Introduction
Bladder cancer is the most ubiquitous malignant
urinary tumor, and is susceptible to recurrence, invasion and
metastasis. However, there are only a few studies describing the
mechanisms of invasion and metastasis (1). Investigations into the
epithelial-mesenchymal transition (EMT) have provided an increased
understanding of the invasion and metastasis of cancer (2–4). The
EMT is a biological process, which occurs under specific conditions
and interacts with the basement membrane, in which epithelial cells
gradually transform into cells with mesenchymal properties. Cancer
cells undergoing the EMT process are depolarized and transformed,
which enables them to invade and metastasize, accompanied by the
inhibition of apoptosis and degradation of the extracellular matrix
(5–7). In a previous study by our group, it
was demonstrated that the Notch signaling pathway had an
anti-tumorigenic function in superficial bladder cancer (8). The expression levels of Notch 1 and
Jagged 1 in bladder cancer were significantly lower compared with
those in normal bladder mucosa, while the expression levels of
Notch 1 and Jagged 1 in invasive bladder cancer were higher
compared with those in superficial bladder cancer (9). Based on these previous studies, the
present study hypothesized that the Notch signaling pathway is
important in the invasion and metastasis of bladder cancer. In the
preset study, the Notch signaling pathway was completely inhibited
in the bladder cancer cell lines T24, 5637 and J82 using a
γ-secretase inhibitor (GSI). The cellular morphology, drug
resistance and invasiveness were subsequently analyzed. The effect
of the Notch signaling pathway on the invasion and drug resistance
of bladder cancer was assessed by measuring the alterations in the
expression levels of the molecular biomarkers associated with EMT,
with the aim of determining whether Notch signaling modified
urinary tumor invasion and drug resistance by affecting the
EMT.
Materials and methods
Cell culture
T24 and 5637 cell lines (Shanghai Cell Bank, Chinese
Academy of Sciences, Shanghai, China) were cultured in RPMI-1640
medium (Beijing Solarbio Science & Technology, Beijing, China),
supplemented with 10% fetal bovine serum (FBS; Beijing TransGen
Biotech Co., Ltd., Beijing, China) and the J82 cell line (Shanghai
Cell Bank, Chinese Academy of Sciences) was cultured in minimum
essential medium (Beijing Solarbio Science & Technology),
supplemented with 10% FBS. All cells were maintained at 37°C in 5%
CO2. When cells were grown to 90% confluence, the 5
μM γ-secretase inhibitor (GSI; cat. no. #565750; Calbiochem,
Merck, Darmstadt, Germany) was added for 24 or 48 h (only for
western blot).
RNA and protein extraction
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA), according to the
manufacturer’s instructions. The RNA quality was measured by
distinct 18S, 28S and total RNA separated by electrophoresis in
agarose gels and the proper ratio of absorbance at 230/280 nm
(UV-8000 Double Beam UV/VIS model; Metash Instruments, Shanghai,
China).
Total proteins were extracted by
radioimmunoprecipitation assay cell lysis reagent supplemented with
proteinase and phosphatase inhibitors (Beijing Solarbio Science
& Technology) at 4°C for 30 min. The cell extracts were
centrifuged at 12,000 ×g for 20 min at 4°C, and the supernatants
containing total proteins were mixed with an equal volume of 5X SDS
loading buffer (Beijing Solarbio Science & Technology). The
samples were heated to 95°C for 5 min.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA (1 μg) was used for RT using a
Reverse Transcription system (PrimeScript™ RT Reagent kit; Takara
Bio, Inc., Shiga, Japan), according to manufacturer’s instructions.
RT-qPCR was performed using SYBR® Premix Ex TaqTM II (TliRNase H
Plus; Takara Bio, Inc.) on the ABI PRISM® 7500 real-time PCR system
(Applied Biosystems, Life Technologies, Thermo Fisher Scientific,
Waltham, MA, USA), with β-actin as an internal control.
