy was accompanied
by the reduce of MDSCs, which suggested that it may dampen the
immune response or alleviate immunosuppression. By contrast, the
triple therapy significantly augmented B cell recruitment,
amplifying the impact of recruitment in the combination group of
anti-PD-1 with anti-PTP1B (Fig.
2B). In the tumor-associated macrophages (TAMs), although the
anti-PD-1 + anti-TNFR2 group, the anti-PD-1 + anti-PTP1B group and
the triple therapy group exhibited an increasing trend, the
proportion of M1 TAMs in the PBS group, the anti-PD-1 group and the
anti-PD-1 + anti-TNFR2 group was lower compared with that in the
triple therapy group. Particularly, the level of the
anti-tumorigenic CD86+M1 TAMs was found to be
significantly higher in the triple therapy group compared with the
anti-PD-1 + anti-TNFR2 group (Fig.
2C). Additionally, the triple therapy significantly expanded
the percentage of antigen-presenting dendritic cells (DCs)
(Fig. 2D), which promotes antitumor
immunity, enhanced by the triple therapy. Notably, although the
combination of anti-PD-1 with either anti-TNFR2 or anti-PTP1B
slightly suppressed MDSCs and promoted macrophages, compared with
these therapies, the triple therapy exhibited optimal efficacy for
immune activation in NSCLC tumors in mice.
Triple therapy distinctly increases
the population of CD8+T cells and the expression of
IFN-γ
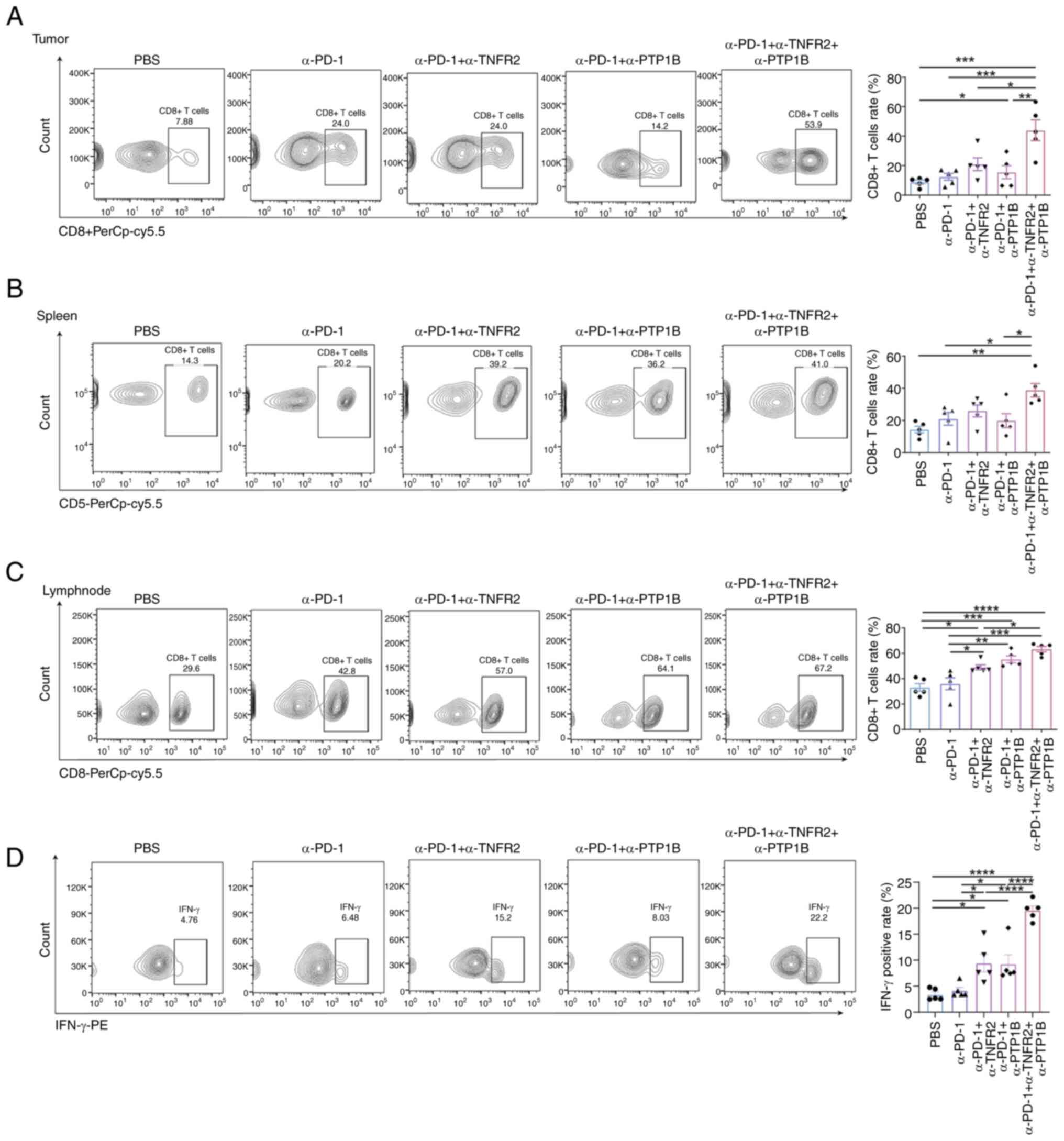
Although nearly all immune cells have the potential
to influence immune compartments in order to enhance antitumor
immunity, focus was directed towards CD8+T cells due to
their predominant representation as tumor-infiltrating lymphocytes
within cancerous tumors. Flow cytometric analysis of mouse tumors,
spleens and draining lymph nodes was conducted for each
immunotherapy group. The findings unveiled a remarkable surge in
