Introduction
Head and neck squamous cell carcinoma (HNSCC),
including oral cancer, is the sixth most common malignancy in
humans worldwide. Oral cancer (OC) is one of the most frequent
types of HNSCC. Approximately 95% of OCs are squamous cell
carcinomas (OSCC) (1).
Histologically, OSCCs are derived from the epithelium lining of the
oral cavity and can occur at various sites in the oral cavity,
including the lips, hard palate, gum and tongue (2), with a preference on the tongue and
floor of the mouth. Each year, ∼405, 000 new cases of oral cancer
(OSCC) are diagnosed and the number is still accumulating in many
countries. In Taiwan, OSCC is the sixth leading cause of cancer
death. Approximately 5,400 new cases are identified and 2,200
deaths per year and the incidence of OSCC has increased 6-fold
during the past decade.
The main causes of oral cancer includes tobacco and
alcohol consumption (3), diets
poor in vitamin A and carotenoids, indoor air pollution and poor
oral hygiene (4,5). The occurrence of oral cancer in
Taiwan is closely related to betel quid chewing, cigarette smoking
and alcohol consumption (6). The
standard treatments for patients with oral cancer include surgery,
radiotherapy and chemotherapy (7).
Despite the improvement in surgery and chemotherapy during the last
20 years (8), oral cancer remains
a disease with poor prognosis and a low survival rate (9). In patients identified with an
advanced stage of the disease, there is a high incidence of
invasion to adjacent tissues, of metastasis to lymph node and
distant areas and of recurrence during the patient’s lifetime
(10,11). As compare to 90% of patients
without metastasis, the 5-year survival rate for patients with
lymph node metastasis at presentation is significantly reduced to
25-40% (12). Additionally, lymph
node metastasis occurs in ∼40% of patients with oral cancer.
Therefore, there is an urgent need to identify agents that can
inhibit the invasion and metastasis of oral cancers.
The active components in natural products such as
polyphenolic and isothiocyanate (ITC)-containing compounds are the
intensive target of research for their promising cancer
preventative and therapeutic properties (13,14)
and low toxicity to cells (15).
Epidemiological investigations have reported an inverse relation
between the dietary intake of fruits and vegetables, including
cruciferous vegetables and the risk of various types of
malignancies (16–18). The anti-carcinogenic effects of
cruciferous vegetables such as broccoli (18) and watercress (19) have been ascribed to certain
chemicals with the isothiocyanate (-N═C═S) functional group
(19,20). Isothiocyanates are produced from
the hydrolysis of the inactive precursor glucosinolates by
myrosinase in cruciferous vegetables when the plant tissues are
crushed or masticated (19–21).
These isothiocyanates, which consist of phenethyl ITC (PEITC),
allyl ITC (AITC), benzyl ITC (BITC) (22) and sulforaphane (SFN), have been
shown to have potential cancer chemopreventive activity in a number
of experimental models, including cancer of the esophagus, mammary
gland, lung, liver, pancreas, fore-stomach, colon, small intestine
and bladder of mice, rats, other rodents and colon cancer in humans
(23–25).
PEITC, a member of isothiocyanate, possesses a
variety of biological activities such as the induction of phase II
detoxification enzymes, the inhibition of cytochrome P450 (CYP)
enzymes (26), arrest of cell
cycle (27) and stimulation of
apoptosis (28–31), inhibition of nuclear factor-κB
(NF-κB)-regulated gene expression (32) and activation of Atg5-mediated
autophagy (33) in different
cancer cell lines. PEITC also suppresses the pulmonary neoplasia
induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butone in the lung
of A/J mouse (34,35), prevents the formation of colonic
aberrant crypt foci induced by azoxymethane (36) and reduces the number and size of
polyps in ApcMin/+ mice (23). The incidence and burden (affected
area) of poorly differentiated tumors in the dorsolateral prostate
of transgenic adenocarcinoma of mouse prostate (TRAMP) model mice
were reduced when given 3 μmole PEITC/kg of diet (37). Further, PEITC is currently in
clinical trials for lung cancer (38). These effects suggest a potential
role of PEITC in the suppression of tumorigenesis.
It has been shown that PEITC inhibited the migration
and invasion of human gastric cancer AGS cells (39) and colon cancer HT29 cells (40). In addition, PEITC treatment reduced
angiogenesis and cell motility of human umbilical vein endothelial
cells and PC-3 prostate cancer cells (41). However, the effects and underlying
mechanism of PEITC on the metastasis of oral squamous cell
carcinomas are still not clear. In this study, we demonstrated that
PEITC acted on the phosphorylation of EGFR and sequentially
inactivated the PI3K/AKT kinase cascade, repressed the
NF-κB-mediated signaling and hence reduced expression of matrix
metalloproteases (MMPs), finally leading to the inhibition of OSCC
invasion.
Materials and methods
Chemicals and reagents
Phenethyl isothiocyanate (PEITC) was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Antibodies against AKT, EGFR,
ERK, IκB, JNK, MMP-2, MMP-9, p38, PI3K, TIMP-1, TIMP-2, β-actin and
GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). Antibodies against phospho-AKT (S308), phospho-AKT (S473),
phospho-EGFR (Y845), phospho-EGFR (Y992), phospho-EGFR (Y1068),
phospho-ERK (Thr202/Tyr204), phospho-IKK, phospho-IκB, phospho-JNK
(Thr183/Tyr185), phospho-PDK1, phospho-PI3K and phospho-p38
(Thr183/Tyr185) were obtained from Cell Signaling Technology
(Danvers, MA, USA). HRP-conjugated secondary antibodies such as
rabbit anti-mouse IgG, goat anti-rabbit IgG and donkey anti-goat
IgG were purchased from Santa Cruz Biotechnology. MTT
(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide)
and epidermal growth factor (EGF) were obtained from Sigma-Aldrich.
DMEM medium, fetal bovine serum (FBS), L-glutamine,
penicillin-streptomycin and trypsin-EDTA were purchased from Gibco
BRL (Invitrogen Life Technologies, Carlsbad, CA).
Cell culture
Human OSCC SAS cell line was cultured in DMEM medium
supplemented with 10% of fetal bovine serum, 100 U/ml penicillin,
100 μg/ml streptomycin and 2 mM glutamine and incubated at
37°C in a humidified chamber with 5% CO2(42).
Cell invasion assay
The membrane of each transwell insert was washed
with 1X PBS and pre-coated with Matrigel (2 mg/ml, 20 μl; BD
Matrigel™ Invasion chamber). SAS cells (2×104) were
seeded into the chamber of the insert and incubated with 0.5 ml of
complete DMEM medium in each transwell. Cells were treated with EGF
(100 ng/ml) and various concentrations of PEITC (0, 0.5, 1 and 2
μM) for 48 h and then cells inside the chamber were removed
by a cotton swab. Invaded cells were fixed with 4% formaldehyde in
PBS and stained with 0.1% of hematoxylin, captured and the number
of invaded cells was counted (43,44).
Cell viability assay
SAS cells (2×104) were seeded into the
96-well plate and treated with EGF (100 ng/ml) and PEITC (0, 0.5, 1
and 2 μM) for 48 h. Medium was removed and replaced with
fresh DMEM medium containing MTT (0.5 mg/ml) and cultured at 37°C
incubator for an additional 4 h. Medium was again removed and 200
μl of DMSO was added into each well to dissolve the formazan
crystals and the absorbance of each well was measured at 570 nm
with a reference wavelength at 620 nm on an ELISA reader. The data
of control sample (0 μM of PEITC) was set as 100% and the
relative cell viability of drug-treated samples was calculated
accordingly. Cell morphology was recorded by using a phase-contrast
microscope (43,44).
Gelatin zymography assay
SAS cells (1×106) were seeded into
12-well plate for 48 h and treated with EGF (100 ng/ml) and various
concentrations of PEITC (0, 0.5, 1 and 2 μM) in serum-free
DMEM medium for an additional 48 h. Culture medium was spun at 1000
× g for 10 min at 4°C, supernatant was collected and protein
concentration was determined as described below. 5 μg of
total proteins were mixed with 2X sample buffer (0.125 M Tris-HCl,
pH 6.8, 4% SDS, 20% glycerol, 0.01% bromophenol blue) and
electrophoresed in an 8% SDS-polyacrylamide gel with 1% gelatin.
