Introduction
The renin-angiotensin system (RAS) is crucial for
cardiovascular regulation (1). In
the RAS, angiotensin-converting enzyme (ACE) metabolizes
angiotensin I (Ang I) to form angiotensin II (Ang II), which exerts
direct trophic actions on cardiac cells through the AT1 receptor,
inducing cardiomyocyte hypertrophy and fibroblast proliferation
(1). Local Ang II production is of
key importance in the pathophysiology of the RAS in the heart
(2). Gradual increases in cardiac
Ang II levels have been demonstrated in experimental models and
clinically during the development of heart failure (3).
In addition to the ACE/Ang II/AT1 receptor axis, the
RAS possesses a counter-regulatory axis comprising ACE2,
Ang-(1-7) and the Mas receptor (4). Ang-(1-7) is
now recognized as a critical component of the RAS, as it exerts a
vast array of actions, many of which oppose the actions of Ang II
(5). Ang-(1-7) is
generated directly from Ang II by ACE2 with high efficiency
(5-7) and also directly from Ang I by neutral
endopeptidase and prolylendopeptidase (8,9).
Recently it was indicated that the heart and blood vessels are the
main targets for the actions of Ang-(1-7)(10).
These actions include biochemical and functional alterations that
result in vasodilation and improved cardiac function (11). It is now well established that the
G protein-coupled receptor Mas, is a functional binding site for
Ang-(1-7)(10,11).
The Mas receptor is present in human cardiomyocytes (12) and mediates most of the known
cardioprotective effects of Ang-(1-7),
such as vasodilatation, anti-fibrosis, -hypertrophic and
-proliferative effects (4). The
Ang-(1-7)/Mas axis has been considered a
potential target for the development of novel cardiovascular
treatment agents (11).
Endothelin-1 (ET-1) is a potent endothelial
cell-derived venous and arterial vasoconstrictor peptide that
functions as a circulating hormone and a paracrine factor in the
regulation of cardiovascular mechanisms (13). ET-1 is responsible for a variety of
cell events, such as contraction, proliferation and apoptosis.
These effects occur following the activation of the endothelin
receptors ETA and ETB, which are present on cardiomyocytes,
fibroblasts, smooth muscle cells, endothelial cells, and glomerular
and tubular cells of the kidney (13). ET-1 levels are increased in
patients with heart disease, particularly in acute myocardial
infarction or congestive heart diseases, as well as in renal
dysfunction (14). In heart
failure, ET-1 levels have been demonstrated to increase in parallel
with the functional capacity and severity of the disease (15,16).
According to previous studies (?), ET-1 and the
Ang-(1-7)/Mas axis are important for cardiac
dysfunction. To the best of our knowledge, the present study
examined, for the first time, the effects of ET-1 on Mas expression
in cultured human cardiomyocytes, aiming to provide indepth
insights into the function of ET-1 and the Ang-(1-7)/Mas
axis in cardiac pathophysiology.
Materials and methods
Reagents
ET-1, Ang-(1-7),
actinomycin D, and kinase inhibitors LY294002, Go6983, PD098059 and
PD169316, were purchased from Sigma (St. Louis, MO, USA).
125I-Sodium iodide (carrier free, 100 mCl/ml) was
purchased from Amersham Biosciences (Piscataway, NJ, USA). In
addition, TRIzol reagent for RNA isolation and the SYBR-Green
Master Mix were purchased from Invitrogen Life Technologies
(Carlsbad, CA, USA) and Applied Biosystems (Foster City, CA, USA),
respectively. Anti-MAS1 (N-15) antibody was purchased from Santa
Cruz Biotechnology, Inc. (sc-54682; Santa Cruz, CA, USA).
Furthermore, anti-phospho-p38 (Thr180/Tyr182) (#9212) and anti-p38
(#8690) antibodies were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). To silence the p38 MAPK gene expression,
siRNA oligonucleotides with the following sequence were designed;
p38 siRNA, 5′-GAAGCTCTCCAGACCATTT-3′. Human Mas promoter-luciferase
reporter construct (#S718157) and LightSwitch assay reagents were
purchased from SwitchGear Genomics (Menlo Park, CA, USA).
Lipofectamine 2000 transfection reagent was purchased from
Invitrogen.
Cell culture and treatment
Human adult cardiomyocytes (no. 6210) and
cardiomyocyte medium (CMM no. 6201), were purchased from ScienCell
Research Laboratories (Carlsbad, CA, USA). The cells were treated
with ET-1 at different concentrations (1, 5, 10, 20 and 30 nM) for
varied time periods (0.5, 1.5, 3, 4.5 or 6 h) in the presence or
absence of BQ123 (1 μM) or BQ788 (1 μM). Actinomycin D and all
kinase inhibitors were dissolved in dimethyl sulfoxide (DMSO; final
concentration, 0.05%). For kinase inhibitor treatment, human
cardiomyocytes were pretreated with the kinase inhibitor for 30 min
and then incubated with the kinase inhibitor and ET-1 (30 nM) for
4.5 h. Human cardiomyocytes treated with ET-1 (30 nM) and DMSO
(0.05%) were used as a control in the experiments. For actinomycin
D treatment, the cells were pretreated with actinomycin D (1 mg/ml)
for 30 min, and then cultured for 1.5, 3 or 4.5 h in medium
containing actinomycin D (1 mg/ml) with or without ET-1 (30 nM).