the abundance of CD8+T cells within tumors subjected to
the combination therapy of anti-PD-1 and anti-PTP1B, as well as the
triple therapy groups. Notably, compared with PBS, anti-PD-1
monotherapy and dual therapy groups, triple therapy exhibited a
significant enhancement in the proportion of CD8+T cells
(Fig. 3A). The triple therapy
elicited a significant expansion of CD8+T cells in the
spleen, surpassing the combination of anti-PD-1 and anti-PTP1B or
monotherapy with anti-PD-1 alone (Fig.
3B). Moreover, combined with α-TNFR2 or α-PTP1B augmented the
expansion of CD8+T cells caused by α-PD-1. And the
triple therapy triple therapy exhibited a significantly potentiated
effect on the immune response of lymph nodes (Fig. 3C). This suggests a localized and
systemic recruitment of immune response. Importantly, a significant
elevation of IFN-γ was observed in the context of immunotherapy.
Specifically, the administration of α-TNFR2 or α-PTP1B
significantly enhanced the positive rate of IFN-γ induced by
treatment with α-PD-1 in mice. Furthermore, the triple therapy
demonstrated a significant amplification in CD8+T cells
compared with the double treatment groups, thereby enhancing the
ratio of IFN-γ on CD8+T cells (Fig. 3D). These findings strongly indicated
that triple therapy exerts powerful immunomodulatory effects in
mice with NSCLC; this combination therapy significantly enhances
the recruitment of CTLs, promoting both localized and systemic
immunostimulation.
Triple therapy suppresses the
immunosuppression of Tregs
TNFR2 dominant expressed on regulatory T cells
(Tregs), which are predominantly immunosuppressive (19,20),
and the elevated expression of this factor serves as a promising
prognostic indicator in cancers enriched with CD8+T
cells. It was recently demonstrated by the authors that the
combination of anti-TNFR2 therapy can enhance immune memory against
