Introduction
P-selectin (CD62P) is a member of the selectin
family of cell adhesion molecules located in the platelet granule
and Weibel-Palade body of endothelial cells. P-selectin mediates
the rolling of blood cells on the surface of the endothelium and
initiates the attachment of leukocytes circulating in the blood to
platelets, endothelial cells and other leukocytes at sites of
tissue injury and inflammation (1). These processes are possible since
P-selectin glycoprotein ligand-1 (PSGL-1), the ligand for
P-selectin, is present on the surface of platelets and endothelial
cells which undergo sulfation of a specific tyrosine residue at the
N-terminal for P-selectin recognition (2,3).
Soluble P-selectin, lacking the cytosolic/transmembrane domain, has
been identified as circulating in plasma in healthy controls and
diseased cohorts. An increased level of soluble P-selectin
(sP-selectin) is a major predictive factor of cardiovascular events
relating to platelet turnover and its activation and function.
P-selectin plays a key role in diseases associated with injury and
arterial thrombosis. Increased expression of P-selectin is observed
in coronary artery disease, acute myocardial infarction (AMI),
stroke and peripheral artery diseases (4,5).
Inflammation plays a significant role in numerous
chronic diseases, including atherosclerosis-associated
cardiovascular disease. The adhesion of immune cells plays a
critical role in the inflammatory response and indeed the
pathophysiology of inflammatory diseases. P-selectin is critical in
the progression of atherosclerosis as evidenced by knockout animal
models, where P-selectin knockout mice crossed with ApoE-deficient
mice exhibited significantly reduced atherosclerosis and leukocyte
recruitment in the plaque. sP-selectin also has pro-atherogenic and
pro-thrombotic effects [as reviewed in (6)]. More than 80% of acute myocardial
infarcts are the result of coronary atherosclerosis with
superimposed luminal thrombus and its rupture. Uncommon causes of
myocardial infarction include coronary spasm, coronary embolism and
thrombosis in nonatherosclerotic normal vessels (7). These findings led us to hypothesize
that sP-selectin-associated AMI is induced by coronary
atherosclerosis formation and rupture.
As the primary adhesion molecule in initiating cell
activation and cell adhesion to platelets and endothelial cells,
and having a critical role in inducing chronic inflammatory and
cardiovascular diseases, P-selectin is an attractive therapeutic
target in vascular diseases. However, the basic tenet of targeting
P-selectin may be complicated by the presence of a soluble form of
P-selectin; thus, the targeting of P-selectin remains a strong
clinical candidate for developing novel therapeutic strategies
(8). Percutaneous coronary
intervention (PCI) and thrombolysis are the two main therapeutic
strategies for AMI (9). Since the
two strategies are used to remove the obstruction of the coronary
artery to prevent AMI, there is a possibility that PCI or
thrombolysis could inhibit sP-selectin to a certain extent.
sP-selectin is increased in AMI and atherosclerosis patients, which
proves only that sP-selectin is correlated with the onset of AMI.
However, it remains unclear whether sP-selectin also affects the
severity of AMI.
In the present study, we investigated P-selectin in
50 patients and the role of sP-selectin by administration of
P-selectin antagonist in two mouse models (atherosclerosis model
and neointimal formation model). We demonstrated that sP-selectin
only promoted atherosclerotic plaque and its rupture, and
neointimal formation and bleeding, then induced the onset of AMI
but did not affect its severity.
Materials and methods
Human sample collection
For human analysis, baseline plasma samples were
obtained from 50 male cohorts from the Fourth Affiliated Hospital
of Harbin Medical University (Harbin, China). Ages ranged from
42–67 years. These comprised 15 control cohorts, who had no any
cardiovascular disease; 15 chest pain cohorts who were diagnosed
with angina; 10 cohorts who were diagnosed with AMI and finally
underwent PCI therapy; and 10 cohorts who were diagnosed with AMI
and finally underwent thrombolysis therapy. Written informed
consent was obtained from the patients. For the 20 AMI cohorts, the
therapeutic choice of PCI or thrombolysis therapy was voluntary.
None of the individuals involved in this experiment suffered from
any sP-selectin-induced disease, such as chronic obstructive
pulmonary disease, rheumatoid arthritis and dermatitis, in which
sP-selectin is reportedly increased (10–12).
Human background and plasma
measurement
For all cohorts, the body mass index (BMI) and waist
circumference (WC) were measured as a routine examination.
Pericardial adipose tissue (PCAT) volume was measured using
64-slice computed tomography and then calculated
three-dimensionally using contrast-enhanced images, as reported
previously (9).
Plasma sP-selectin, total cholesterol (TC),
low-density lipoprotein (LDL), high-density lipoprotein (HDL),
tumor necrosis factor (TNF)-α, adiponectin, creatine kinase (CK)-MB
and cardiac troponin I (cTnl) were measured by a specific ELISA kit
(MyBioSource, Inc., San Diego, CA, USA). The basic data are shown
in Table I.
 | Table ICharacteristics of patients. |
Table I
Characteristics of patients.