The cells treated with DMSO (0.05%) were used as a control.
qPCR
RNA was prepared using TRIzol reagent followed by
purification with TURBO DNA-free ™ kit (Ambion, Austin, TX,
USA). The cDNAs were synthesized using SuperScript II reverse
transcriptase (Invitrogen). qPCR was performed on an ABI Prism 7700
Sequence Detection System, with use of the fluorescent dye
SYBR-Green Master Mix (Applied Biosystems) following the
manufacturer’s instructions. The results were normalized against
that of the housekeeping gene glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) in the same sample. The primers used
were as follows: Human Mas, 5′-TTCCGGATGAGAAGAAATCC-3′ (forward)
and 5′-ATGGCCAGAAGAAAGCTCAT-3′ (reverse); and human GAPDH,
5′-GTCAGTGGTGGACCTGACCT-3′ (forward) and 5′-TGCTGTAGCCAAATTCGTTG-3′
(reverse). The mRNA level of treated cells was shown as fold
changes to that of untreated control cells (designated as 1). Each
experiment was repeated three times in triplicate. Results are
expressed as the mean ± standard deviation.
Luciferase assay
Human cardiomyocytes were transfected with human Mas
promoter-luciferase reporter constructs (SwitchGear Genomics) using
Lipofectamine 2000 transfection reagent (Invitrogen) and then
treated with ET-1 (10 or 30 nM) for 4.5 h. Luciferase assays were
performed 24 h later with LightSwitch assay reagents (SwitchGear
Genomics) according to the manufacturer’s instructions. Each
experiment was repeated three times in duplicate. Untreated human
cardiomyocytes were used as a control.
Western blot analysis
Human cardiomyocytes were lysed in 250 μl of 2X
sodium dodecyl sulphate (SDS) loading buffer (62.5 mM Tris HCl, pH
6.8, 2% SDS, 25% glycerol, 0.01% bromphenol blue and 5%
2-mercaptoethanol) and incubated at 95ºC for 10 min. An equal
volume of proteins (100 μg) for each sample was separated by 8–15%
SDS-polyacrylamide gel and blotted onto a polyvinylidene difluoride
microporous membrane (Millipore, Billerica, MA, USA). The membranes
were incubated for 1 h at a 1:1000 dilution of the primary antibody
and then washed and revealed using secondary antibodies with
horseradish peroxidase conjugate (1:5000, 1 h). Peroxidase was
revealed with a GE Healthcare enhanced chemiluminescence kit.
Proteins were quantified prior to being loaded onto the gel.
(125I)Ang-(1-7)
binding assay
Human cardiomyocytes in 12-well plates were rinsed
two times with DMEM and equilibrated on ice with incubation buffer
(DMEM containing 0.2% bovine serum albumin and a protease inhibitor
cocktail, pH 7.4) for 30 min. Subsequently, the plates were
incubated at 4ºC for 60 min with incubation buffer containing 0.5
nmol/l 125I-Ang-(1-7)(17).
Incubation was stopped by rinsing the cells three times with
ice-cold phosphate-buffered saline. Cells were solubilized by
incubation with 0.1 mol/l NaOH for 60 min and the radioactivity was
measured. Non-specific binding was determined in the presence of 10
μmol/l unlabeled Ang-(1-7), which was no higher than 15%. Specific
binding was calculated by the subtraction of non-specific binding
from total binding. The disintegrations per min (dpm) data were
normalized against the cell number (per 20000 cells) and shown as a
percentage of that of untreated control cells (designated as 100%).
Each experiment was repeated three times in triplicates. Results
are expressed as the mean ± standard deviation.
Statistical analysis
Statistical analysis was performed with SPSS
software, for Windows version 10.0 (SPPS, Inc., Chicago, IL, USA).
Data values were expressed as the mean ± standard deviation.
Comparison of means among multiple groups was performed with
one-way analysis of variance (ANOVA) followed by post-hoc pairwise
comparisons using Tukey’s tests. The significance level of this
study was set at a two-tailed P=0.05.
Results
Relative Mas mRNA levels in human
cardiomyocytes in the presence of ET-1 with or without ET receptor
blockers
Human cardiomyocytes were treated with ET-1 at
different concentrations (1, 5, 10, 20 and 30 nM) for varied time
periods (0.5, 1.5, 3, 4.5 or 6 h). The Mas mRNA levels were
examined using qPCR. The Mas mRNA level of treated cells was shown
as fold changes to that of untreated control cells (designated as
1). As shown in Table I, ET-1
concentration of 5–20 nM, decreased the Mas mRNA level in a
statistically significant dose- and time-dependent manner within
4.5 h of treatment, which was completely eliminated by selective
ETA receptor blocker BQ123, but not by the selective ETB receptor
blocker BQ788. ET-1 at 1 nM had no significant effects on the Mas
mRNA levels at all time points. Additionally, treatment for 0.5 h
with ET-1 at 1–30 nM also showed no significant effects on the Mas
mRNA level.
 | Table IRelative Mas mRNA levels in human
cardiomyocytes in the presence of endothelin-1 (ET-1) with or
without ET receptor blockers. |
Table I
Relative Mas mRNA levels in human
cardiomyocytes in the presence of endothelin-1 (ET-1) with or
without ET receptor blockers.