tumors and bolster secondary prevention by suppressing Tregs
(21). Another study reported that
anti-TNFR2 can induce TAM infiltration while promoting
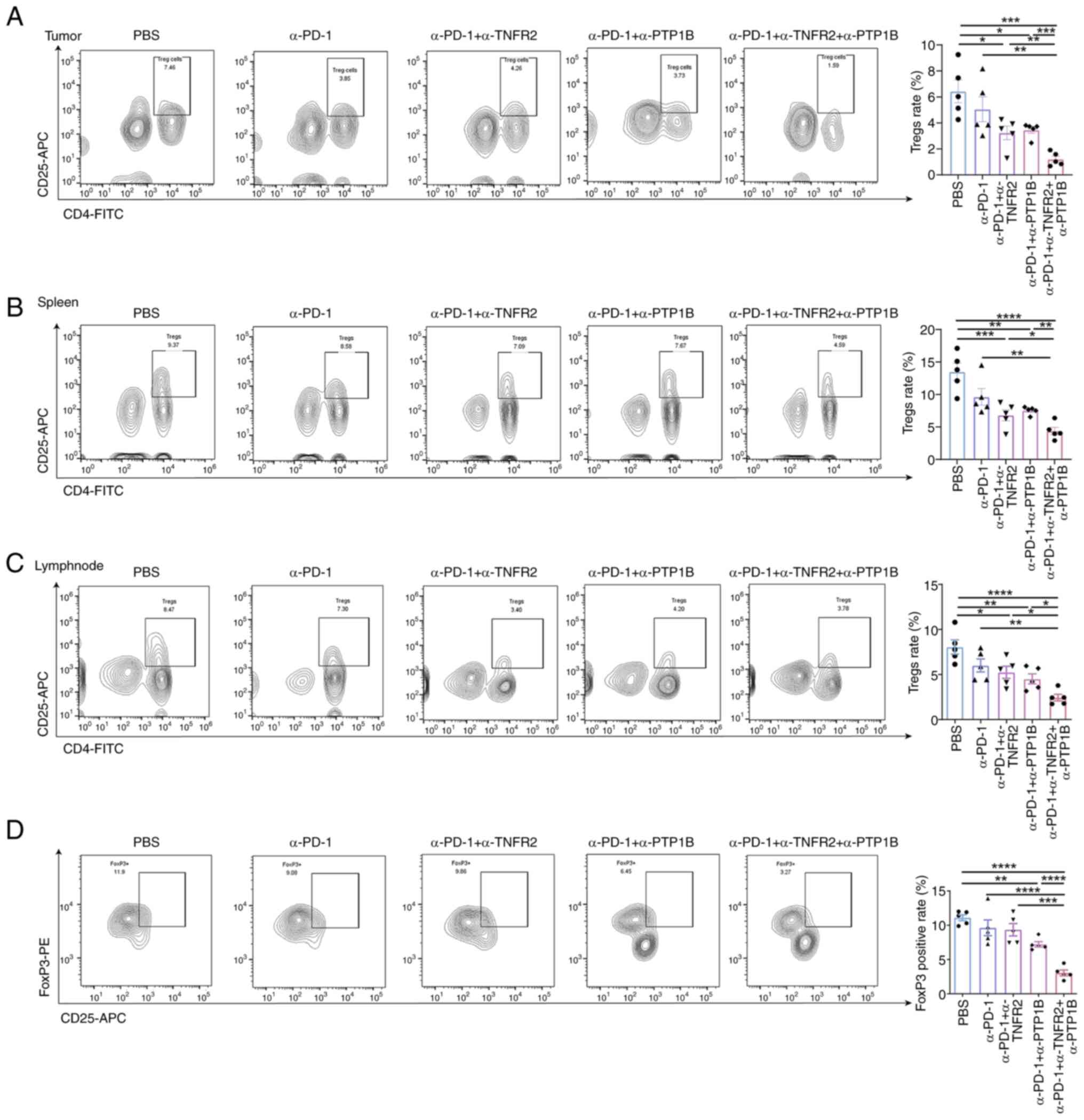
CD8+T cell activation in TIME (19). To investigate the impact of triple
therapy on immunosuppression, the distribution of
CD4+CD25+Tregs was analyzed in mouse spleens,
lymph nodes and tumors; FoxP3 expression in blood samples was also
assessed using flow cytometry. Anti-PD-1 combined with anti-PTP1B
or anti-TNFR2 groups demonstrated a reduction in the rate of
CD4+CD25+ Tregs in tumors, spleens and lymph
nodes. The triple therapy exhibited the most significant degree of
suppression. It was observed that the combination of two or three
therapies significantly decreased the proportion of
CD4+CD25+ Tregs. Compared with single and
double treatments, the triple therapy evidently diminished the
percentage of Tregs in mice tumors (Fig. 4A). Notably, it was found that
combination therapies reduced the rate of Tregs in spleens. The
triple therapy significantly enhanced the reduction of Tregs
(Fig. 4B). In addition, there was a
similar response observed in the decrease of Tregs in lymph nodes,
as shown in Fig. 4C. Compared with
the PBS group, the combination of anti-PD-1 and anti-PTP1B
exhibited a significant reduction in Foxp3 levels in the
bloodstream, while the triple therapy demonstrated a significant
decrease in the proportion of FoxP3 on
CD4+CD25+ Tregs (Fig. 4D). As expected, the triple therapy
had the most pronounced inhibition effect compared with other
treatments that included PBS, anti-PD-1 with or without anti-PTP1B
or anti-TNFR2. The results demonstrated that the combination
immunotherapy effectively suppressed immunosuppression in
NSCLC-bearing mice, enhancing effector T cell-mediated antitumor
responses.