Gel was incubated with 2.5% Triton X-100 at room temperature for 30
min to remove residual SDS and then incubated in Zymogen developing
buffer (50 mM Tris, pH 7.5, 200 mM NaCl, 5 mM CaCl2, 1
μM ZnCl2, 0.02% Brij-35; Bio-Rad Laboratories,
Hercules, CA, USA) at 37°C overnight. Gel was then washed
extensively with water and stained with 0.5% Coomassie blue G-250
(0.5% Coomassie blue G-250, 50% methanol and 10% acetic acid) for 2
h and de-stained in de-staining solution (50% methanol and 10%
acetic acid) until clear zones were evident. The gel was scanned by
a scanning digitizing system and digitized by using free Image J
software (NIH) (43,44).
Preparation of whole cell lysate
SAS cells were challenged with EGF (100 ng/ml) and
treated with various concentrations of PEITC for the specified time
and cells were collected for the preparation of whole cell lysate
using iced-cold RIPA buffer (50 mM Tris-base, 150 mM NaCl, 0.1%
SDS, 1% sodium deoxycholate, 1% NP-40, pH 7.5) supplemented with
protease inhibitors including leupeptin (17 mg/ml), sodium
orthovana-date (10 mg/ml), phenylmethanesulfonyl fluoride (10
mg/ml). Cells were completely re-suspended in extraction buffer and
kept in ice for 30 min with occasional mixing and cell lysate were
collected by a spin at 12,000 × g for 10 min at 4°C. The protein
concentrations present in the samples were measured by using
Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad) (43,45).
Western blotting
The obtained whole cell lysate was resolved in
sodium dodecyl sulfate-polyacrylamide gel and transferred onto
polyvinylidene fluoride (PVDF) membrane (Millipore) by using the
iBlot Dry Blotting Transfer System (Invitrogen/Life Technologies).
The transferred membranes were blocked in 5% non-fat milk (prepared
in Tris-buffered saline supplemented with 0.1% Tween-20; TBST) at
ambient temperature for 1 h and incubated with primary antibody at
4°C overnight. Membranes were washed with TBST three times for 10
min before incubated with HRP-coupled secondary antibody for 1 h.
Protein signals were visualized by enhanced chemiluminescence (ECL)
and exposed to Bio-MAX MR X-ray film (Eastman Kodak, Rochester, NY,
USA) (43,46,47).
Quantitative real-time PCR analyses
SAS cells were treated with 0, 1 and 2 μM of
PEITC and EGF (100 ng/ml) for 24 h and cells were collected. Total
RNAs were isolated using the Qiagen RNeasy mini Kit. cDNAs were
synthesized using the High Capacity cDNA Reverse Transcription kit
according to the supplier’s brochure (Applied Biosystems). For the
quantitative PCR reaction, 1 μl of cDNAs were mixed with 2X
SYBR Green PCR Master Mix (Applied Biosystems) and 200 nM of
forward and reverse primers (see below for detailed sequences). PCR
reaction was performed on an Applied Biosystems 7300 Real-Time PCR
system in triplicate according to the following conditions: 2 min
at 50°C, 10 min at 95°C and 40 cycles of 15 sec at 95°C, 1 min at
60°C. Fold changes of the gene expression were derived using the
comparative CT method (40,48).
The used primer pairs were: human MMP-2-forward,
5′-CCCCAGACAGGTGATCTTGAC-3′; human MMP-2-reverse,
5′-GCTTGCGAGGGAAGAAGT TG-3′; human MMP-9-forward, 5′-CGCTGGGCTTAGAT
CATTCC-3′; human MMP-9-reverse, 5′-AGGTTGGATACAT CACTGCATTAGG-3′;
human GAPDH-forward, 5′-ACACC CACTCCTCCACCTTT-3′; human
GAPDH-reverse, 5′-TAGC CAAATTCGTTGTCATACC-3′ (49).
Immunofluorescence staining
SAS cells were seeded onto slides overnight and
incubated with EGF (100 ng/ml) and PEITC (0, 1 and 2 μM) for
6 h. Cells were fixed in 4% formaldehyde at room temperature for 15
min, washed with PBS and permeabolized with 0.1% Triton X-100 in
PBS for 15 min, then incubated with primary antibodies at 4°C
overnight. After extensive washes, cells were incubated with
FITC-conjugated secondary antibodies for 2 h. Cells were sealed and
images were acquired with a fluorescence microscope (Nikon) and
processed in Photoshop 7.0 software (50).
Statistical analysis
One-way ANOVA followed by Student’s t-test was used
to evaluate the differences between treated and experimental
groups. p<0.05 was considered to define a statistically
significant difference (49,51).
Results
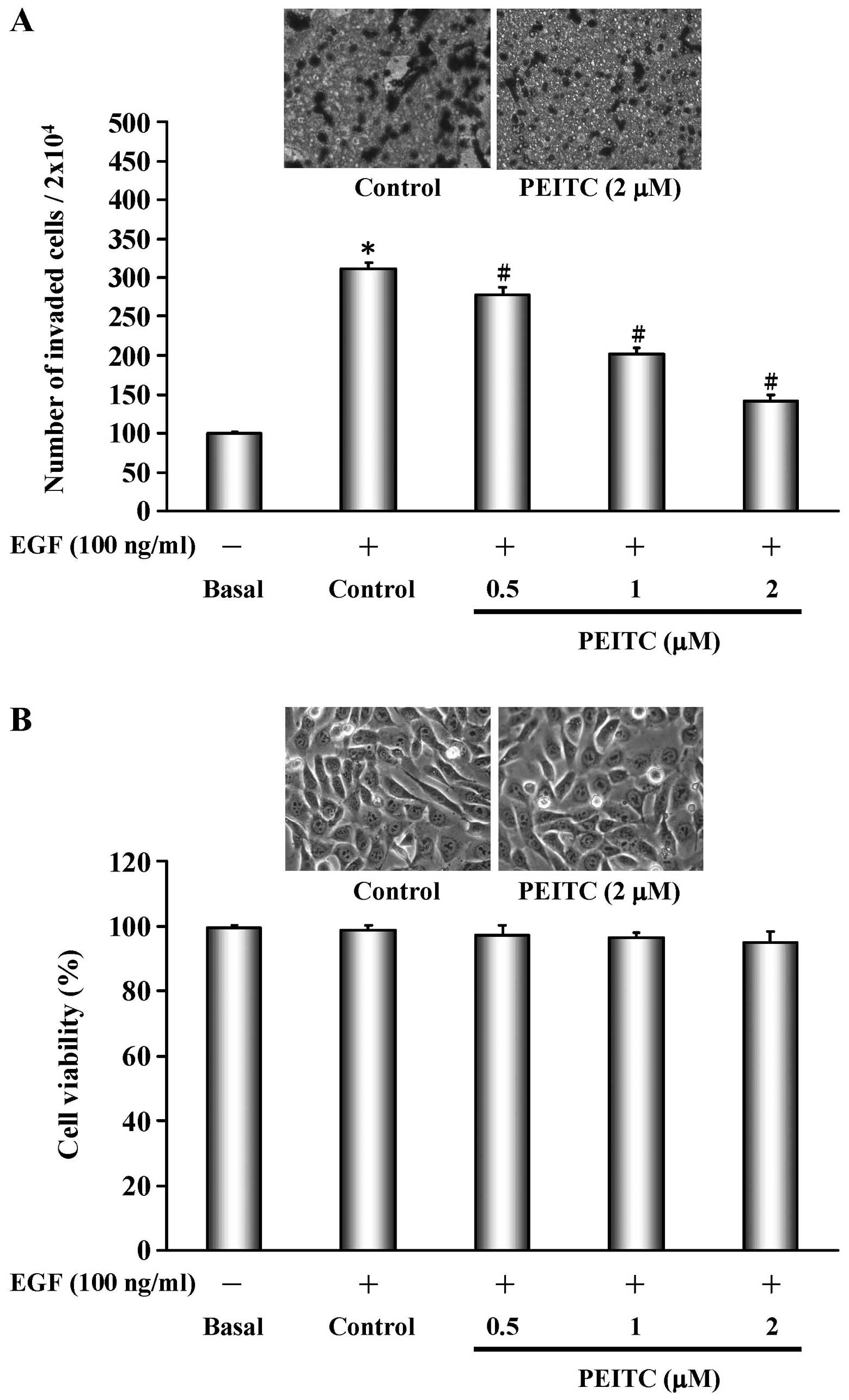
PEITC inhibits EGF-stimulated invasion of
SAS cells
We therefore determined the effects of PEITC on
EGF-stimulated SAS cells. EGF-treatment increased the invasion of
SAS cells, as revealed by Matrigel invasion assay (Fig. 1A). Treatment of EGF-stimulated SAS
cells with PEITC decreased the invasion of cells in a
concentration-dependent manner (Fig.