| Control (n=15) | Chest pain
(n=15) | PCI (n=10) | Thrombolysis
(n=10) |
|---|
| Gender | Male | Male | Male | Male |
| Age | 49±7 | 52±8 | 55±12 | 56±9 |
| sP-selectin
(ng/ml) | 101±20 | 120±29 | 156±57 | 166±76 |
| BMI | 24.4±3.4 | 26.7±3.6 | 30.2±4.0 | 28.1±3.6 |
| WC (cm) | 80±9.7 | 81±8.5 | 92.3±9.3 | 97.1±8.5 |
| PCAT | 154±14 | 162±16 | 169±16 | 162±17 |
| TC (mmol/l) | 4.02±0.66 | 4.39±0.73 | 5.29±1.30 | 5.62±1.51 |
| LDL (mmol/l) | 2.14±0.64 | 2.98±0.77 | 3.44±1.03 | 3.43±1.25 |
| HDL (mmol/l) | 1.51±0.28 | 1.15±0.39 | 1.07±0.24 | 1.15±0.34 |
| TNF-α (ng/ml) | 0.83±0.41 | 0.73±0.35 | 1.02±0.38 | 1.22±0.39 |
| Adiponectin
(μg/ml) | 7.06±1.77 | 7.39±1.5 | 5.89±1.67 | 6.52±2.45 |
| CK-MB (U/l) | - | - | 54.8±14.4 | 44.2±13.8 |
| cTnl (μg/l) | - | - | 2.23±1.08 | 2.88±0.75 |
Animal models
To generate the atherosclerotic formation and plaque
rupture models, male ApoE−/− mice (Biocytogen
Co., Ltd., Beijing, China), aged 12 weeks, were fed a
high-cholesterol diet for 5 and 10 weeks (n=12 each), as described
previously (13,14).
To generate the neointimal hyperplasia and
neointimal bleeding model, the bilateral femoral arteries of
16-week-old C57BL/6N mice were subject to transluminal wire injury
as described previously (15). The
neointimal hyperplasia mice were fed a normal diet and the
neointimal bleeding mice were fed a high-cholesterol diet for 5
weeks. All mice had ad libitum access to water. The present
study was approved by the ethics committee of Harbin Medical
University.
Antibody administration
In the atherosclerotic group, each mouse was given a
single bolus of 100 μg RB40.34 mAb (Thermo Fisher Scientific Inc.,
Waltham, MA, USA) via IP injection once a week for 5 weeks.
In the neointimal hyperplasia group, three hours
before femoral arterial injury, each mouse was given a single bolus
of 100 μg RB40.34 mAb via IP injection with the operator blinded to
treatment (16).
Tissue staining and the evaluation of
plaque and neointimal hyperplasia
Anesthetized mice were perfused with
phosphate-buffered saline (PBS) through the left cardiac ventricle,
and then tissues were fixed by perfusion of 4% paraformaldehyde
containing PBS. For neutral lipid visualization, sections were
rinsed in 60% isopropanol and incubated in a saturated, filtered
solution of Oil Red O in 60% isopropanol for 1 h. Elastica van
Gieson staining was performed as reported previously (17).
Statistical analysis
Data are expressed as the means ± standard error of
the mean of triplicate runs. Each experiment was repeated at least
three times. Student’s t-test and analysis of variance were used to
assess differences, and P<0.05 was considered to indicate a
statistically significant difference.
Results
sP-selectin is increased in obesity and
hyperlipidemia patients
Since sP-selectin, which induces the adhesion of
platelet and endothelium cells, is reported to be an inflammatory
factor, and obesity is a well-known cause of classic chronic
systemic inflammation, the BMI and WC indices are often used to
evaluate obesity (18). Therefore,
we firstly investigated the correlation of sP-selectin and obesity
using these two obesity indices in all 50 patients, regardless of
their AMI status. sP-selectin levels were found to be 90±14, 128±35
and 57±54 ng/ml, respectively, in the three BMI subgroups
(BMI<25, 25<BMI<30 and BMI>30; Fig. 1A), in which 25 and 30 were
excluded, and sP-selectin levels were 108±27 and 158±59 ng/ml,
respectively, in the two WC subgroups (WC<80 cm and WC>80 cm;
Fig. 1B; excluding 80 cm). In
previous studies, PCAT was reported to have a higher correlation
with coronary heart disease than abdominal obesity (13,20).
We therefore supposed that sP-selectin was also correlated with
PCAT. It was demonstrated that sP-selectin was 148±55 ng/ml in PCAT
areas over 160 cm2, which was higher than the 110±41
ng/ml measured in areas less than 160 cm2 (Fig. 1C) 160 cm2 itself was not
assessed.