| Time (h) |
|---|
|
|
|---|
| ET-1 (nm) | 0.5 | 1.5 | 3 | 4.5 | 6 |
|---|
| 1 | 0.99±0.02 | 1.01±0.03 | 0.98±0.02 | 0.98±0.03 | 0.96±0.04 |
| 5 | 0.99±0.02 | 0.87±0.03a,d | 0.59±0.04a,d,e | 0.38±0.05a,d,e,f | 0.38±0.04a,d,e,f |
| 10 | 0.98±0.05 | 0.52±0.05a,b,d | 0.37±0.03a,b,d,e | 0.26±0.03a,b,d,e,f | 0.25±0.02a,b,d,e,f |
| 20 | 0.97±0.04 | 0.39±0.05a,b,c,d | 0.25±0.03a,b,c,d,e | 0.14±0.05a,b,c,d,e,f | 0.14±0.03a,b,c,d,e,f |
| 30 | 0.96±0.03 | 0.35±0.04a,b,c,d | 0.20±0.04a,b,c,d,e | 0.11±0.02a,b,c,d,e,f | 0.10±0.04a,b,c,d,e,f |
| 30+BQ123 (1
μM) | 1.05±0.04 | 1.05±0.05 | 1.07±0.03 | 1.08±0.07 | 1.10±0.03 |
| 30+BQ788 (1
μM) | 0.96±0.06 | 0.38±0.05a,b,c,d | 0.25±0.04a,b,c,d,e | 0.15±0.03a,b,c,d,e,f | 0.13±0.05a,b,c,d,e,f |
Effects of ET-1 on human Mas promoter
activities and on Mas mRNA level
To evaluate the effects of ET-1 on Mas mRNA
stability, human cardiomyocytes were pretreated with transcription
inhibitor actinomycin D (1 mg/ml) for 30 min and then cultured for
1.5, 3 or 4.5 h in medium containing actinomycin D (1 mg/ml), with
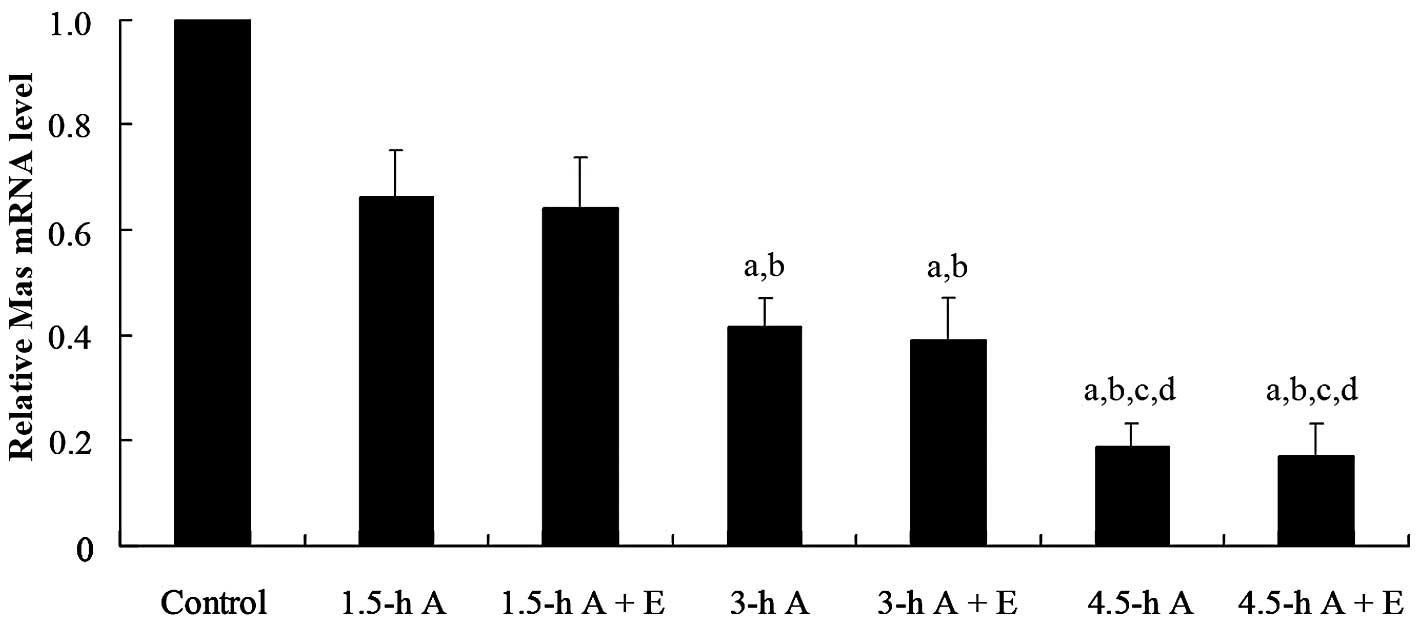
or without ET-1 (30 nM). qPCR assays showed that the Mas mRNA level
significantly decreased with time following actinomycin D treatment
(Fig. 1). In the presence of
actinomycin D, ET-1 had no significant effects on the Mas mRNA
level at any of the time points (Fig.