1A). From Fig. 1A, we have
known that PEITC inhibited the invasion of EGF-stimulated SAS
cells, a result that could be due to the inhibition of PEITC on the
viability of EGF-stimulated cells. To test this, we treated
EGF-stimulated SAS cells with different concentrations (0, 0.5, 1
and 2 μM) of PEITC and performed MTT cell viability assay.
The result showed that PEITC at 0.5–2 μM of concentrations
did not inhibit the viability of EGF-stimulated SAS cells, as
compared to cells without drug treatment (Fig. 1B). The cell morphology was
comparable between treatments with 0 and 2 μM of PEITC (see
inserts in Fig. 1B).
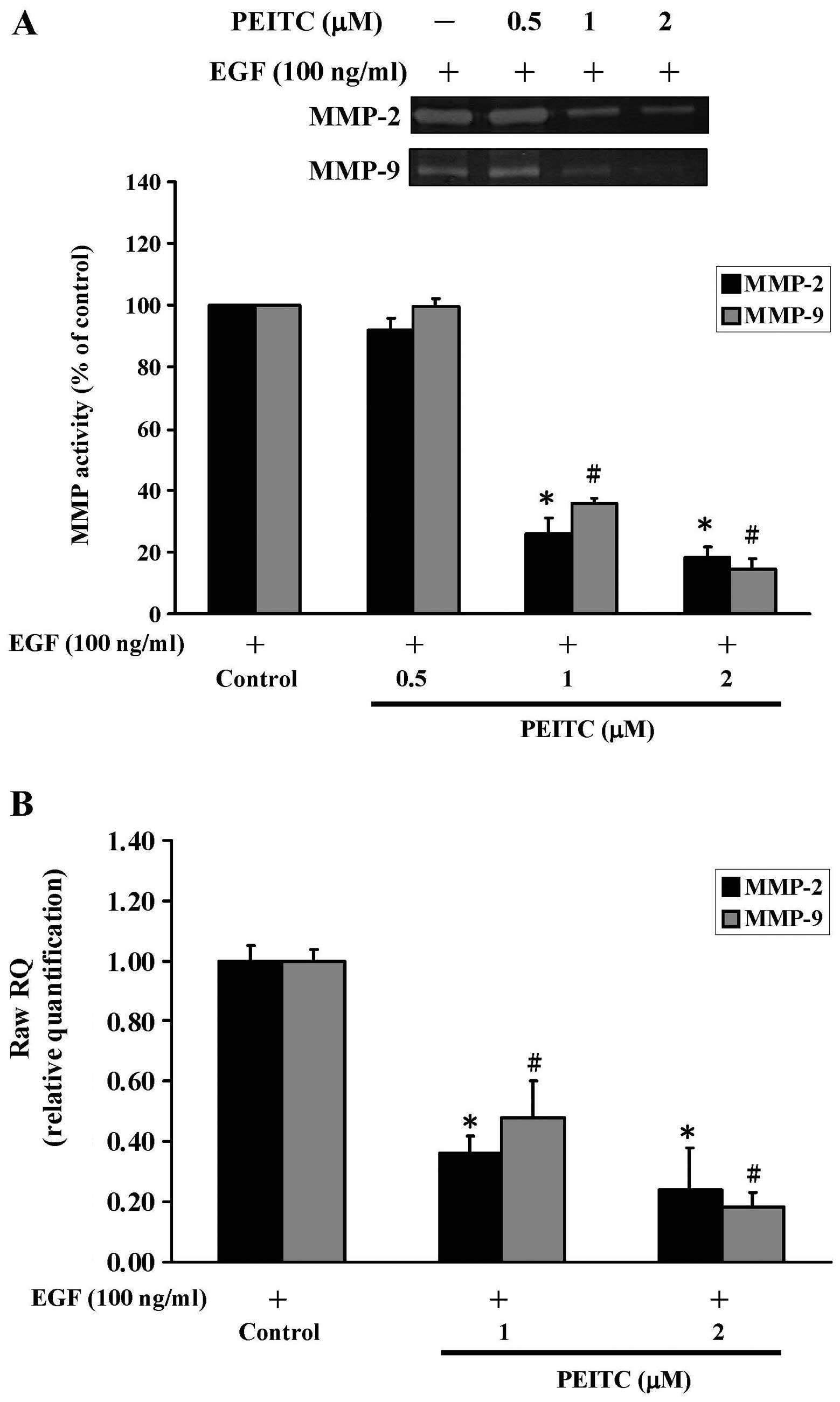
PEITC inhibits the enzymatic activities
and gene expression of MMP-2 and MMP-9 in EGF-stimulated SAS
cells
Studies have shown that matrix metalloproteinases
including MMP-2 (gelatinase A) and MMP-9 (gelatinase B) are
expressed in oral cancers. These two MMPs are closely linked to the
malignant potential of tumor cells and are also important for tumor
invasion and metastasis (44,52).
To evaluate the effects of PEITC on the enzymatic activities of
MMP-2 and MMP-9, we treated EGF-stimulated SAS cells with different
concentrations (0, 0.5, 1 and 2 μM) of PEITC and assessed
the enzymatic activities of MMP-2 and MMP-9 by gelatin zymography.
As shown in Fig. 2A, treatments of
cells with PEITC suppressed the enzymatic activities of both MMP-2
and MMP-9 in a concentration-dependent manner. Pronounced
inhibition of the activities was observed at concentrations >1
μM of PEITC (see columns 3 and 4 and inserts of Fig. 2A). To address whether the
inhibition of MMP-2 and MMP-9 was at the transcriptional level, we
treated EGF-stimulated cells with PEITC (0, 1 and 2 μM) and
the effects of PEITC were analyzed by quantitative RT-PCR. As shown
in Fig. 2B, treatment of PEITC
significantly decreased the gene expression of both MMP-2 and MMP-9
in a concentration-dependent fashion. Taken together, these data
suggested that both gelatinases (MMP-2 and MMP-9) are involved in
the EGF-induced invasion of SAS cells.
PEITC inhibits the protein expression of
MMP-1, MMP-2 and increased the protein expression of TIMP-1 and
TIMP-2 in EGF-stimulated SAS cells
Since PEITC inhibited the enzymatic activities and
gene expression of MMP-2 and MMP-9 in EGF-stimulated cells, we
infered that PEITC could inhibit the protein expression of both
metalloproteinases. As shown in Fig.
3A, treatment of EGF-stimulated SAS cells with PEITC (1 and 2
μM) reduced the protein expression of MMP-2 and MMP-9. It is
well known that tissue inhibitor of metalloproteinases (TIMPs),
TIMP-1 and TIMP-2, can bind and inhibit the enzymatic activities of
MMP-2 and MMP-9 (53). We
therefore examined the protein expression of TIMP-1 and TIMP-2 in
EGF-stimulated cells treated with PEITC (0, 1 and 2 μM). The
result showed that treatment with PEITC significantly increased the
protein expression of TIMP-1 and TIMP-2 (Fig. 3A). These data suggested that PEITC
can suppress the gene expression and protein expression of MMP-2
and MMP-9 and increase the protein expression of TIMP-1 and TIMP-2,
leading to the decrease in the enzymatic activities of MMP-2 and
MMP-9 in EGF-stimulated SAS cells.