Triple therapy enhances the expression
of PD-L1
The engagement of PD-1 with its ligands,
predominantly PD-L1, leads to the attenuation of crucial molecules,
resulting in the inhibition of proliferation, activation and
cytokine production in immune cells within the TIME. Therefore, to
elucidate the local protein expression characteristics within
tumors and unravel the pivotal role of anti-PTP1B in combination
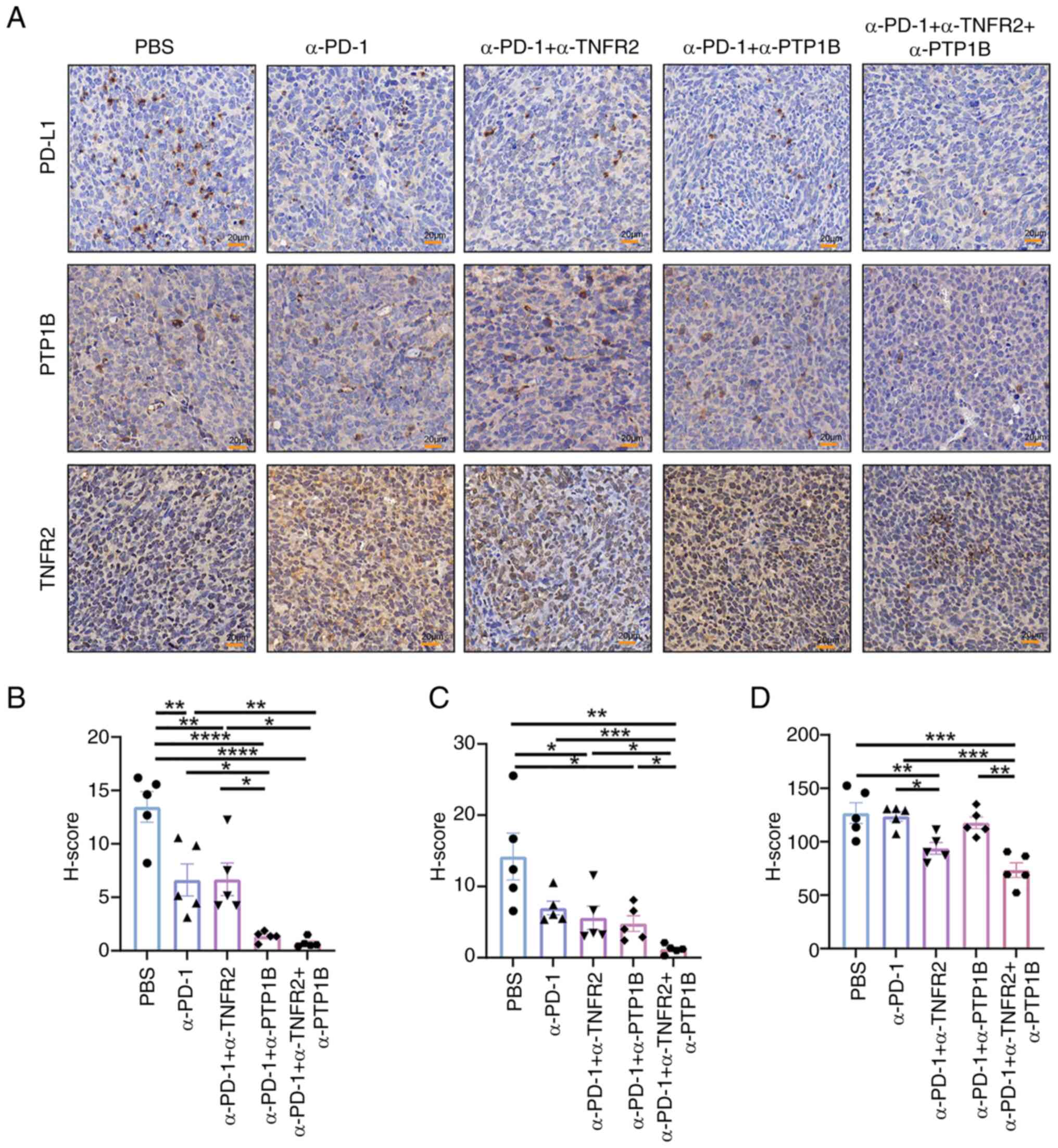
therapies, the levels of PD-L1, PTP1B and TNFR2 were evaluated in
tumor tissues. The results revealed that the combination of
anti-PD-1 with either anti-PTP1B or anti-TNFR2 significantly
decreases the H-score value of PD-L1 in tumor tissue. Both dual
therapy and triple therapy effectively suppressed the expression of
PD-L1 (Fig. 5A). Notably, anti-PD-1
combined with anti-PTP1B or anti-TNFR2 reduced the expression of
PTP1B, and the triple therapy resulted in a significant reduction
compared with other groups (Fig.
5B). The implication here is that the administration of
anti-TNFR2 or anti-PTP1B treatments leads to a downregulation of
PD-L1 and PTP1B expression, thereby potentially amplifying the
immune response through an augmentation in both quantity and
functionality of CD8+T cells (Fig. 5C). Moreover, compared with the PBS
and anti-PD-1 cohort, the combination of anti-PD-1 with anti-TNFR2
exhibited a significant reduction in TNFR2 expression. The
anti-PD-1 + anti-PTP1B group exhibited a significant decrease
compared with the triple therapy, indicating that the expression of
TNFR2 was reduced by anti-TNFR2 treatment. However, there was no
significant effect observed between the anti-PD-1 + anti-TNFR2
group and the triple therapy, suggesting that anti-PTP1B may not
influence TNFR2 expression (Fig.