1). The results suggest that ET-1 decreases Mas expression at
the transcriptional level rather than decreasing Mas mRNA stability
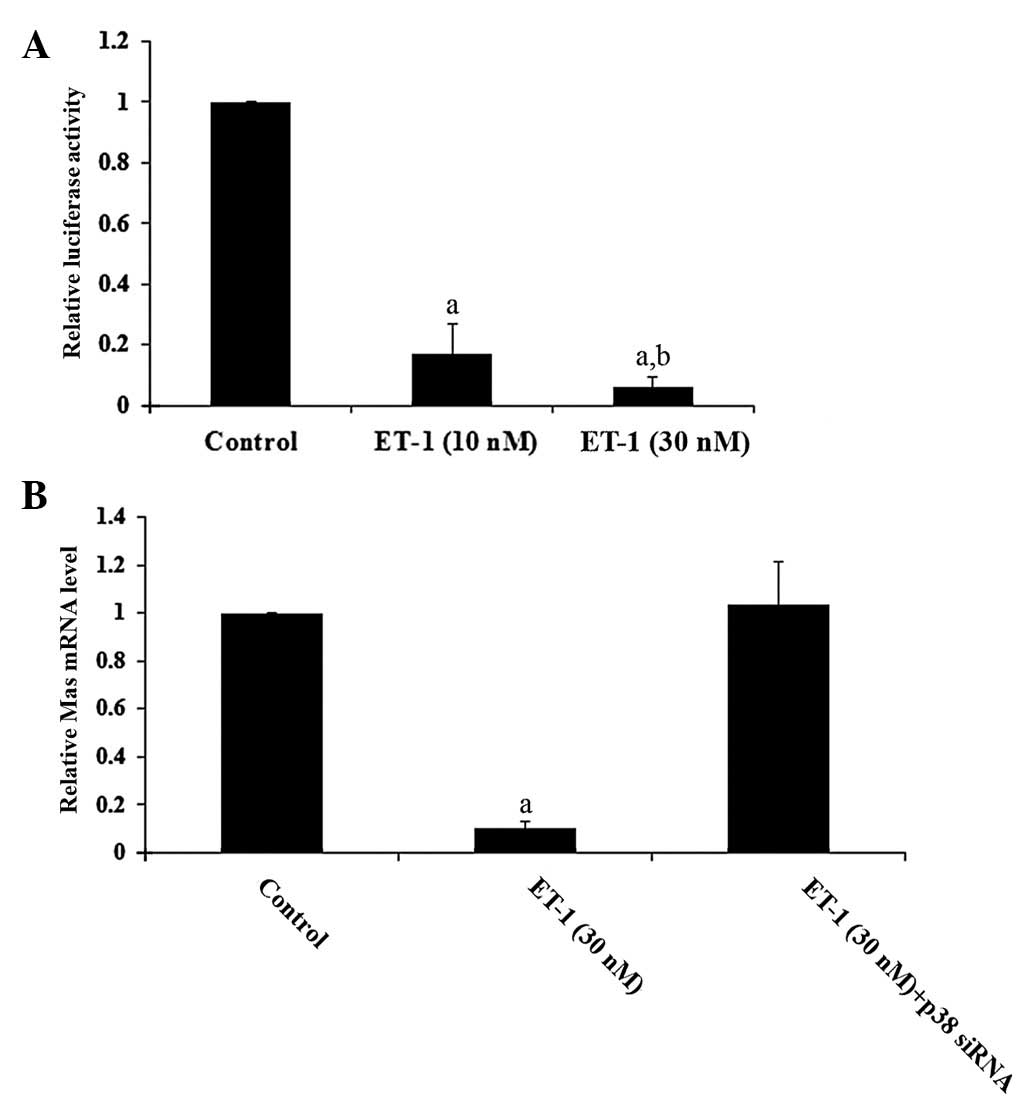
at the post-transcriptional level. This was confirmed by luciferase
assays, which showed that ET-1 had a dose-dependent inhibitory
effect on human Mas promoter activity (Fig. 2A).
To determine the signaling pathways involved in the
inhibitory effects of ET-1 on Mas gene transcription, we examined
the Mas mRNA levels in human cardiomyocytes treated with ET-1 (30
nM) with or without different kinase inhibitors, for 4.5 h. As
shown in Table II, inhibition of
protein kinase C (Go6983; 250 nM), MAPK (PD098059; 25 μM) and p38
kinase (LY294002; 25 μM), had no significant effects on the Mas
mRNA level. By contrast, the inhibition of p38 MAPK by selective
inhibitor PD169316 (25 μM) completely eliminated the inhibitory
effects of ET-1 on Mas gene transcription. We also employed siRNA
to knock down p38 MAPK in human cardiomyocytes. As shown in
Fig. 2B, while ET-1 (30 nM)
decreased the Mas mRNA level by >90%, there were no significant
effects on cardiomyocytes transfected with p38 (MAPK) siRNA,
confirming that p38 MAPK signaling is key to the inhibitory effects
of ET-1 on Mas gene transcription.
| Table IIRelative Mas mRNA levels in human
cardiomyocytes in the presence of endothelin-1 (ET-1) with or
without kinase inhibitors. |
Table II
Relative Mas mRNA levels in human
cardiomyocytes in the presence of endothelin-1 (ET-1) with or
without kinase inhibitors.