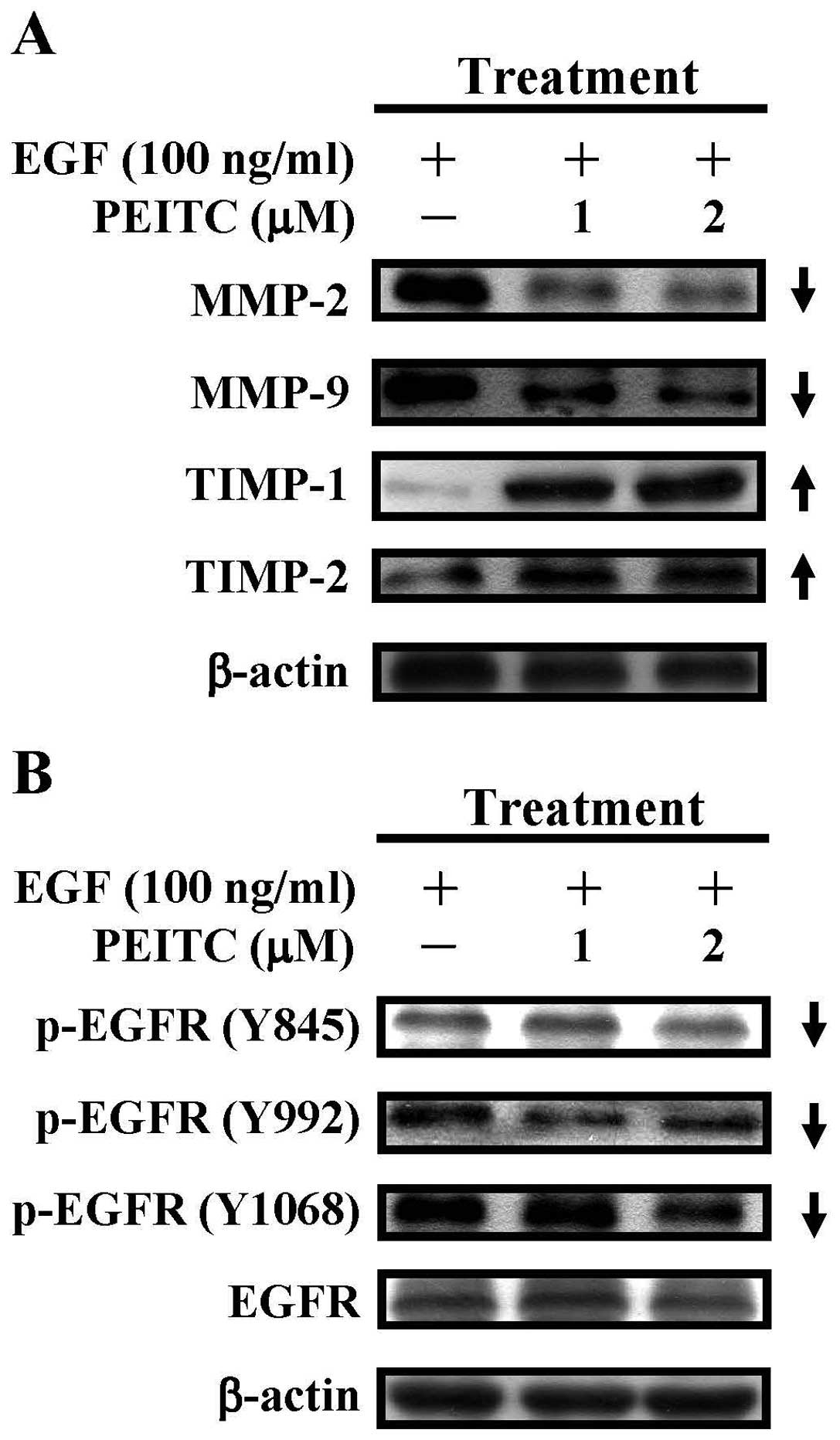 | Figure 3.PEITC inhibits the expression of
metalloproteases MMP-2, MMP-9 and induces the expression of tissue
inhibitor of metalloproteinases TIMP-1 and TIMP-2 through
inactivation of the epidermal growth factor receptor (EGFR) in
EGF-stimulated SAS cells. (A) The effects of PEITC on the
expression of MMP-2, MMP-9, TIMP-1 and TIMP-2 proteins in
EGF-stimulated SAS cells. Cells were treated with EGF (100 ng/ml)
and PEITC (0, 1 and 2 μM) for 48 h and cells were harvested
for western blot analyses with MMP-2, MMP-9, TIMP-1 and TIMP-2
antibodies, respectively. The β-actin served as the loading
control. (B) The effects of PEITC on the activation of the
epidermal growth factor receptor (EGFR) in EGF-stimulated SAS
cells. Cells were treated with EGF (100 ng/ml) and PEITC (0, 1 and
2 μM) for 6 h and cell lysates were subjected to western
blot analyses and detected with p-EGFR (Y845), p-EGFR (Y992),
p-EGFR (Y1068) and EGFR antibodies, respectively. The β-actin
served as the loading control. |
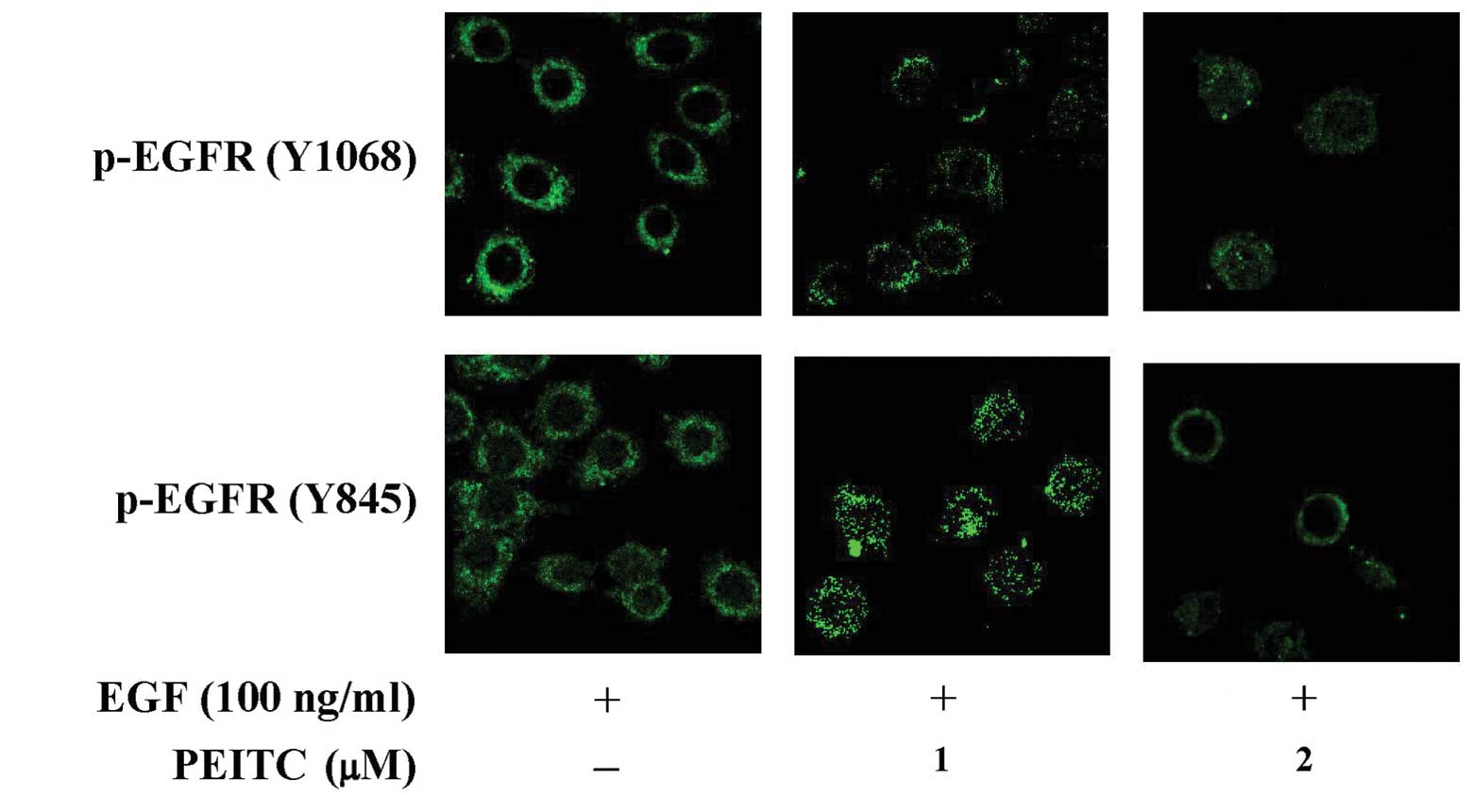
PEITC suppressed the activation of
EGFR
Binding of EGF to its cognate receptor EGFR results
in the activation of EGFR that involves the autophosphorylation of
EGFR and activation of intracellular signaling pathways, such as
activation of PI3K/AKT, mitogen-activated protein kinases (MAPKs)
and the signal transducer and activators of transcription (STATs)
pathways, leading to cell proliferation and survival, invasion,
metastasis and angiogenesis (54–56).