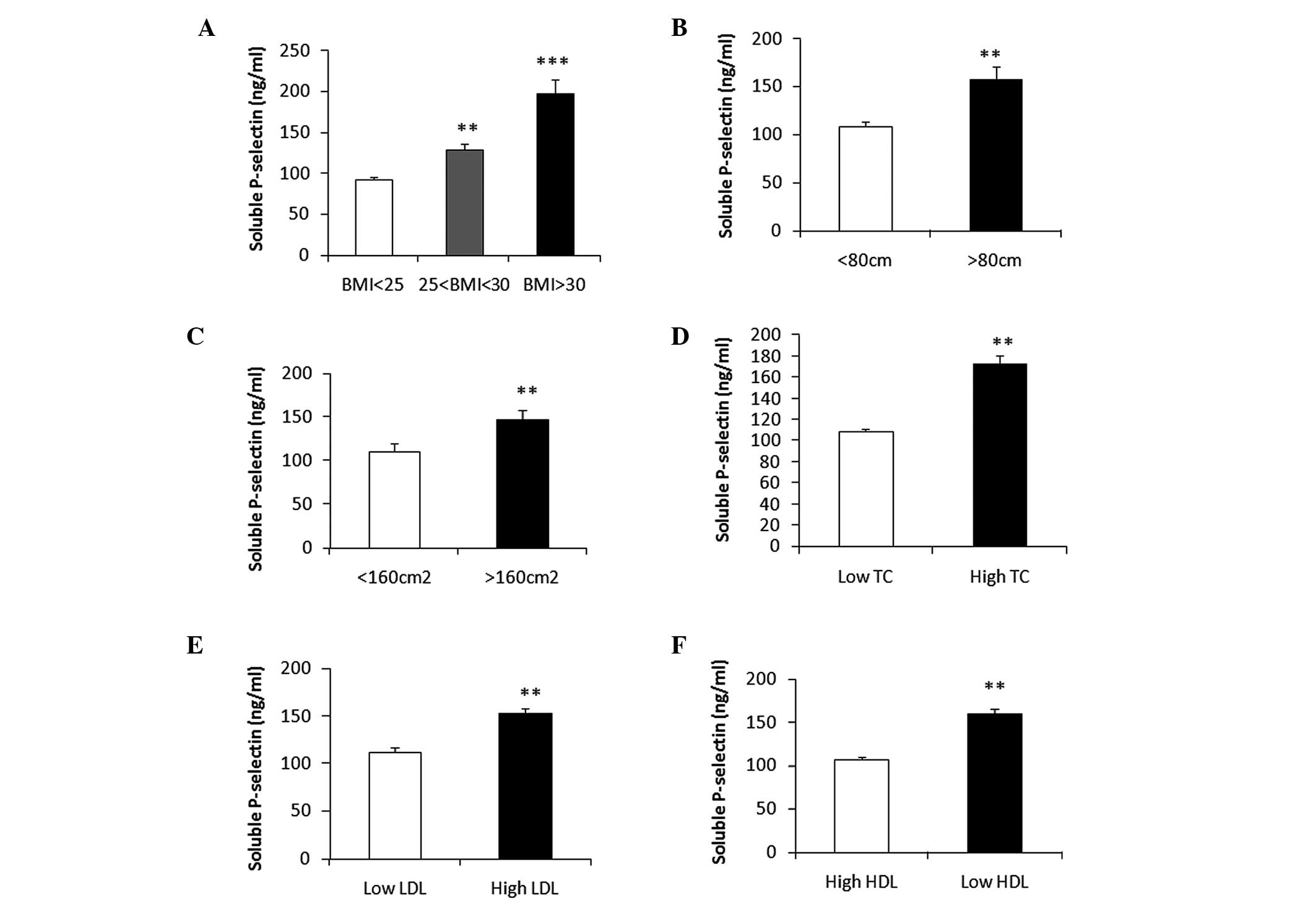 | Figure 1Soluble P-selectin (sP-selectin) is
increased in patients with obesity and hyperlipidemia. Plasma
concentration of sP-selectin in (A) three body mass index (BMI)
groups (BMI<25, n=17; 25<BMI<30, n=22; BMI>30, n=11),
(B) two waist circumference (WC) groups (WC<80 cm, n=29;
WC>80 cm, n=21) and (C) two pericardial adipose tissue (PCAT)
area groups (PCAT<160 cm2, n=22; PCAT>160
cm2, n=28), in all the controls, angina patients and
acute myocardial infarction patients before percutaneous coronary
intervention or thrombolysis therapy. Plasma concentration of
soluble P-selectin in low and high (D) total cholesterol (TC), (E)
low-density lipoprotein (LDL) and (F) high-density lipoprotein
(HDL) groups (TC: low TC<5 ng/ml, high TC>5 ng/ml; LDL: low
LDL<3 ng/ml, high LDL>3 ng/ml; HDL: low HDL<1.05 ng/ml;
high HDL>1.05 ng/ml). *P<0.05 and
**P<0.01, compared with the white bar; data are
presented as the mean ± SEM. |
Obesity may lead to hyperlipidemia, which is a high
risk factor for myocardial infarction, so next we confirmed the
correlation between sP-selectin and hyperlipidemia. We revealed
that sP-selectin levels were significantly higher in high TC
(6.37±0.24 mmol/l) patients than in low TC (4.18±0.11 mmol/l)
patients (173±15 vs. 109±5 ng/ml; Fig.
1D), sP-selectin levels were significantly higher in high LDL
(3.83±0.12 mmol/l) patients than in low LDL (2.12±0.16 mmol/l)
patients (153±10 vs.113±9 ng/ml; Fig.
1E), and sP-selectin levels were significantly higher in low
HDL (0.94±0.04 mmol/l) patients than in high HDL (1.49±0.05 mmol/l)
patients (160±12 vs. 107±6 ng/ml; Fig.