5D). The findings suggested that the collaboration between
anti-PTP1B and anti-PD-1 might modulate immune activity by altering
PD-L1 expression, while the synergistic modulation of PTP1B and
suppression of TIME might be achieved by anti-TNFR2.
Discussion
In the present study, compelling evidence was
presented demonstrating that the triple therapy exerts a
significant inhibitory effect on tumor growth in a NSCLC model
through redistribution of immune cells. Particularly, it
predominantly enhanced immunoreactivity and attenuated
immunosuppressive effects, which was achieved through a significant
increase in CD8+T cells, reduction of Tregs, weakened
PD-L1 and TNFR2 within the TIME of NSCLC mice, thus providing
evidence in favor of the immunocompetence impact of the triple
therapy and suggesting potential therapeutic perspectives.
The reactivation of cytotoxic T cells for the
eradication of cancer cells is accomplished through the utilization
of antibodies that impede the interaction between PD-1 and PD-L1
(22). The majority of patients
with cancer, however, fail to respond to PD-1/PD-L1 blockade due to
either elevated or diminished expression of PD-L1, resulting in
T-cell exhaustion and enabling tumor cells to elude immune assault.
Consequently, this leads to a compromised immune response and
unfavorable clinical prognosis for individuals with cancer.
Therefore, it is imperative to devise innovative strategies
targeting the modulation of immune key molecules' expression and
the expansion of immune cells in order to enhance the effectiveness
of immunotherapies.
PTP1B, encoded by PTPN1, is a pivotal member of the
PTP superfamily, which has been implicated in the pathogenesis of
obesity, diabetes, various cancers and cardiovascular diseases
(23). PTPN1 was upregulated in
hepatocellular carcinoma, and its silencing suppressed
proliferation and induced apoptosis of hepatocellular cells
(24). The expression of PTP1B is
significantly elevated in breast cancer, where it is considered to
play a pivotal role in promoting tumorigenesis (25). The present study revealed that PTP1B
expression is increased in effector/memory CD8+T cells
infiltrating tumors, which is associated with their cytotoxic
activity and expansion through JAK/STAT5 signaling coordination
(26). Notably, the deletion or
inhibition of PTP1B can effectively suppress CD4+T
cells, natural killer cells and DCs, while simultaneously promoting
the recruitment of Tregs, TAMs, as well as MDSCs. The augmentation
significantly enhances the efficacy of CAR-T cells, without
triggering autoimmune response or systemic inflammation (15). Therefore, a targeted approach
towards PTP1B holds great potential as a promising therapeutic
strategy for solid tumors. In the present study, the inhibition of
PTP1B significantly decreased the expression of PD-L1 and TNFR2 on
lung cancer cell lines, concomitant with the enhancement of
CD8+T cells and reducing of CD4+T cells,
indicating its pivotal role in modulating both tumor and immune
responses in lung cancer. A similar study demonstrated that the
inhibition of PTP1B/PTPN2 in T cells enhanced antitumor immunity in
immunogenic tumors, while also exerting direct effects on both
tumor cells and T cells in cold tumors (27). Due to the presence of PTP1B, the
JAK/STAT signaling cascade is closely linked to cytokine-receptor
activation in immune cells, which is crucial for the function of
activated monocyte-derived DCs (28). The anti-PTP1B may exert a profound
influence on the immune system, effectively modulating both cancer
cells and their TIME. Through the administration of a synergistic
combination of anti-PTP1B and anti-TNFR2, along with PD-1 blockade
in vivo, there was a remarkable inhibition observed in tumor
volume in mice. This treatment regimen resulted in significantly
prolonged survival for all the mice involved, without any
accompanying side effects. Furthermore, flow cytometry was employed
to validate the efficacy of this combination treatment. The triple
therapy demonstrated superior efficacy in modulating the immune
system distribution in murine models of lung carcinoma, inhibited
immune suppressor cells and activated immune effector cells,
reorienting the TIME towards an antitumor response. Numerous
signaling pathways may be associated with these actions. The
GM-CSF-STAT3-TRAF3-PTP1B signaling axis operates in inflammatory
macrophages and monocytes, collectively regulating the expansion of
MDSCs (29). The suppression of M1
cytokines was no longer observed in macrophages derived from
PTP1B-KO mice (30). The total
ablation of PTP1B can also lead to the hyperactivation of STAT4,
STAT1 and Src kinase (28). The
expression of PD-L1 in lung cancer cell lines exhibits a positive
correlation with MET phosphorylation (31). The PD-L1-associated signaling
pathway attenuates macrophage p38α activity by inducing PTP1B
(30). The phosphatases suppressor
of cytokine signaling 3 (SOCS3), is a crucial gene in tumorigenesis
(32); it can synergistically
interact with PTP1B to sustain the activation of the JAK-STAT
pathway signaling or leptin signaling pathway (33), facilitating immune evasion from
CD8+T-cell-mediated cytotoxicity (34). The findings present promising
prospects for potential therapeutic strategies against cancer.