Thus, we determined the effects of PEITC on the activation of EGFR
proteins by examining the tyrosine phosphorylation of EGFR in
EGF-challenged SAS cells. As shown in Fig. 3B, PEITC treatment (1 and 2
μM) inhibited the tyrosine phosphorylation of EGFR at Y845,
Y992 and Y1068 concentration-dependently, while the protein levels
of total EGFR remained largely unchanged. Immuno-fluorescent
staining with anti-p-EGFR (Y1068) and anti-p-EGFR (Y845) also
showed that tyrosine-phosphorylated EGFRs at Y1068 and Y845 were
dramatically reduced in EGF-challenged cells treated with PEITC
(Fig. 4). These data suggested
that PEITC treatment suppressed the activation of EGFR in
EGF-challenged SAS cells.
PEITC decreases the protein
phosphorylation of PI3K, AKT, IKK and IκBα in EGF-stimulated SAS
cells
Since treatment with PEITC suppressed the activation
of EGFR in EGF-challenged cells (Figs.
3B and 4), we next examined
the effects of PEITC on downstream PI3K/AKT and NF-κB signaling
pathways. PEITC treatment (1 and 2 μM) reduced the protein
phosphorylation of PDK1, PI3K (p85) and profoundly reduced the
protein phosphorylation of AKT (S308) and AKT (S473) in
EGF-challenged cells (Fig. 5A,
panels 1, 2, 4 and 5). Total proteins of PI3K (p85) was not changed
during drug treatment, although total proteins of AKT were slightly
reduced at 2 μM of PEITC. In addition, PEITC treatment (1
and 2 μM) decreased the protein phosphorylation of IKK and
of IκBα, while increased the protein stability of IκBα. These data
indicated that PI3K/AKT and NF-κB signaling pathway downstream of
EGFR signaling is downregulated by PEITC treatment.
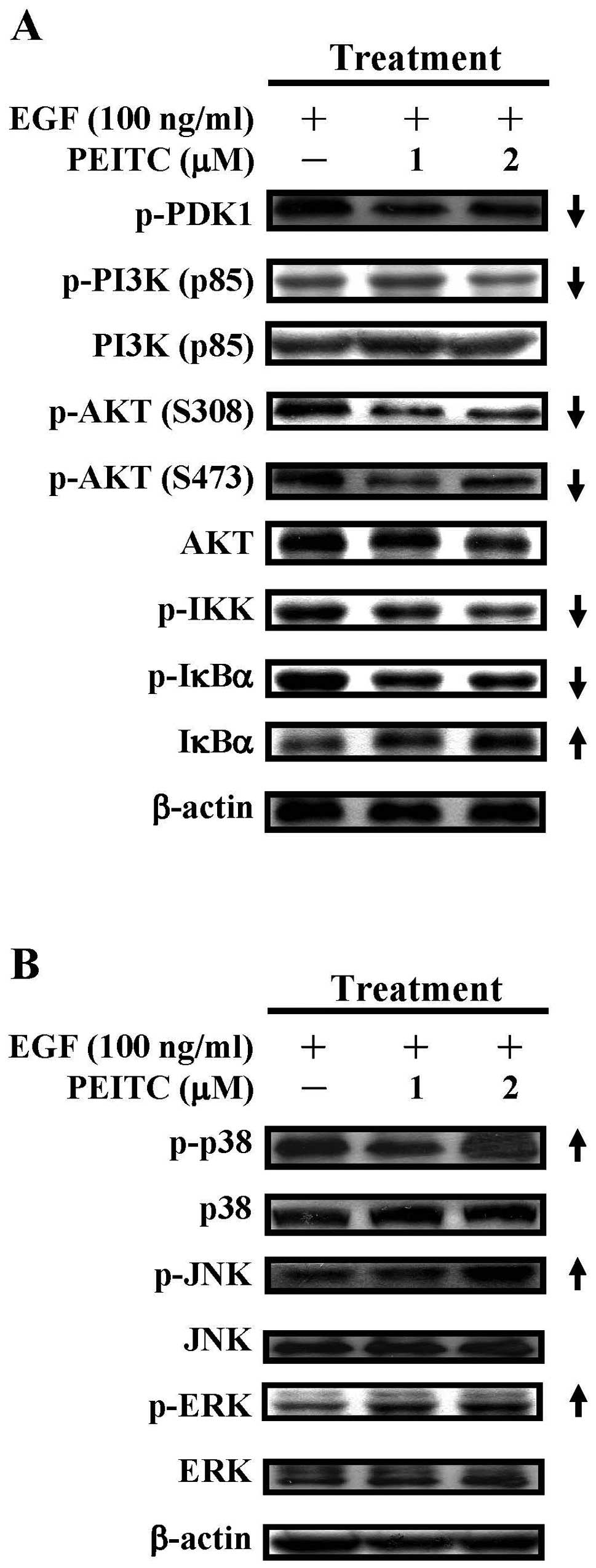 | Figure 5.The effects of PEITC on the PI3K/AKT
and MAPK signaling pathways in EGF-stimulated SAS cells. Cells were
treated with EGF (100 ng/ml) as well as PEITC (0, 1 and 2
μM) for 6 h and cell lysates were prepared for the western
blot analyses of (A) phosphorylated PDK1, PI3K (p85),
phosphorylated PI3K (p85), AKT, phosphorylated AKT (S308 and S473),
phosphorylated IKK, IκBα and phosphorylated IκBα and (B) p38,
phosphorylated p38, JNK, phosphorylated JNK, ERK and phosphorylated
ERK respectively. The β-actin served as the loading control. |
The effects of PEITC on the MAPK
signaling pathway in EGF-stimulated SAS cells
We also determined the effects of PEITC on the MAPK
signaling pathways-p38, JNK and ERK signaling pathways downstream
of EGFR activation. PEITC treatment (1 and 2 μM) increased
the protein phosphorylation of p38 at higher drug concentration (2
μM) (Fig. 5B, top panel).
PEITC treatment (1 and 2 μM) also increased the protein
phosphorylation of JNK and ERK in a concentration-dependent manner.
The protein expression of p38, JNK and ERK appeared not to be
affected (Fig. 5B, panels 2, 4 and
6), suggesting that the MAPK signaling pathway can be activated
after PEITC treatment.
Discussion
Oral squamous cell carcinoma (OSCC) is a leading
cause of cancer deaths in the world, characterized by poor
prognosis and a low survival rate in spite of advances in treatment
with surgery and radiotherapy. The main cause of death in OSCC is
metastasis which primarily occurs through the lymphatic system.
Once it has metastasized to the lymph nodes, the overall mortality
rate of the disease is high and the 5-year overall survival rate
does not exceed 50%, which is one of the lowest rates for all major
cancers (10,57). Therefore, identification of new
drugs for the chemotherapy of OSCC metastasis is highly
desirable.