1F). In conclusion, sP-selectin levels were significantly
higher in individuals with obesity (abdominal and pericardial
obesity) and hyperlipidemia.
sP-selectin is positively correlated with
chronic inflammation
As we have confirmed that sP-selectin is increased
in individuals with obesity and hyperlipidemia, and it has been
reported that obesity and hyperlipidemia lead to a chronic
inflammation state (21), we
hypothesized that sP-selectin was also correlated with chronic
inflammation. To confirm this, we measured the serum TNF-α and
adiponectin levels. It was demonstrated that sP-selectin was
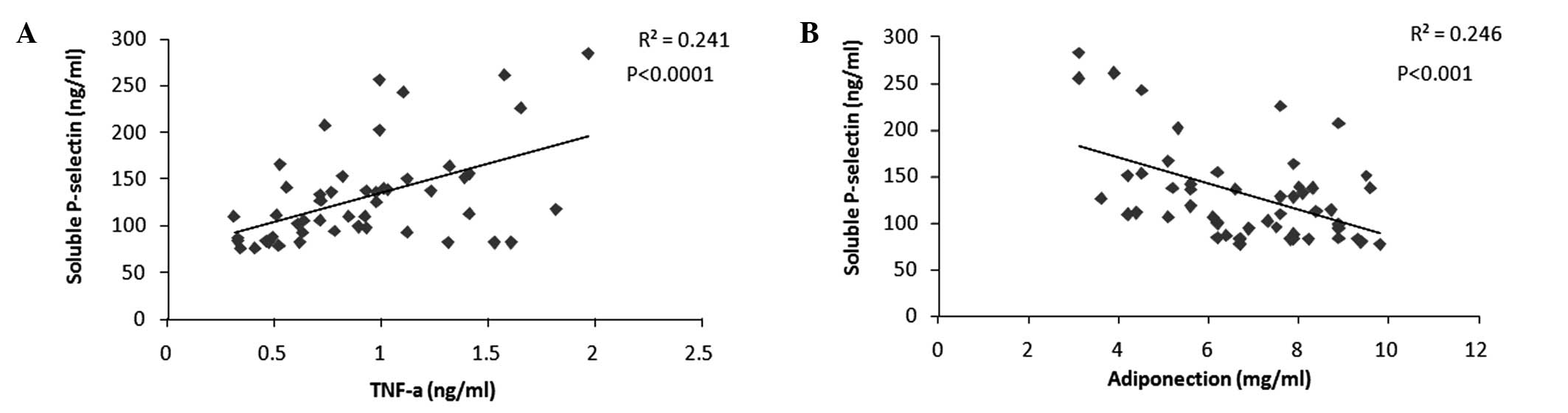
positively correlated with serum TNF-α (Fig. 2A) and negatively correlated with
serum adiponectin (Fig. 2B).
The above results suggest that chronic inflammation
associated with obesity and hyperlipidemia may induce an increase
in sP-selectin levels. The increased sP-selectin may play a
significant role in certain inflammation-induced diseases.
sP-selectin is correlated with AMI onset
but not AMI severity
Obesity, hyperlipidemia and chronic inflammation
have been shown to be risk factors of AMI (17). It has been reported that
sP-selectin may lead to cardiovascular diseases (4). Therefore, we firstly confirmed that
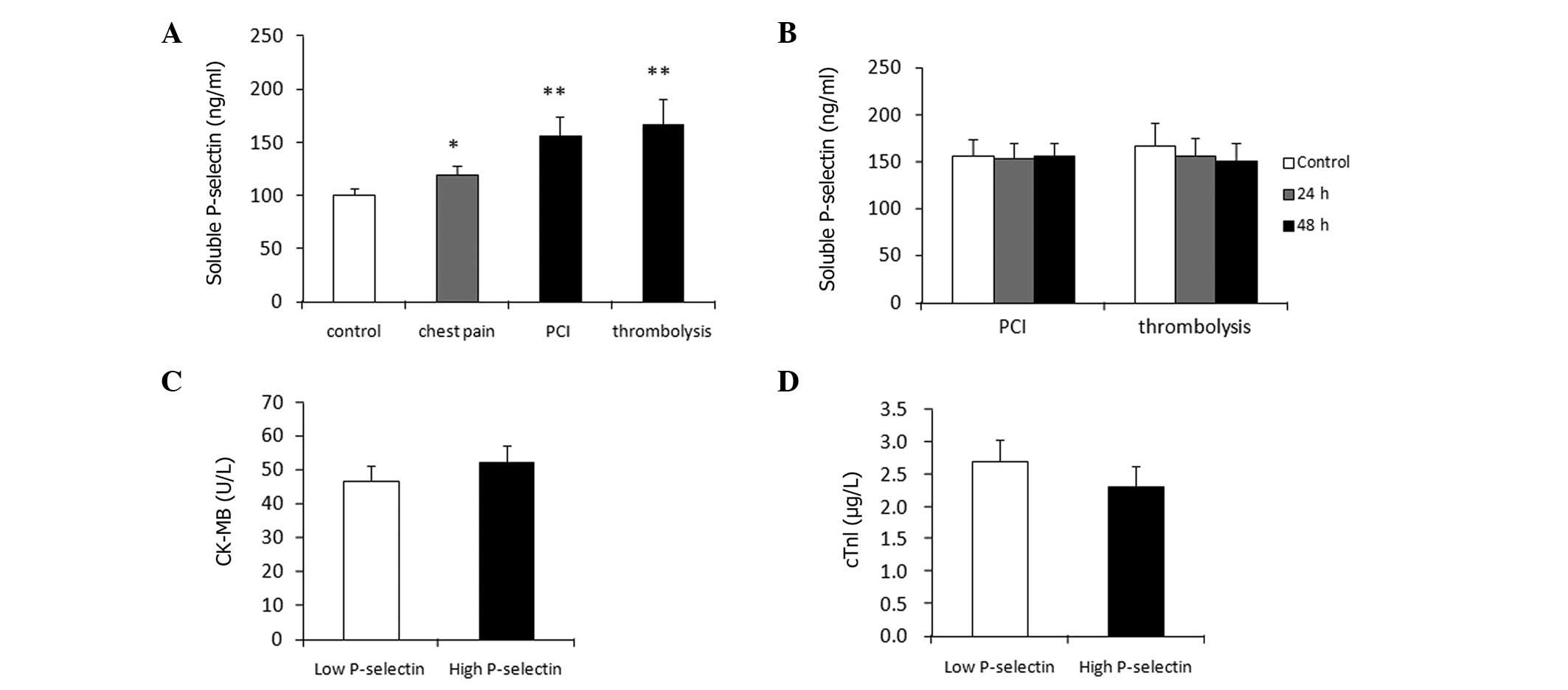
sP-selectin was significantly higher in PCI or thrombolysis therapy
patients than in chest pain or control patients (Fig. 3A). Next, we considered whether PCI
or thrombolysis could decrease the sP-selectin level. To
investigate this we measured the sP-selectin levels 24 and 48 h
after PCI or thrombolysis treatment, and compared it with those
before treatment. sP-selectin was not found to be affected by PCI
or thrombolysis (Fig. 3B).