| Treatment | Relative Mas mRNA
level |
|---|
| Control | 0.11±0.02a |
| +LY294002 (25
μM) | 0.21±0.12a |
| +Go6983 (250
nM) | 0.18±0.06a |
| +PD098059 (25
μM) | 0.20±0.08a |
| +PD169316 (25
μM) | 1.11±0.07 |
Western blot analysis
Western blot analysis showed that ET-1 treatment for
4.5 h dose-dependently decreased the Mas protein level in human
cardiomyocytes, which was blocked by BQ123 and PD169316, but not by
BQ788 (Fig. 3). Similarly, human
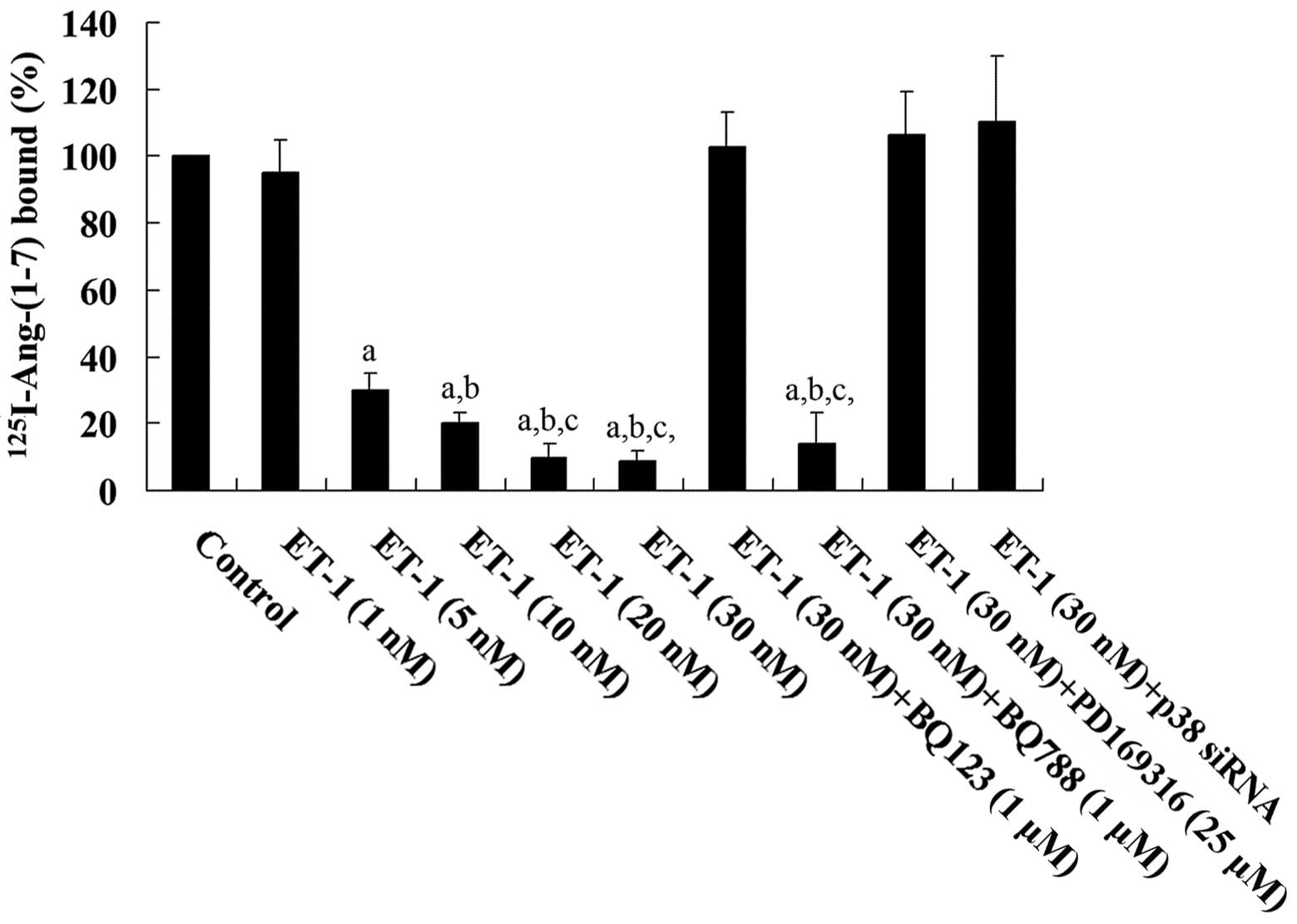
cardiomyocytes treated with ET-1 (30 nM) for 4.5 h showed a
dose-dependent decrease in Ang-(1-7)
binding on the cell membrane, which was blocked by BQ123 and
PD169316, but not by BQ788 (Fig.
4). A previous study demonstrated that the phosphorylation and
activation of p38 MAPK is involved in ET-1-induced biological
effects (18). As shown in
Fig. 5, western blot analysis
showed that ET-1 treatment for 4.5 h, dose-dependently increased
phosphorylated p38 MAPK (Thr180/Tyr182) levels in human
cardiomyocytes, which was blocked by BQ123, PD169316 and p38 MAPK
siRNA, but not by BQ788. Fig. 5
also shows that the p38 siRNA knocked down >70% of endogenous
p38 MAPK expression. Taken together, the results suggest that ET-1
significantly decreased the density of ligand-binding Mas receptor
on the cell membrane of human cardiomyocytes by downregulating Mas
gene transcription via the ETA receptor by a p38 MAPK-dependent
mechanism.