The epidermal growth factor receptor (EGFR) belongs
to the HER/ErbB protein family of receptor tyrosine kinases. The
EGFR gene encodes a 170-kDa transmembrane glycoprotein with its
tyrosine kinase domain located within the cytoplasmic region.
Ligand binding induces the activation of tyrosine kinase activity
that triggers intracellular signaling cascades, including the
Ras-Raf-mitogen-activated protein kinase pathway, the
phosphatidylinositol 3-kinase-AKT pathway and the signal transducer
and activators of transcription pathway, which contribute to cell
proliferation and survival (58).
Dysregulation in the signaling of EGFR and other members of the
tyrosine kinase receptor family has been linked to cell
transformation, autonomous cell growth, angiogenesis, invasion and
metastases in a number of cancers (59). Up to 90% of head and neck squamous
cell carcinoma (HNSCC) patients are identified with EGFR
overexpression, which is considered to be involved in tumorigenesis
and metastasis (60).
Overexpression of EGFR in HNSCC is often associated with the
simultaneous increase in its ligands such as the transforming
growth factor α (54,61), which will lead to excessive
activation of EGFR signaling either in an autocrineor
paracrine-dependent manner. Thus, EGFR appears to be a promising
therapeutic target for oral cancer metastasis (44). Our data indicated that PEITC can
inhibit the EGF-induced invasion of SAS cells through the
inactivation of EGFR and downstream signaling, including the
suppression in the phosphorylation cascade of PI3K, PDK1 and AKT
and hence the reduction of phosphorylated IKK, the decrease in the
phosphorylation of IκB and the simultaneous increase in the
stability of IκB and the block in the release and activation of
NF-κB. These results are consistent with previous studies using
prostate cancer PC-3 cells (32,62).
Matrix metalloproteases (MMPs) are responsible for
the degradation of the extracellular matrix and facilitating
spreading and metastasis of tumor cells. They are strongly blocked
by the endogenous tissue inhibitors of metalloproteinases (TIMP-1,
-2, -3 and -4). The expression of MMP-2 and MMP-9 was shown to be
associated with tumor invasion and lymph node metastasis of oral
cancer (63). The expression can
also be regulated by NF-κB as their promoters possess NF-κB binding
sites. As expected, the activities and expression of MMP-2 and
MMP-9 were downregulated after PEITC treatment in EGF-stimulated
SAS cells (Figs. 2 and 3A). These results could be due to the
inactivation of NF-κB caused by the disruption in the EGFR
signaling after PEITC treatment, leading to the failure in the
expression of MMP-2 and MMP-9. Concomitantly, the increase in the
expression of MMP-2 and -9 inhibitors, TIMP-1 and -2 proteins, was
observed (Fig. 3A). These data
suggested that PEITC suppressed the invasion and metastasis of
EGFR-overexpressed oral cancers by reducing the expression and
activities of MMP-2 and MMP-9 through the interference with the
phosphorylation of EGFR and downstream signaling.
The other major downstream pathway regulated by EGFR
is MAPK. Our finding that the phosphorylation of p38, JNK and ERK
was increased after PEITC treatment in SAS cells is contradictory
to the previous reports that PEITC can suppress the MAPK activation
to inhibit the invasion and metastasis of HT-29 colon cancer cells
and AGS gastric cancer cells (39,40).
These discrepancies could be due to cell type-specific effects.
Alternatively, it has been reported that PEITC can induce cell
apoptosis through the activation of MAPK (29,64–68)
and this raises the possibility that PETIC may trigger apoptosis in
addition to the suppression of invasion and metastasis in our
system. However, our data showed that the cell viability was not
affected after PEITC treatment (1–2 μM) (Fig. 1B). This suggests that the
activation of MAPK by PEITC may elicit the downstream signaling and
drive specific genes expression to repress the invasion and
metastasis of OSCC, but not to cause apoptosis. Further detailed
mechanism needs to be elucidated.
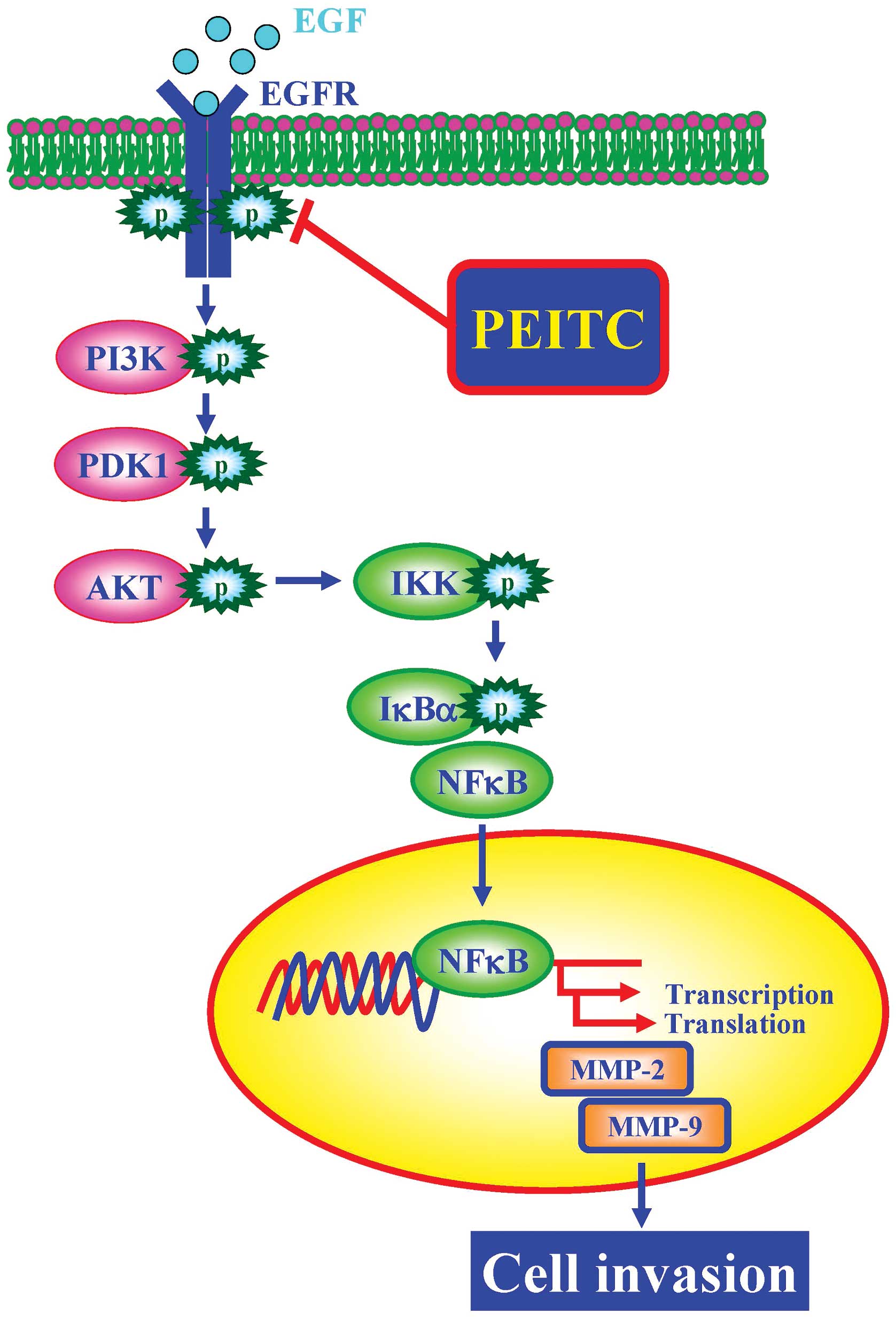
In conclusion, the signaling pathway underlying the
effects of PEITC on the invasion of EGF-stimulated SAS cells is
shown in Fig. 6. PEITC suppressed
the phosphorylation and activation of EGFR and inhibited the
sequential phosphorylation and activation of PI3K, PDK1 and AKT,
resulting in the reduced expression of phosphorylated IKK and hence
the reduced protein phosphorylation and increased protein stability
of IκBα, which in turn suppressed the expression and enzymatic
activities of MMP-2 and MMP-9, contributing to the inhibition of
invasion in EGF-challenged SAS cells. Our data suggested that PEITC
will be a promising therapeutic agent for the treatment of oral
cancer metastasis.