It was known that sP-selectin could induce AMI
onset, but it was unclear whether sP-selectin affected AMI
severity. In order to elucidate this, we divided all the PCI and
thrombolysis therapy patients into two groups according to their
sP-selectin levels: A low sP-selectin (55±4 ng/ml) group and a high
sP-selectin (107±8 ng/ml) group. Subsequently, we compared the
CK-MB and cTnl levels prior to any treatment. We demonstrated that
in all AMI patients, there was no difference in CK-MB and cTnl
levels between the low sP-selectin and high sP-selectin groups
(Fig. 3C and D). These results
demonstrate that sP-selectin did not affect AMI severity.
sP-selectin induces atherosclerotic
plaque and rupture, and neointimal formation and bleeding
We have confirmed that sP-selectin induces AMI onset
but does not affect AMI severity. AMI is the result of coronary
atherosclerosis and its rupture; hence, we next investigated
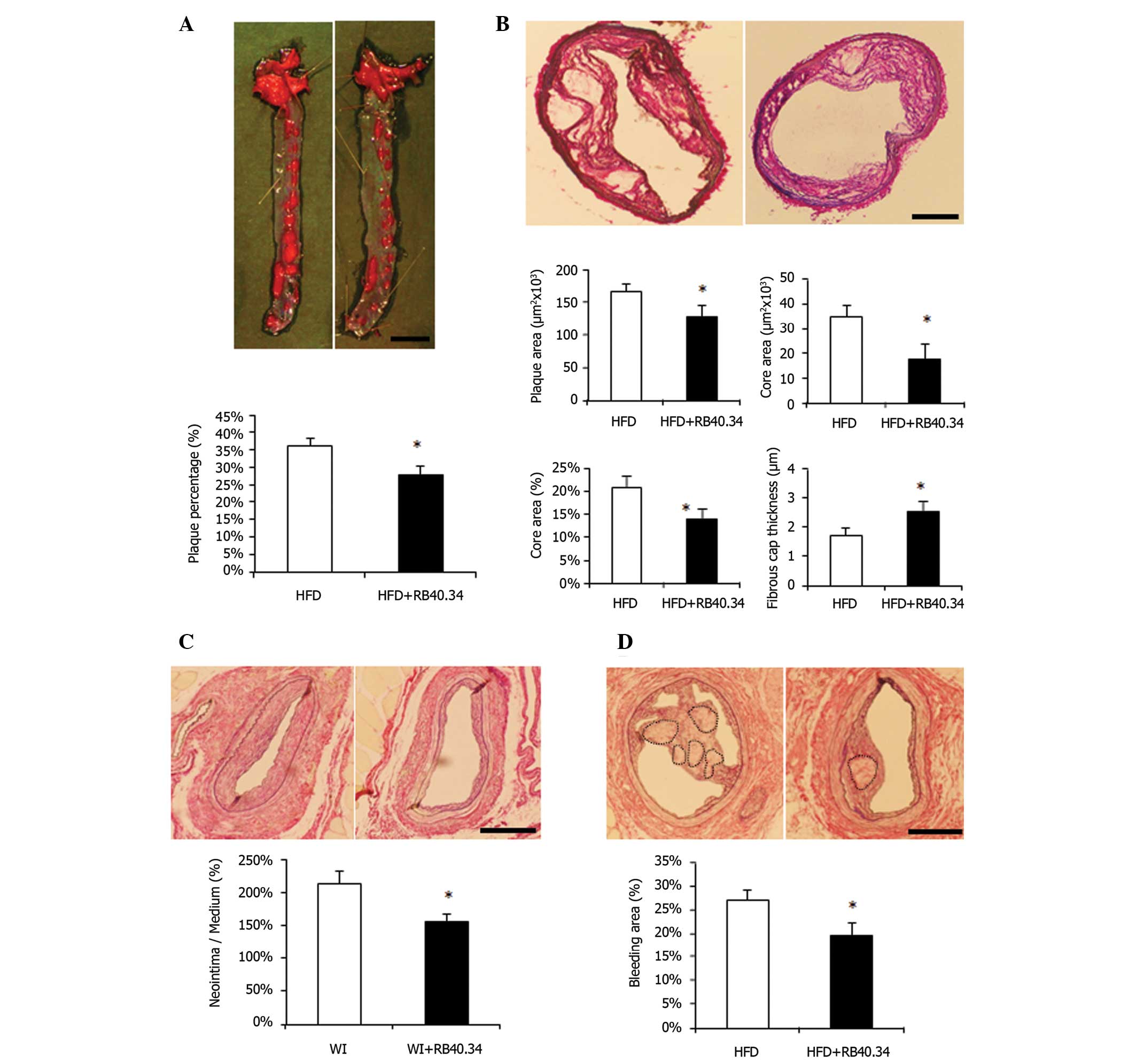
whether sP-selectin was capable of inducing atherosclerotic plaque
formation. In order to investigate this, ApoE−/− mice
were used to create the atherosclerotic plaque model. Next, we
administered RB40.43, a blocking monoclonal antibody of
sP-selectin, to block sP-selectin. As a result, the RB40.43
administration group mice were found to exhibit decreased
atherosclerotic plaque formation (Fig.