The triple therapy resulted in an augmentation of
CD8+T cell proportion and a reduction in Tregs,
accompanied with the enhancement of IFN-γ and alleviation of FoxP3.
The findings suggested that the inhibition of PTP1B may confer an
enhanced augmenting effect on NSCLC immunotherapy when combined
with anti-PD-1 and anti-TNFR2, potentially leading to a more
efficacious treatment outcome (17,18).
TNFR2, expressed in various tumor cells and Tregs (35), modulates the TGF-β signaling pathway
that constitutes a significant proportion within tumors (36), regulating the immune inflammatory
signal (37). TNFR2 plays a crucial
role in promoting immune evasion, contributing to tumor initiation
and progression (21). The
combination of PD-1 and TNFR2 synergistically enhances efficacy;
however, it results in infiltration of immunotoxin cells within the
spleen, indicating suboptimal outcomes. Interestingly, as a pivotal
negative regulator for restraining allergic responses,
PTP1B-deficient mice exhibited augmented chemotaxis, chemokinesis
and trans-endothelial migration in splenic leukocytes (38). Meanwhile, PTP1B is likely to exert
inhibitory effects on the vascular endothelial cells through its
dephosphorylation of vascular endothelial growth factor receptor 2,
a pivotal regulator governing tumor angiogenesis (39,40).
Therefore, a triple therapy approach consisting of PD-1 blockade,
PTP1B inhibition and anti-TNFR2 immunotherapy was employed to
enhance the sensitivity of immunotherapy. A significant
amplification in the population of CD8+ T cells was
identified, accompanied by a simultaneous reduction in the
abundance of Tregs within spleens, lymph nodes and tumors. These
findings suggested that triple therapy can modulate the equilibrium
of the TIME to favorably enhance immunotherapy sensitivity and
elicit systemic immune responses devoid of any associated immune
response-related side effects.
Additionally, there are certain limitations to the
present study. Since only one cell type was used to explore the
immune microenvironment of NSCLC, further investigation of multiple
cell types may be more convincing for the efficacy of combination
therapy. At the same time, further exploration of the intracellular
mechanism of immune cells is of reference significance to clarify
the effect of this combination therapy on a variety of tumor cells,
which has a guiding role in enhancing the efficacy of immunotherapy
for other tumors suitable for immunotherapy. Collectively, the
combination of PD-1 blockades and TNFR2 inhibition, with assistance
from a PTP1B antibody, activates the immune system and inhibits
tumor growth in mice with lung cancer, which may achieve by
promoting the immunocompetent cells, and suppressing the
immunosuppression. These actions promote survival and prognosis in
lung cancer mice. The combination immunotherapy regimen further
enhances the efficacy of triple therapy and inhibits lung cancer
tumor progression.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Non-profit Central
Research Institute Fund of Chinese Academy of Medical Sciences
(grant no. 2019PT320003), the National Natural Science Foundation
of China (grant no. 82060308), the National Key Laboratory of
Respiratory Diseases of China (grant no. SKLRD-OP-202208), the
Science and Technology Plan Project of Guizhou in China (grant nos.
GPPH-NSFC-2020-6, GPPH-NSFC-2020-7 and GCC [2022]037-1), the
Research Fund Projects of Guizhou Immunotherapy Research Talent
Base in China (grant no. RCJD2018-11), the China Scholarship
Council (grant no. 202306670011), the Science and Technology Fund
project of Guizhou Health Commission (grant no. gzwkj2024-084) and
the Qianxinan Prefecture Science and Technology Plan Project (grant
no. 2023-3-5).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HG and HY wrote the original draft. YiN, YuN, HY,
QJ, LL, HL, QW, LZ, SCh and MW wrote, reviewed and edited the
manuscript. YiN, YuN, HY and XZ performed the conceptualization.
HY, XZ, QJ and LL performed the data analysis. QW, LZ, SCh, LL and
MW performed the formal analysis. YiN, YuN, HY, QJ, HL, QW, LZ,
SCh, MW and XZ designed the methodology, created the animal models
and performed treatment experiments. QJ, LL, QW and MW performed
the project administration. LZ, SCh and HG provided study materials
and reagents. YiN and XZ performed the supervision. YiN and HY
performed the validation. HY and YuN generated the figures. XZ, YiN
and HG provided the funding. HG, HY and YiN confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All experiments involving animals were conducted
according to the ethical policies and procedures approved (approval
no. 2305196) by the Animal Research Ethics Committee of Guizhou
Medical University (Guiyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boyero L, Sánchez-Gastaldo A, Alonso M,
Noguera-Uclés JF, Molina-Pinelo S and Bernabé-Caro R: Primary and
acquired resistance to immunotherapy in lung cancer: Unveiling the
mechanisms underlying of immune checkpoint blockade therapy.