Acknowledgements
This study was supported in part by
research grants from the National Science Council of the Republic
of China (NSC 101-2313-B-039-008) awarded to J.-S. Yang. This study
was also supported in part by grant from China Medical University
CMU101-S-27 awarded to J.-S. Yang and CMU98-N1-16 to H.-J.
Chen.
References
|
1.
|
Noguti J, De Moura CF, De Jesus GP, et al:
Metastasis from oral cancer: an overview. Cancer Genomics
Proteomics. 9:329–335. 2012.
|
|
2.
|
Sarode SC, Sarode GS, Karmarkar S and
Tupkari JV: A new classification for potentially malignant
disorders of the oral cavity. Oral Oncol. 47:920–921. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Seethala RR, Gooding WE, Handler PN, et
al: Immunohistochemical analysis of phosphotyrosine signal
transducer and activator of transcription 3 and epidermal growth
factor receptor autocrine signaling pathways in head and neck
cancers and meta-static lymph nodes. Clin Cancer Res. 14:1303–1309.
2008. View Article : Google Scholar
|
|
4.
|
Franceschi S, Bidoli E, Baron AE, et al:
Nutrition and cancer of the oral cavity and pharynx in north-east
Italy. Int J Cancer. 47:20–25. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Velly AM, Franco EL, Schlecht N, et al:
Relationship between dental factors and risk of upper aerodigestive
tract cancer. Oral Oncol. 34:284–291. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Ko YC, Huang YL, Lee CH, Chen MJ, Lin LM
and Tsai CC: Betel quid chewing, cigarette smoking and alcohol
consumption related to oral cancer in Taiwan. J Oral Pathol Med.
24:450–453. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Ichimiya Y, Fuwa N, Kamata M, et al:
Treatment results of stage I oral tongue cancer with definitive
radiotherapy. Oral Oncol. 41:520–525. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Xi S and Grandis JR: Gene therapy for the
treatment of oral squamous cell carcinoma. J Dent Res. 82:11–16.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Pereira MC, Oliveira DT, Landman G and
Kowalski LP: Histologic subtypes of oral squamous cell carcinoma:
prognostic relevance. J Can Dent Assoc. 73:339–344. 2007.PubMed/NCBI
|
|
10.
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
11.
|
Okura M, Aikawa T, Sawai NY, Iida S and
Kogo M: Decision analysis and treatment threshold in a management
for the N0 neck of the oral cavity carcinoma. Oral Oncol.
45:908–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Greenberg JS, Fowler R, Gomez J, et al:
Extent of extracapsular spread: a critical prognosticator in oral
tongue cancer. Cancer. 97:1464–1470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Stan SD, Kar S, Stoner GD and Singh SV:
Bioactive food components and cancer risk reduction. J Cell
Biochem. 104:339–356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Stan SD, Singh SV and Brand RE:
Chemoprevention strategies for pancreatic cancer. Nat Rev
Gastroenterol Hepatol. 7:347–356. 2010.PubMed/NCBI
|
|
15.
|
Keum YS, Jeong WS and Kong AN:
Chemopreventive functions of isothiocyanates. Drug News Perspect.
18:445–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ambrosone CB, McCann SE, Freudenheim JL,
Marshall JR, Zhang Y and Shields PG: Breast cancer risk in
premenopausal women is inversely associated with consumption of
broccoli, a source of isothiocyanates, but is not modified by GST
genotype. J Nutr. 134:1134–1138. 2004.PubMed/NCBI
|
|
17.
|
Greenwald P, Clifford CK and Milner JA:
Diet and cancer prevention. Eur J Cancer. 37:948–965. 2001.
View Article : Google Scholar
|
|
18.
|
Conaway CC, Yang YM and Chung FL:
Isothiocyanates as cancer chemopreventive agents: their biological
activities and metabolism in rodents and humans. Curr Drug Metab.
3:233–255. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Hecht SS: Inhibition of carcinogenesis by
isothiocyanates. Drug Metab Rev. 32:395–411. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Fahey JW, Zalcmann AT and Talalay P: The
chemical diversity and distribution of glucosinolates and
isothiocyanates among plants. Phytochemistry. 56:5–51. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Hayes JD, Kelleher MO and Eggleston IM:
The cancer chemopreventive actions of phytochemicals derived from
glucosinolates. Eur J Nutr. 47(Suppl 2): 73–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kumar A and Sabbioni G: New biomarkers for
monitoring the levels of isothiocyanates in humans. Chem Res
Toxicol. 23:756–765. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Khor TO, Cheung WK, Prawan A, Reddy BS and
Kong AN: Chemoprevention of familial adenomatous polyposis in
Apc(Min/+) mice by phenethyl isothiocyanate (PEITC). Mol Carcinog.
47:321–325. 2008.
|
|
24.
|
Moy KA, Yuan JM, Chung FL, et al: Urinary
total isothiocyanates and colorectal cancer: a prospective study of
men in Shanghai, China. Cancer Epidemiol Biomarkers Prev.
17:1354–1359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Hecht SS: Chemoprevention by
isothiocyanates. J Cell Biochem. (Suppl 22): 195–209. 1995.
View Article : Google Scholar
|
|
26.
|
Zhang Y: Cancer-preventive
isothiocyanates: measurement of human exposure and mechanism of
action. Mutat Res. 555:173–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Xiao D, Johnson CS, Trump DL and Singh SV:
Proteasome-mediated degradation of cell division cycle 25C and
cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced
G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells.
Mol Cancer Ther. 3:567–575. 2004.
|
|
28.
|
Huong le D, Shim JH, Choi KH, et al:
Effect of beta-phenylethyl isothiocyanate from cruciferous
vegetables on growth inhibition and apoptosis of cervical cancer
cells through the induction of death receptors 4 and 5. J Agric
Food Chem. 59:8124–8131. 2011.PubMed/NCBI
|
|
29.
|
Yan H, Zhu Y, Liu B, et al:
Mitogen-activated protein kinase mediates the apoptosis of highly
metastatic human non-small cell lung cancer cells induced by
isothiocyanates. Br J Nutr. 106:1779–1791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Chen YR, Han J, Kori R, Kong AN and Tan
TH: Phenylethyl isothiocyanate induces apoptotic signaling via
suppressing phosphatase activity against c-Jun N-terminal kinase. J
Biol Chem. 277:39334–39342. 2002. View Article : Google Scholar
|
|
31.
|
Hwang ES and Lee HJ: Effects of
phenylethyl isothiocyanate and its metabolite on cell-cycle arrest
and apoptosis in LNCaP human prostate cancer cells. Int J Food Sci
Nutr. 61:324–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Xu C, Shen G, Chen C, Gelinas C and Kong
AN: Suppression of NF-kappaB and NF-kappaB-regulated gene
expression by sulforaphane and PEITC through IkappaBalpha, IKK
pathway in human prostate cancer PC-3 cells. Oncogene.
24:4486–4495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Bommareddy A, Hahm ER, Xiao D, et al: Atg5
regulates phenethyl isothiocyanate-induced autophagic and apoptotic
cell death in human prostate cancer cells. Cancer Res.