4A). AMI was caused by unstable plaque and rupture of plaque.
Notably, inhibition of sP-selectin also suppressed necrotic core
formation, and thus enhanced fibrous cap thickness (Fig. 4B). These results indicated that
sP-selectin could induce atherosclerotic plaque formation and its
rupture.
Neointimal formation has been reported as the
endothelial initial remodeling of atherosclerosis, and prevention
of injury-induced neointimal formation also reduces spontaneous
atherosclerosis (23). For this
reason, we considered whether sP-selectin was capable of inducing
neointimal formation. To investigate this, normal wild-type mice
were used to create a wire-injured neointimal formation model, and
RB40.43 was administered. As a result, the mice exhibited weak
neointimal formation compared with the control group (Fig. 4C). As neointimal hyperplasia also
exhibited neointimal bleeding in certain model mice (16), we used a high-fat diet (HFD) to
induce potential neointimal bleeding. We revealed that, in the HFD
group, half of the mice exhibited neointimal bleeding; however, in
the RB40.34 administration group, only 27% of mice exhibited
neointimal bleeding (data not shown). Among the bleeding mice, the
RB40.34 administration group exhibited a smaller degree of bleeding
compared with the HFD group, and we also demonstrated that
inhibition of sP-selectin by RB40.34 reduced the bleeding area
(Fig. 4D). These results indicate
that sP-selectin induced neointimal formation and bleeding.
Discussion
P-selectin is a transmembrane protein present in the
alpha granules of platelets and the Weibel-Palade bodies of
endothelial cells. P-selectin expression in platelets and
endothelial cells has been shown to be elevated in disorders
associated with cardiovascular diseases (24). Animal models have demonstrated the
significant roles of P-selectin in the process of these
cardiovascular diseases. For example, increased P-selectin
expression has been demonstrated in active atherosclerotic plaque;
in contrast, animals lacking P-selectin have a decreased tendency
to form atherosclerotic plaque. Increased levels of soluble
P-selectin in the plasma have also been demonstrated in a variety
of cardiovascular disorders, including coronary artery disease,
hypertension and atrial fibrillation, with some correlation with
prognosis [as reviewed in (4)].
However, it was only known that sP-selectin was capable of inducing
AMI, and not whether sP-selectin also affected severity in patients
with existing AMI.
It is reported that fibrotic inactive plaque lacks
sP-selectin expression (25),
which indicated the possibility that a lack of sP-selectin would
lead to relatively stable plaque, a relatively safe state against
the AMI onset for atherosclerosis patients. Although AMI onset was
positively correlated with the formation of atherosclerotic plaque,
in which patients the sP-selectin levels were elevated, not all
patients with atherosclerotic plaque will progress to AMI. Although
unstable plaque is not synonymous with AMI (26), unstable plaque notably led to more
patients with AMI (27).
Therefore, we assumed that sP-selectin was a risk factor in
promoting unstable plaque formation, and furthermore inducing the
onset of AMI. However, in individuals already suffering AMI, AMI
severity has become the most significant index for prognosis
(28). Therefore, in this study,
we investigated the role of sP-selectin in the onset and severity
of AMI.
Obesity, hyperlipidemia and systemic inflammation
are reported as risk factors for AMI and other cardiovascular
diseases (29,30). Hence, in the present study, we
firstly confirmed that sP-selectin was increased in obesity and
hyperlipidemia patients, and also correlated with systemic
inflammation (Figs. 1 and 2). These results were consistent with
those of previous studies (31–33).
We additionally reported that sP-selectin levels were increased in
patients with a higher PCAT area. However, the reason for the
association between sP-selectin and PCAT area remains unknown.
Notably, sP-selectin is also increased in patients with type 1 and
type 2 diabetes mellitus (34,35),
which also has systemic chronic inflammatory status, so this
evidence also supports the inflammation-promotion role of
sP-selectin. It is a limitation of the present study that the
diabetes mellitus background of patients was not investigated. The
abovementioned studies and our data indicate that obesity,
hyperlipidemia and systemic chronic inflammation are the basic risk
factors to induce sP-selectin.
The present study also confirmed that sP-selectin
was increased in AMI patients (Fig.
3A), which was consistent with a previous study (36). Hence, methods of reducing the
sP-selectin level have become a significant means of preventing
AMI. PCI and thrombolysis are extremely effective therapies;
however, neither PCI nor thrombolysis were capable of reducing
sP-selectin (Fig. 3B), and
sP-selectin did not affect the severity of AMI before patients
accepted PCI or thrombolysis (Fig.