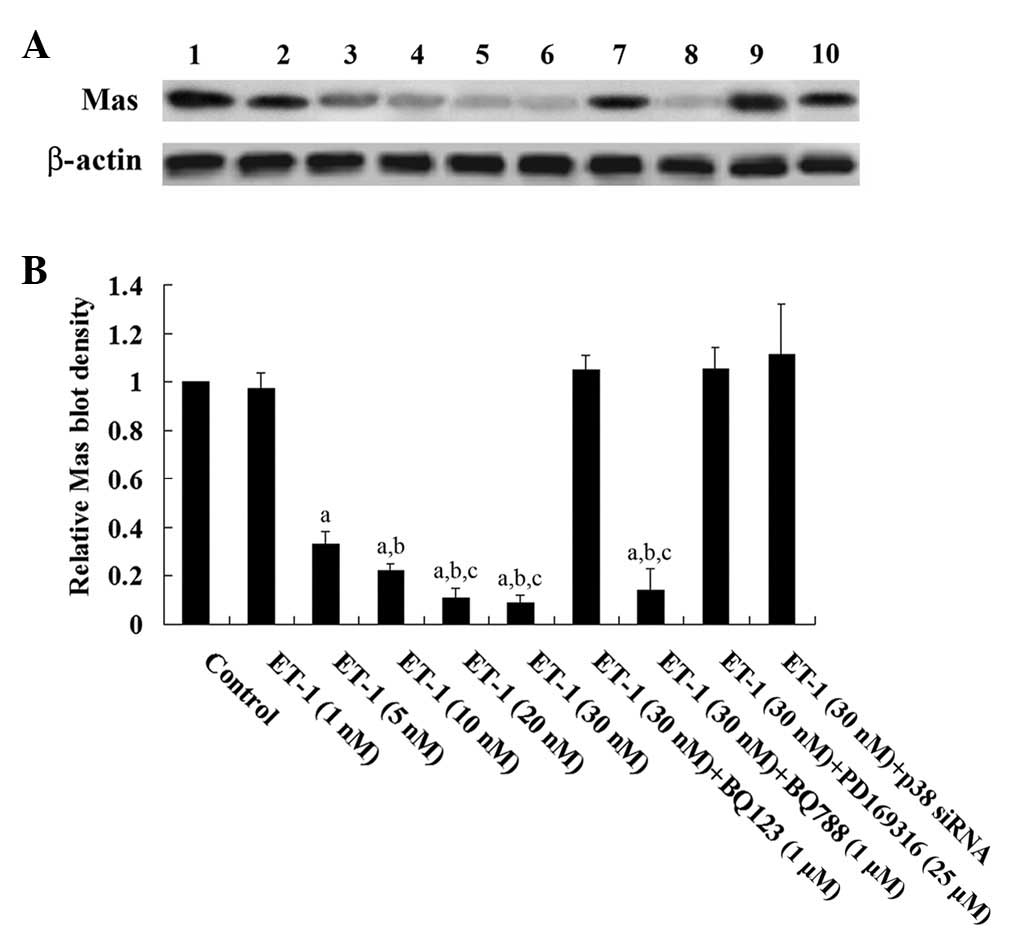 | Figure 3Western blot analysis of Mas
expression in human cardiomyocytes. (A) Human cardiomyocytes were
treated with ET-1 (1, 5, 10, 20 and 30 nM) with or without BQ123 (1
μM), BQ788 (1 μm), PD169316 (25 μm) or p38 mitogen-activated
protein kinase siRNA for 4.5 h. Twenty-four hours later, cell
lysates were subject to western blot analysis for Mas expression.
Lysates from untreated human cardiomyocytes were used as a control
(lane 1). Concentrations of ET-1 were as follows: Lane 2, 1 nM;
lane 3, 5 nM; lane 4, 10 nM; lane 5, 20 nM); lane 6, 30 nM; lane 7,
30 nM and BQ123 (1 μM); lane 8, 30 nM and BQ788 (1 μM); lane 9, 30
nM and PD169316 (25 μM) and lane 10, 30 nM and p38 siRNA. β-actin
blotting was used as a loading control. (B) Mas and β-actin blots
were measured by densitometry. The density of the Mas blot was
normalized against that of β-actin to obtain a relative density,
which was expressed as fold changes to the relative Mas density of
untreated control cells (designated as 1). aP<0.05
compared with untreated control cells; bP<0.05
compared with 5 nm ET-1 treatment andcP<0.05
compared with 10 nm ET-1 treatment. |
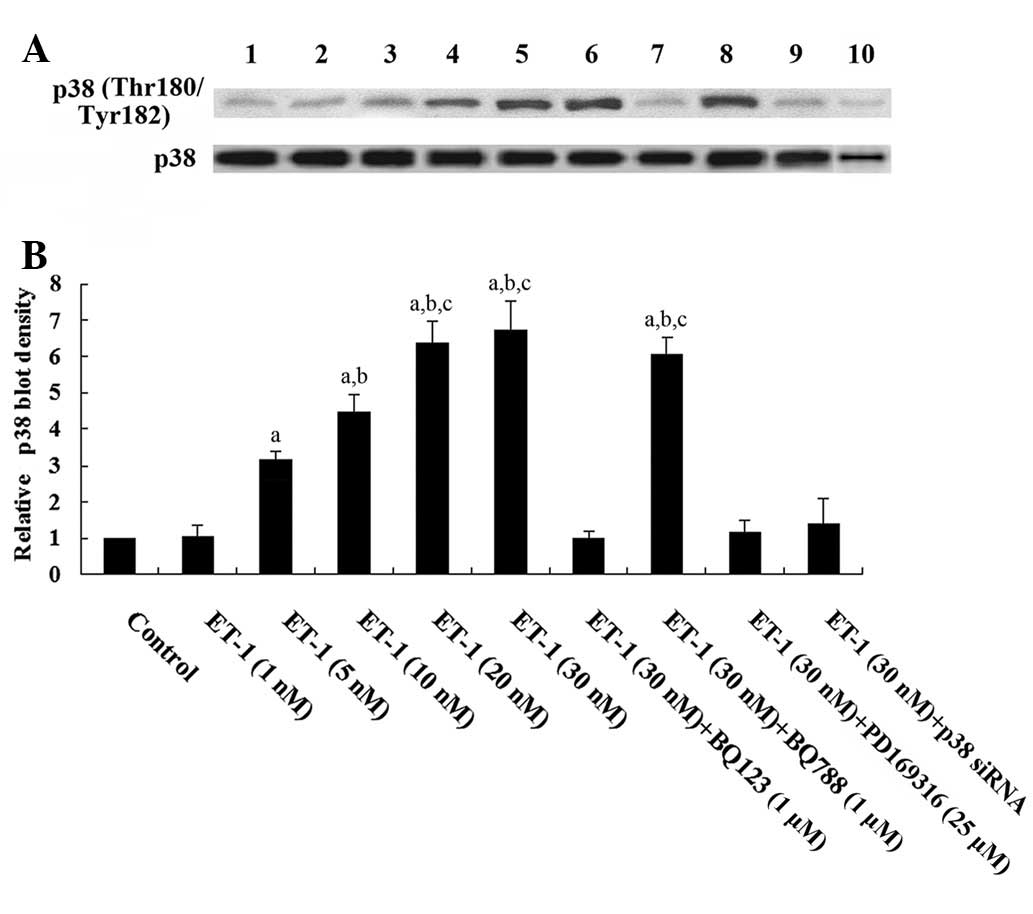 | Figure 5Western blot analysis of
phosphorylated p38 mitogen-activated protein kinase (MAPK) levels
in human cardiomyocytes. (A) Human cardiomyocytes were treated with
ET-1 (1, 5, 10, 20 and 30 nM) with or without BQ123 (1 μM), BQ788
(1 μM), PD169316 (25 μM) or p38 MAPK siRNA for 4.5 h. Lysates from
untreated human cardiomyocytes were used as a control (lane 1).
Concentrations of ET-1 were as follows: Lane 2, 1 nM; lane 3, 5 nM;
lane 4, 10 nM; lane 5, 20 nM; lane 6, 30 nM; lane 7, 30 nM and
BQ123 (1 μM); lane 8, 30 nM and BQ788 (1 μM); lane 9, 30 nM and
PD169316 (25 μM); lane 10, 30 nM and p38 siRNA. β-actin blotting
was used as a loading control. (B) Phosphorylated p38
(Thr180/Tyr182) and total p38 MAPK levels were measured by
densitometry. The density of the phosphorylated p38 (Thr180/Tyr182)
(pp38) MAPK blot was normalized against that of total p38 MAPK
levels to obtain a relative density, which was expressed as fold
changes to that of untreated control cells (designated as 1).
aP<0.05 compared with untreated control cells;
bP<0.05 compared with 5 nM ET-1 treatment and
cP<0.05 compared with 10 nM ET-1 treatment. |
Discussion
Chronic myocardial stimulation by Ang II, the
effector of the RAS, results in cardiac dysfunction (1). It is now accepted that the