Cancers (Basel). 12:37292020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilky BA: Immune checkpoint inhibitors:
The linchpins of modern immunotherapy. Immunol Rev. 290:6–23. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patil NS, Nabet BY, Müller S, Koeppen H,
Zou W, Giltnane J, Au-Yeung A, Srivats S, Cheng JH, Takahashi C, et
al: Intratumoral plasma cells predict outcomes to PD-L1 blockade in
non-small cell lung cancer. Cancer Cell. 40:289–300.e4. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yi M, Zheng X, Niu M, Zhu S, Ge H and Wu
K: Combination strategies with PD-1/PD-L1 blockade: Current
advances and future directions. Mol Cancer. 21:282022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schoenfeld JD, Giobbie-Hurder A,
Ranasinghe S, Kao KZ, Lako A, Tsuji J, Liu Y, Brennick RC, Gentzler
RD, Lee C, et al: Durvalumab plus tremelimumab alone or in
combination with low-dose or hypofractionated radiotherapy in
metastatic non-small-cell lung cancer refractory to previous
PD(L)-1 therapy: An open-label, multicentre, randomised, phase 2
trial. Lancet Oncol. 23:279–291. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao B, Zhao H and Zhao J: Efficacy of
PD-1/PD-L1 blockade monotherapy in clinical trials. Ther Adv Med
Oncol. 12:17588359209376122020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Liu F, Chen X, Zhao C, Li X, Zhou
C, Hu J, Chu Q and Jiang T: Outcome differences between PD-1/PD-L1
inhibitors-based monotherapy and combination treatments in NSCLC
with brain metastases. Exp Hematol Oncol. 12:562023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bakke J and Haj FG: Protein-tyrosine
phosphatase 1B substrates and metabolic regulation. Semin Cell Dev
Biol. 37:58–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bai KH, Zhu MJ, Zhang YY, Li XP, Chen SL,
Wang DW and Dai YJ: Multi-omics analyses of tumor-associated
immune-infiltrating cells with the novel immune checkpoint protein
tyrosine phosphatase 1B (PTP1B) in extracellular matrix of
brain-lower-grade-glioma (LGG) and uveal-melanoma (UVM). Front
Immunol. 13:10538562022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilson NS and Huntington ND: Small
molecule. Big biology. Dual phosphatase inhibitor enters the
immunotherapy fray. Immunol Cell Biol. 102:8–11. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wiede F, Lu KH, Du X, Zeissig MN, Xu R,
Goh PK, Xirouchaki CE, Hogarth SJ, Greatorex S, Sek K, et al: PTP1B
is an intracellular checkpoint that limits T-cell and CAR T-cell
antitumor immunity. Cancer Discov. 12:752–773. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zappasodi R, Merghoub T and Wolchok JD:
Emerging concepts for immune checkpoint blockade-based combination
therapies. Cancer Cell. 34:6902018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu L, Zhang X, Chen X, Yang D, Nie Y, Pan
R, Li L, Wang C, Gui H, Chen S, et al: Anti-TNFR2 enhanced the
antitumor activity of a new HMGN1/3M-052 stimulated dendritic cell
vaccine in a mouse model of colon cancer. Biochem Biophys Res
Commun. 653:106–114. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nie Y, Yang D, Trivett A, Han Z, Xin H,
Chen X and Oppenheim JJ: Development of a curative therapeutic
vaccine (TheraVac) for the treatment of large established tumors.
Sci Rep. 7:141862017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Lao M, Xu J, Duan Y, Yang H, Li
M, Ying H, He L, Sun K, Guo C, et al: Combination cancer
immunotherapy targeting TNFR2 and PD-1/PD-L1 signaling reduces
immunosuppressive effects in the microenvironment of pancreatic
tumors. J Immunother Cancer. 10:e0039822022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bourebaba L, Serwotka-Suszczak A,
Bourebaba N, Zyzak M and Marycz K: The PTP1B inhibitor
trodusquemine (MSI-1436) improves glucose uptake in equine
metabolic syndrome affected liver through anti-inflammatory and
antifibrotic activity. Int J Inflam. 2023:38030562023.PubMed/NCBI
|
|
21
|
Jing Q, Wan Q, Nie Y, Luo J, Zhang X, Zhu
L, Gui H, Li L, Wang C, Chen S, et al: Ansofaxine hydrochloride
inhibits tumor growth and enhances Anti-TNFR2 in murine colon
cancer model. Front Pharmacol. 14:12860612023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Dang F, Ren J and Wei W:
Biochemical Aspects of PD-L1 regulation in cancer immunotherapy.