3C). Therefore, it was concluded that sP-selectin only induced
AMI onset, but did not affect its severity. The plasma level of
sP-selectin was increased in patients with unstable angina(Fig. 3A). Transient coronary arterial
spasm could activate coronary arterial endothelium cells, and also
lead to a high inflammatory status followed by transient myocardial
ischemia. Activated coronary arterial endothelium cells and
inflammation induce an increase in sP-selectin (37). However, although PCI and
thrombolysis reduce systemic inflammation through enlargement of
the narrow coronary arterial lumen and breakdown of apolipoprotein
by thrombolysis (38), sP-selectin
was not reduced. It is possible that in PCI patients, although
stenosis was improved and systemic inflammation was decreased, the
catheter would injure the coronary arterial endothelium cells,
which would lead to a potential increase in inflammation.
Alternatively, the reduction in sP-selectin may take a relatively
long time following PCI or thrombolysis treatments, but in the
present study we only investigated up to 48 h post-treatment. AMI
severity depends on the location of coronary arterial stenosis and
the hospitalization time. In the present study, we investigated 15
PCI and 15 thrombolysis patients, and the hospitalization time for
the two groups of patients was almost the same (data not shown).
Therefore, sP-selectin has no correlation with the location of
coronary arterial stenosis.
As superimposed luminal thrombus and rupture of
coronary atherosclerosis are main causes of AMI, we believed
atherosclerosis plaque formation to be a further cause of AMI,
induced by sP-selectin. Since neointimal formation was thought to
be the initial pathological change of atherosclerosis, we
hypothesized that sP-selectin could induce atherosclerosis plaque
and neointimal formation (Fig. 4A and
C). Increased plaque is an increased risk factor for coronary
artery stenosis, and once the stenosis reaches 70%, obstructive
coronary artery disease may be defined. Once patients are diagnosed
with obstructive coronary artery disease, the stability of the
plaque is the critical issue to address, as unstable plaque leads
directly to AMI. In the present study, we demonstrated that
sP-selectin inhibitor not only reduced plaque rupture but also
decreased the neointimal bleeding incidence rate and the bleeding
area (Fig. 4B and D).
In conclusion, in the current study, we demonstrated
that an excess of sP-selectin, induced by systemic inflammation and
hyperlipidemia, leads to neointimal hyperplasia and atherosclerosis
plaque formation with unstable neointimal bleeding and plaque
rupture, and then leads to the onset of AMI. However, in
individuals already suffering AMI, a further increase of
sP-selectin may have no effect on severity.
References
|
1
|
Geng JG, Chen M and Chou KC: P-selectin
cell adhesion molecule in inflammation, thrombosis, cancer growth
and metastasis. Curr Med Chem. 11:2153–2160. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Polek A, Sobiczewski W and Matowicka-Karna
J: P-selectin and its role in some diseases. Postepy Hig Med Dosw
(Online). 63:465–470. 2009.
|
|
3
|
Yang J, Furie BC and Furie B: The biology
of P-selectin glycoprotein ligand-1: its role as a selectin
counterreceptor in leukocyte-endothelial and leukocyte-platelet
interaction. Thromb Haemost. 81:1–7. 1999.PubMed/NCBI
|
|
4
|
Blann AD, Nadar SK and Lip GY: The
adhesion molecule P-selectin and cardiovascular disease. Eur Heart
J. 24:2166–2179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ramacciotti E, Blackburn S, Hawley AE, et
al: Evaluation of soluble P-selectin as a marker for the diagnosis
of deep venous thrombosis. Clin Appl Thromb Hemost. 17:425–431.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Woollard KJ and Chin-Dusting J: P-selectin
antagonism in inflammatory disease. Curr Pharm Des. 16:4113–4118.
2010. View Article : Google Scholar
|
|
7
|
Burke AP and Virmani R: Pathophysiology of
acute myocardial infarction. Med Clin North Am. 91:553–572. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woollard KJ and Chin-Dusting J:
Therapeutic targeting of p-selectin in atherosclerosis. Inflamm
Allergy Drug Targets. 6:69–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aversano T, Aversano LT, Passamani E, et
al: Thrombolytic therapy vs primary percutaneous coronary
intervention for myocardial infarction in patients presenting to
hospitals without on-site cardiac surgery: a randomized controlled
trial. JAMA. 287:1943–1951. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferroni P, Basili S, Martini F, et al:
Soluble P-selectin as a marker of platelet hyperactivity in
patients with chronic obstructive pulmonary disease. J Investig
Med. 48:21–27. 2000.PubMed/NCBI
|
|
11
|
Ertenli I, Kiraz S, Arici M, et al:
P-selectin as a circulating molecular marker in rheumatoid
arthritis with thrombocytosis. J Rheumatol. 25:1054–1058.
1998.PubMed/NCBI
|
|
12
|
Tamagawa-Mineoka R, Katoh N, Ueda E,
Masuda K and Kishimoto S: Platelet-derived microparticles and
soluble P-selectin as platelet activation markers in patients with
atopic dermatitis. Clin Immunol. 131:495–500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Konishi M, Sugiyama S, Sugamura K, et al:
Association of pericardial fat accumulation rather than abdominal
obesity with coronary atherosclerotic plaque formation in patients
with suspected coronary artery disease. Atherosclerosis.
209:573–578. 2010. View Article : Google Scholar
|
|
14
|
Zhou J, Lhotak S, Hilditch BA and Austin
RC: Activation of the unfolded protein response occurs at all
stages of atherosclerotic lesion development in apolipoprotein
E-deficient mice. Circulation. 111:1814–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson JL and Jackson CL: Atherosclerotic
plaque rupture in the apolipoprotein E knockout mouse.