ACE2/Ang-(1-7)/Mas axis is able to counteract most of
the deleterious actions of the ACE/Ang II/AT1 receptor axis,
particularly in pathological conditions (11). ET-1, an important circulating
hormone and a paracrine factor, is also involved in cardiac
dysfunction (19). The present
study was the first to demonstrate that ET-1 downregulates Mas
expression in human cardiomyocytes.
ET-1 is released from cardiomyocytes and affects
cardiac functions by either autocrine or paracrine mechanisms
(20). A selective ETA receptor
blocker, but not ETB receptor blocker, completely eliminated the
effects of ET-1, indicating that ET-1 inhibited Mas expression via
the ETA receptor. In addition, a selective p38 MAPK inhibitor or
siRNA completely blocked the inhibitory effects of ET-1, indicating
that ET-1 downregulates Mas expression by a p38 MAPK-dependent
mechanism via the ETA receptor. Additional studies are needed to
elaborate on the mechanisms of how ET-1 regulates Mas expression
through p38 MAPK signaling.
The present study demonstrated that 10 nM ET-1
inhibited Mas gene transcription by ~75% within 4.5 h, suggesting
that ET-1 is a strong negative regulator of the ACE2/Ang-(1-7)/Mas
axis. This finding supports results of a previous study whereby
ET-1 treatment at 10 nM for 12 h reduced ACE2 mRNA by ~60% in
cardiomyocytes (21). According to
the findings of the present study, Mas is regulated more readily
and may serve as a better therapeutic target compared with ACE2 in
cardiomyocytes, as it responds to the ET-1 treatment more rapidly
and to a greater extent. Thus alterations in the Mas expression
level may be an acute cardiac response to the pathophysiological
conditions, such as elevated plasma ET-1 levels.
Mas mediates most of the known cardioprotective
effects of Ang-(1-7)(4).
Elevated ET-1 levels may promote cardiac dysfunction by inhibiting
ACE2 (21) and Mas expression,
this will result in an increased concentration of Ang II, a
recognized mediator of cardiomyocyte and cardiac dysfunction
(1); a decreased concentration of
Ang-(1-7), the protective peptide for
cardiomyocytes, mainly by antagonizing the effects of Ang II; and a
decreased Ang-(1-7)/Mas signaling due to a markedly reduced
Mas expression. As the inhibitory effects of ET-1 on Mas expression
is mediated through the ETA receptor, a selective ETA blocker may
be beneficial for patients with cardiac dysfunction, which would be
an interesting topic for future in vivo studies.