Trends Biochem Sci. 43:1014–1032. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharma B, Xie L, Yang F, Wang W, Zhou Q,
Xiang M, Zhou S, Lv W, Jia Y, Pokhrel L, et al: Recent advance on
PTP1B inhibitors and their biomedical applications. Eur J Med Chem.
199:1123762020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yuan F, Gao Q, Tang H, Shi J and Zhou Y:
Ophiopogonin-B targets PTP1B to inhibit the malignant progression
of hepatocellular carcinoma by regulating the PI3K/AKT and AMPK
signaling pathways. Mol Med Rep. 25:1222022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bentires-Alj M and Neel BG:
Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced
breast cancer. Cancer Res. 67:2420–2424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ocaranza P, Gaete X, Román R, Morales F,
Íñiguez G and Cassorla F: Phosphotyrosine phosphatases in
GH-stimulated skin fibroblasts from children with idiopathic short
stature. J Pediatr Endocrinol Metab. 26:833–840. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang S, Tran E, Du X, Dong J, Sudholz H,
Chen H, Qu Z, Huntington ND, Babon JJ, Kershaw NJ, et al: A small
molecule inhibitor of PTP1B and PTPN2 enhances T cell anti-tumor
immunity. Nat Commun. 14:45242023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Penafuerte C, Feldhammer M, Mills JR,
Vinette V, Pike KA, Hall A, Migon E, Karsenty G, Pelletier J,
Zogopoulos G and Tremblay ML: Downregulation of PTP1B and TC-PTP
phosphatases potentiate dendritic cell-based immunotherapy through
IL-12/IFNγ signaling. Oncoimmunology. 6:e13211852017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu S, Lalani AI, Jin J, Sant'Angelo D,
Covey LR, Liu K, Young HA, Ostrand-Rosenberg S and Xie P: The
adaptor protein TRAF3 is an immune checkpoint that inhibits
myeloid-derived suppressor cell expansion. Front Immunol.
14:11679242023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ubil E, Caskey L, Holtzhausen A, Hunter D,
Story C and Earp HS: Tumor-secreted Pros1 inhibits macrophage M1
polarization to reduce antitumor immune response. J Clin Invest.
128:2356–2369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu S, Sun Z, Hu W, Yin S, Zhao C and Hu H:
PD-L1 positively regulates MET phosphorylation through inhibiting
PTP1B. Cancer Sci. 112:1878–1887. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao S, Gang J, Yu M, Xin G and Tan H:
Computational analysis for identification of early diagnostic
biomarkers and prognostic biomarkers of liver cancer based on GEO
and TCGA databases and studies on pathways and biological functions
affecting the survival time of liver cancer. BMC Cancer.
21:7912021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao S, Tan H and Gang J: Inhibition of
hepatocellular carcinoma cell proliferation through regulation of
the Cell Cycle, AGE-RAGE, and Leptin signaling pathways by a
compound formulation comprised of andrographolide, wogonin, and
oroxylin A derived from Andrographis Paniculata(Burm.f.) Nees. J
Ethnopharmacol. 329:1180012024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu R, Wang C, Li Z, Xiao J, Li C, Wang X,
Kong P, Cao J, Huang F, Li Z, et al: SOX2 promotes resistance of
melanoma with PD-L1 high expression to T-cell-mediated cytotoxicity
that can be reversed by SAHA. J Immunother Cancer. 8:e0010372020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qu Y, Wang X, Bai S, Niu L, Zhao G, Yao Y,
Li B and Li H: The effects of TNF-α/TNFR2 in regulatory T cells on
the microenvironment and progression of gastric cancer. Int J
Cancer. 150:1373–1391. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao S, Tan H and Li D: Oridonin suppresses
gastric cancer SGC-7901 cell proliferation by targeting the
TNF-alpha/androgen receptor/TGF-beta signalling pathway axis. J
Cell Mol Med. 27:2661–2674. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Medler J, Kucka K and Wajant H: Tumor
necrosis factor receptor 2 (TNFR2): An emerging target in cancer
therapy. Cancers (Basel). 14:26032022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Berdnikovs S, Pavlov VI, Abdala-Valencia
H, McCary CA, Klumpp DJ, Tremblay ML and Cook-Mills JM: PTP1B
deficiency exacerbates inflammation and accelerates leukocyte
trafficking in vivo. J Immunol. 188:874–884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang J, Li L, Li J, Liu Y, Zhang CY,
Zhang Y and Zen K: Protein tyrosine phosphatase 1B impairs diabetic
wound healing through vascular endothelial growth factor receptor 2
dephosphorylation. Arterioscler Thromb Vasc Biol. 35:163–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mabeta P and Steenkamp V: The VEGF/VEGFR
axis revisited: Implications for cancer therapy. Int J Mol Sci.
23:155852022. View Article : Google Scholar : PubMed/NCBI
|