Atherosclerosis. 154:399–406. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sata M, Maejima Y, Adachi F, et al: A
mouse model of vascular injury that induces rapid onset of medial
cell apoptosis followed by reproducible neointimal hyperplasia. J
Mol Cell Cardiol. 32:2097–2104. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Phillips JW, Barringhaus KG, Sanders JM,
et al: Single injection of P-selectin or P-selectin glycoprotein
ligand-1 monoclonal antibody blocks neointima formation after
arterial injury in apolipoprotein E-deficient mice. Circulation.
107:2244–2249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin WY, Lee LT, Chen CY, et al: Optimal
cut-off values for obesity: using simple anthropometric indices to
predict cardiovascular risk factors in Taiwan. Int J Obes Relat
Metab Disord. 26:1232–1238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo L, Ning W, Tan Z, Gong Z and Li X:
Mechanism of matrix metalloproteinase axis-induced neointimal
growth. J Mol Cell Cardiol. 66:116–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Konishi M, Sugiyama S, Sato Y, et al:
Pericardial fat inflammation correlates with coronary artery
disease. Atherosclerosis. 213:649–655. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamminga EA, van der Lely AJ, Neumann HA
and Thio HB: Chronic inflammation in psoriasis and obesity:
implications for therapy. Med Hypotheses. 67:768–773. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hackam DG and Anand SS: Emerging risk
factors for atherosclerotic vascular disease: a critical review of
the evidence. JAMA. 290:932–940. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quarck R, De Geest B, Stengel D, et al:
Adenovirus-mediated gene transfer of human platelet-activating
factor-acetylhydrolase prevents injury-induced neointima formation
and reduces spontaneous atherosclerosis in apolipoprotein
E-deficient mice. Circulation. 103:2495–2500. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Merten M and Thiagarajan P: P-selectin in
arterial thrombosis. Z Kardiol. 93:855–863. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong MK, Mintz GS, Lee CW, et al:
Comparison of coronary plaque rupture between stable angina and
acute myocardial infarction: a three-vessel intravascular
ultrasound study in 235 patients. Circulation. 110:928–933. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee SG, Lee CW, Hong MK, Kim JJ, Park SW
and Park SJ: Change of multiple complex coronary plaques in
patients with acute myocardial infarction: a study with coronary
angiography. Am Heart J. 147:281–286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Finn AV, Nakano M, Narula J, Kolodgie FD
and Virmani R: Concept of vulnerable/unstable plaque. Arterioscler
Thromb Vasc Biol. 30:1282–1292. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Claeys MJ, Bosmans J, Veenstra L, Jorens
P, De Raedt H and Vrints CJ: Determinants and prognostic
implications of persistent ST-segment elevation after primary
angioplasty for acute myocardial infarction: importance of
microvascular reperfusion injury on clinical outcome. Circulation.
99:1972–1977. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Triant VA, Lee H, Hadigan C and Grinspoon
SK: Increased acute myocardial infarction rates and cardiovascular
risk factors among patients with human immunodeficiency virus
disease. J Clin Endocrinol Metab. 92:2506–2512. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosengren A, Wallentin L, Simoons M, et
al: Cardiovascular risk factors and clinical presentation in acute
coronary syndromes. Heart. 91:1141–1147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kvasnicka T, Kvasnicka J, Ceska R, Grauova
B and Vrablik M: Increasing plasma levels of soluble cell adhesion
molecules (sE-selectin, sP-selectin and sICAM-1) in overweight
adults with combined hyperlipidemia. Sb Lek. 102:473–477. 2001.
|
|
32
|
De Pergola G, Pannacciulli N, Coviello M,
et al: sP-selectin plasma levels in obesity: association with
insulin resistance and related metabolic and prothrombotic factors.
Nutr Metab Cardiovasc Dis. 18:227–232. 2008. View Article : Google Scholar
|
|
33
|
Larsson PT, Hallerstam S, Rosfors S and
Wallen NH: Circulating markers of inflammation are related to
carotid artery atherosclerosis. Int Angiol. 24:43–51.
2005.PubMed/NCBI
|
|
34
|
Yngen M, Ostenson CG, Hu H, Li N, Hjemdahl
P and Wallen NH: Enhanced P-selectin expression and increased
soluble CD40 Ligand in patients with Type 1 diabetes mellitus and
microangiopathy: evidence for platelet hyperactivity and chronic
inflammation. Diabetologia. 47:537–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gokulakrishnan K, Deepa R, Mohan V and
Gross MD: Soluble P-selectin and CD40L levels in subjects with
prediabetes, diabetes mellitus, and metabolic syndrome - the
Chennai Urban Rural Epidemiology Study. Metabolism. 55:237–242.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu DY, Zhao SP and Peng WP: Elevated
plasma levels of soluble P-selectin in patients with acute
myocardial infarction and unstable angina. An inverse link to
lipoprotein(a). Int J Cardiol. 64:253–258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Atalar E, Aytemir K, Haznedaroglu I, et
al: Increased plasma levels of soluble selectins in patients with
unstable angina. Int J Cardiol. 78:69–73. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Eberini I, Gianazza E, Breghi L, et al:
Apolipoprotein A-I breakdown is induced by thrombolysis in coronary
patients. Ann Med. 39:306–311. 2007. View Article : Google Scholar : PubMed/NCBI
|