A number of studies have substantially supported the
importance of the ACE2/Ang-(1-7)/Mas
axis in renal function (22). As
ET-1 is also involved in renal dysfunction (14), the regulatory effects of ET-1 on
Mas expression would constitute an interesting topic in the field
of kidney disease.
In conclusion, the present study has demonstrated
that ET-1 downregulates Mas expression at the transcription level
in human cardiomyocytes via the ETA receptor by a p38
MAPK-dependent mechanism. This study provides novel insights into
the function of ET-1 and the Ang-(1-7)/Mas
axis in cardiac pathophysiology.
References
|
1
|
Domenighetti AA, Wang Q, Egger M, Richards
SM, Pedrazzini T and Delbridge LM: Angiotensin II-mediated
phenotypic cardiomyocyte remodeling leads to age-dependent cardiac
dysfunction and failure. Hypertension. 46:426–432. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mazzolai L, Pedrazzini T, Nicoud F,
Gabbiani G, Brunner HR and Nussberger J: Increased cardiac
angiotensin II levels induce right and left ventricular hypertrophy
in normotensive mice. Hypertension. 35:985–991. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Domenighetti AA, Ritchie M, Smyth G,
Pedrazzini T, Proietto J and Delbridge LMD: Gene expression
profiling reveals distinct sets of genes altered during hormonally
and metabolically induced cardiac hypertrophies. J Mol Cell
Cardiol. 37:3032004.
|
|
4
|
Ferreira AJ, Murça TM, Fraga-Silva RA,
Castro CH, Raizada MK and Santos RA: New cardiovascular and
pulmonary therapeutic strategies based on the
Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor
axis. Int J Hypertens. 2012:1478252012.PubMed/NCBI
|
|
5
|
Raizada MK and Ferreira AJ: ACE2: a new
target for cardiovascular disease therapeutics. J Cardiovasc
Pharmacol. 50:112–119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vickers C, Hales P, Kaushik V, et al:
Hydrolysis of biological peptides by human angiotensin-converting
enzyme-related carboxypeptidase. J Biol Chem. 277:14838–14843.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Der Sarkissian S, Huentelman MJ, Stewart
J, Katovich MJ and Raizada MK: ACE2: a novel therapeutic target for
cardiovascular diseases. Prog Biophys Mol Biol. 91:163–198.
2006.PubMed/NCBI
|
|
8
|
Rice GI, Thomas DA, Grant PJ, Turner AJ
and Hooper NM: Evaluation of angiotensin-converting enzyme (ACE),
its homologue ACE2 and neprilysin in angiotensin peptide
metabolism. Biochem J. 383:45–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stanziola L, Greene LJ and Santos RA:
Effect of chronic angiotensin converting enzyme inhibition on
angiotensin I and bradykinin metabolism in rats. Am J Hypertens.
12:1021–1029. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gomes ER, Santos RA and Guatimosim S:
Angiotensin-(1-7)- mediated signaling in cardiomyocytes. Int J
Hypertens. 2012:4931292012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ferreira AJ and Santos RA: Cardiovascular
actions of angiotensin-(1-7). Braz J Med Biol Res. 38:499–507.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang T, Li Z, Dang H, et al: Inhibition
of Mas G-protein signaling improves coronary blood flow, reduces
myocardial infarct size, and provides long-term cardioprotection.
Am J Physiol Heart Circ Physiol. 302:H299–H311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vignon-Zellweger N, Heiden S, Miyauchi T
and Emoto N: Endothelin and endothelin receptors in the renal and
cardiovascular systems. Life Sci. 91:490–500. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shichiri M, Hirata Y, Ando K, et al:
Plasma endothelin levels in hypertension and chronic renal failure.
Hypertension. 15:493–496. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei CM, Lerman A, Rodeheffer RJ, et al:
Endothelin in human congestive heart failure. Circulation.
89:1580–1586. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yazici M, Demircan S, Durna K and Sahin M:
The relation between endothelin-1 levels and myocardial injury in
chronic ischemic heart failure. Heart Vessels. 20:95–99. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gironacci MM, Adamo HP, Corradi G, Santos
RA, Ortiz P and Carretero OA: Angiotensin (1-7) induces MAS
receptor internalization. Hypertension. 58:176–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Windischhofer W, Zach D, Fauler G,
Raspotnig G, Köfeler H and Leis HJ: Involvement of Rho and p38 MAPK
in endothelin-1-induced expression of PGHS-2 mRNA in
osteoblast-like cells. J Bone Miner Res. 17:1774–1784. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohmae M: Endothelin-1 levels in chronic
congestive heart failure. Wien Klin Wochenschr. 123:714–717. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gallagher PE, Ferrario CM and Tallant EA:
Regulation of ACE2 in cardiac myocytes and fibroblasts. Am J
Physiol Heart Circ Physiol. 295:H2373–H2379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Jonge HW, Dekkers DH, Houtsmuller AB,
Sharma HS and Lamers JM: Differential signaling and hypertrophic
responses in cyclically stretched vs endothelin-1 stimulated
neonatal rat cardiomyocytes. Cell Biochem Biophys. 47:21–32.
2007.PubMed/NCBI
|
|
22
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1-7) and
Mas: new players of the renin angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar : PubMed/NCBI
|