69:3704–3712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Wu X, Kassie F and Mersch-Sundermann V:
Induction of apoptosis in tumor cells by naturally occurring
sulfur-containing compounds. Mutat Res. 589:81–102. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Morse MA, Amin SG, Hecht SS and Chung FL:
Effects of aromatic isothiocyanates on tumorigenicity,
O6-methylguanine formation and metabolism of the tobacco-specific
nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J
mouse lung. Cancer Res. 49:2894–2897. 1989.
|
|
36.
|
Zhang Y, Kensler TW, Cho CG, Posner GH and
Talalay P: Anticarcinogenic activities of sulforaphane and
structurally related synthetic norbornyl isothiocyanates. Proc Natl
Acad Sci USA. 91:3147–3150. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Powolny AA, Bommareddy A, Hahm ER, et al:
Chemopreventative potential of the cruciferous vegetable
constituent phenethyl isothiocyanate in a mouse model of prostate
cancer. J Natl Cancer Inst. 103:571–584. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Kelloff GJ, Crowell JA, Hawk ET, et al:
Strategy and planning for chemopreventive drug development:
clinical development plans II. J Cell Biochem. (Suppl 26): 54–71.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Yang MD, Lai KC, Lai TY, et al: Phenethyl
isothiocyanate inhibits migration and invasion of human gastric
cancer AGS cells through suppressing MAPK and NF-kappaB signal
pathways. Anticancer Res. 30:2135–2143. 2010.PubMed/NCBI
|
|
40.
|
Lai KC, Huang AC, Hsu SC, et al: Benzyl
isothiocyanate (BITC) inhibits migration and invasion of human
colon cancer HT29 cells by inhibiting matrix metalloproteinase-2/-9
and urokinase plasminogen (uPA) through PKC and MAPK signaling
pathway. J Agric Food Chem. 58:2935–2942. 2010. View Article : Google Scholar
|
|
41.
|
Xiao D and Singh SV: Phenethyl
isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer
Res. 67:2239–2246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Lu KW, Chen JC, Lai TY, et al: Gypenosides
inhibits migration and invasion of human oral cancer SAS cells
through the inhibition of matrix metalloproteinase-2 -9 and
urokinase-plasminogen by ERK1/2 and NF-kappa B signaling pathways.
Hum Exp Toxicol. 30:406–415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Hour MJ, Tsai SC, Wu HC, et al: Antitumor
effects of the novel quinazolinone MJ-33: Inhibition of metastasis
through the MAPK, AKT, NF-kappaB and AP-1 signaling pathways in
DU145 human prostate cancer cells. Int J Oncol. 41:1513–1519.
2012.PubMed/NCBI
|
|
44.
|
Ohnishi Y, Lieger O, Attygalla M, Iizuka T
and Kakudo K: Effects of epidermal growth factor on the invasion
activity of the oral cancer cell lines HSC3 and SAS. Oral Oncol.
44:1155–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Yang JS, Liu CW, Ma YS, et al: Chlorogenic
acid induces apoptotic cell death in U937 leukemia cells through
caspase- and mitochondria-dependent pathways. In Vivo. 26:971–978.
2012.PubMed/NCBI
|
|
46.
|
Lan YH, Chiang JH, Huang WW, et al:
Activations of both extrinsic and intrinsic pathways in HCT 116
human colorectal cancer cells contribute to apoptosis through
p53-mediated ATM/Fas signaling by Emilia sonchifolia
extract, a folklore medicinal plant. Evid Based Complement Alternat
Med. 2012:1781782012.PubMed/NCBI
|
|
47.
|
Tsai SC, Huang WW, Huang WC, et al:
ERK-modulated intrinsic signaling and G(2)/M phase arrest
contribute to the induction of apoptotic death by allyl
isothiocyanate in MDA-MB-468 human breast adenocarcinoma cells. Int
J Oncol. 41:2065–2072. 2012.
|
|
48.
|
Kao WT, Lin CY, Lee LT, et al:
Investigation of MMP-2 and -9 in a highly invasive A431 tumor cell
sub-line selected from a Boyden chamber assay. Anticancer Res.
28:2109–2120. 2008.PubMed/NCBI
|
|
49.
|
Chen KT, Hour MJ, Tsai SC, et al: The
novel synthesized
6-fluoro-(3-fluorophenyl)-4-(3-methoxyanilino)quinazoline (LJJ-10)
compound exhibits anti-metastatic effects in human osteosarcoma U-2
OS cells through targeting insulin-like growth factor-I receptor.
Int J Oncol. 39:611–619. 2011.
|
|
50.
|
Yang JS, Wu CC, Kuo CL, et al: Solanum
lyratum extracts induce extrinsic and intrinsic pathways of
apoptosis in WEHI-3 murine leukemia cells and inhibit allograft
tumor. Evid Based Complement Alternat Med.
2012:2549602012.PubMed/NCBI
|
|
51.
|
Chiu YJ, Hour MJ, Lu CC, et al: Novel
quinazoline HMJ-30 induces U-2 OS human osteogenic sarcoma cell
apoptosis through induction of oxidative stress and up-regulation
of ATM/p53 signaling pathway. J Orthop Res. 29:1448–1456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Bjorklund M and Koivunen E:
Gelatinase-mediated migration and invasion of cancer cells. Biochim
Biophys Acta. 1755:37–69. 2005.PubMed/NCBI
|
|
53.
|
Bode W, Fernandez-Catalan C, Grams F, et
al: Insights into MMP-TIMP interactions. Ann NY Acad Sci.
878:73–91. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Kalyankrishna S and Grandis JR: Epidermal
growth factor receptor biology in head and neck cancer. J Clin
Oncol. 24:2666–2672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Zimmermann M, Zouhair A, Azria D and
Ozsahin M: The epidermal growth factor receptor (EGFR) in head and
neck cancer: its role and treatment implications. Radiat Oncol.
1:112006. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Rogers SJ, Harrington KJ, Rhys-Evans P,
O-Charoenrat P and Eccles SA: Biological significance of c-erbB
family oncogenes in head and neck cancer. Cancer Metastasis Rev.
24:47–69. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Jemal A, Murray T, Samuels A, Ghafoor A,
Ward E and Thun MJ: Cancer statistics, 2003. CA Cancer J Clin.
53:5–26. 2003. View Article : Google Scholar
|
|
58.
|
Lo HW and Hung MC: Nuclear EGFR signalling
network in cancers: linking EGFR pathway to cell cycle progression,
nitric oxide pathway and patient survival. Br J Cancer. 94:184–188.
2006. View Article : Google Scholar
|
|
59.
|
Yarden Y: The EGFR family and its ligands
in human cancer. signalling mechanisms and therapeutic
opportunities. Eur J Cancer. 37(Suppl 4): S3–S8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Temam S, Kawaguchi H, El-Naggar AK, et al:
Epidermal growth factor receptor copy number alterations correlate
with poor clinical outcome in patients with head and neck squamous
cancer. J Clin Oncol. 25:2164–2170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Chung CH, Ely K, McGavran L, et al:
Increased epidermal growth factor receptor gene copy number is
associated with poor prognosis in head and neck squamous cell
carcinomas. J Clin Oncol. 24:4170–4176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Kim JH, Xu C, Keum YS, Reddy B, Conney A
and Kong AN: Inhibition of EGFR signaling in human prostate cancer
PC-3 cells by combination treatment with beta-phenylethyl
isothiocyanate and curcumin. Carcinogenesis. 27:475–482. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Thomas GT, Lewis MP and Speight PM: Matrix
metalloproteinases and oral cancer. Oral Oncol. 35:227–233. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Xiao D, Choi S, Lee YJ and Singh SV: Role
of mitogen-activated protein kinases in phenethyl
isothiocyanate-induced apoptosis in human prostate cancer cells.
Mol Carcinog. 43:130–140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Huong LD, Shin JA, Choi ES, et al:
beta-Phenethyl isothiocyanate induces death receptor 5 to induce
apoptosis in human oral cancer cells via p38. Oral Dis. 18:513–519.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Yang YM, Conaway CC, Chiao JW, et al:
Inhibition of benzo(a) pyrene-induced lung tumorigenesis in A/J
mice by dietary N-acetylcysteine conjugates of benzyl and phenethyl
isothiocyanates during the postinitiation phase is associated with
activation of mitogen-activated protein kinases and p53 activity
and induction of apoptosis. Cancer Res. 62:2–7. 2002.
|
|
67.
|
Xiao D and Singh SV: Phenethyl
isothiocyanate-induced apoptosis in p53-deficient PC-3 human
prostate cancer cell line is mediated by extracellular
signal-regulated kinases. Cancer Res. 62:3615–3619. 2002.PubMed/NCBI
|
|
68.
|
Hu R, Kim BR, Chen C, Hebbar V and Kong
AN: The roles of JNK and apoptotic signaling pathways in
PEITC-mediated responses in human HT-29 colon adenocarcinoma cells.
Carcinogenesis. 24:1361–1367. 2003. View Article : Google Scholar : PubMed/NCBI
|