Thermocycling was performed in a final volume of 20 μl consisting
of 40 cycles at 95°C for 5 sec then 55°C for 30 sec, following an
initial denaturation step at 95°C for 10 sec. The sequences of the
PCR primers of E-cadherin, N-cadherin, vimentin and smooth muscle
actin are listed in Table I. The
results were analyzed using the 2−ΔΔCt method. ΔCt =
average gene Ct-Δ average of β-actin Ct. ΔΔCt was calculated as
follows: ΔΔCt = ΔCt of sample group-ΔΔCt of control group.
 | Table IPrimer sequences for polymerase chain
reaction amplification. |
Table I
Primer sequences for polymerase chain
reaction amplification.
| Gene | Sequence | Product size
(bp) |
|---|
| E-cadherin | F:
5′-AAGGCACAGCCTGTCGAAGCA-3′
R: 5′-ACGTTGTCCCGGGTGTCATCCT-3′ | 167 |
| N-cadherin | F:
5′-TGCGCGTGAAGGTTTGCCAGT-3′
R: 5′-TGGCGTTCTTTATCCCGGCGT-3′ | 175 |
| Vimentin | F:
5′-ACCGCACACAGCAAGGCGAT-3′
R: 5′-CGATTGAGGGCTCCTAGCGGTT3′ | 132 |
| α-smooth muscle
actin | F:
5′-TGCCCCATGCCATCATGCGT-3′
R: 5′-TGCGGCAGTGGCCATCTCAT-3′ | 182 |
| β-actin | F:
5′-AGCGAGCATCCCCCAAAGTT-3′
R: 5′-GGGCACGAAGGCTCATCATT-3′ | 322 |
Western blot analysis
The protein concentrations were quantified using a
Micro Bicinchoninic Acid Protein Assay kit (Pierce Biotechnology,
Inc., Rockford, IL, USA). Identical quantities of total protein
from each sample (30 μg) were loaded and separated by 12%
SDS-PAGE and transferred onto a polyvinylidene fluoride membrane.
The membranes were incubated with the following primary antibodies
at 4°C overnight: Anti-N-cadherin (1:800; cat. no. ab18203; Abcam,
Cambridge, MA, USA), anti-vimentin (1:500; cat. no. 3877; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-α-smooth muscle
actin (1:1,000; cat. no. P62736; Abgent, Inc., San Diego, CA, USA),
anti-E-cadherin (1:800; cat no. 5296; Cell Signaling Technology,
Inc.), and anti-β-actin (1:1,500; cat. no. ab6276; Abcam). The
membranes were then incubated with horseradish
peroxidase-conjugated AffiniPure goat anti-rabbit lgG(H+L)
secondary antibody (1:10,000; cat. no. ZF-0316-1; ZSGB-BIO,
Beijing, China) or horseradish peroxidase-conjugated AffiniPure
goat anti-mouse lgG(H+L) secondary antibody (1:10,000; cat. no.
ZF-0315-1; ZSGB-BIO) at room temperature for 2 h. The signals were
detected using an enhanced chemiluminescence system (cat. no.
32109; Thermo Fisher Scientific) and the films (cat. no. 6535876;
Eastman Kodak Company, Rochester, NY, USA) were scanned and
analyzed by AlphaEaseFC version 1.1 software (Alpha Innotech, San
Leandro, CA, USA).
MTT assay
Cells in the logarithmic growth phase were
subcultured in 96-well plates at a cell density of 5,000
cells/well. In each experiment, five wells corresponded to each
control group: Mitoxantrone-treated, zero-adjustment wells and
control wells. The mitoxantrone wells contained 1, 4, 16, 64 and
256 ng/ml of drug (Sigma-Aldrich, St. Louis, MO, USA). Following 24
h of drug treatment, 20 μl MTT (Beijing Solarbio Science
& Technology) solution was added to each well and the plate was
incubated for an additional 4 h, prior to the addition of 150
μl dimethylsulfoxide (Beijing Solarbio Science &
Technology) to dissolve the formazan crystals. The absorbance
values were measured at 490 nm (Anthos Zenyth 340rt; Biochrom Ltd.,
Cambridge, UK). The ratio of inhibition was calculated using the
following formula: Inhibition ratio = (1 − Abexperiment
/ Abcontrol) × 100%.
Transwell assay
Transwell plates were coated with 40 μl 50
mg/l Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). Cells (300
μl) in the logarithmic growth phase were added to the
Transwell plates at a final cell density of 1×105
cells/ml. Culture media, supplemented with 20% FBS (800 μl),
was added to each well. The plate was incubated at 37°C and 5%
CO2 for 24 h. The supernatants were subsequently removed
from the Transwells, followed by rinsing, fixation with 2 ml 3.7%
paraformaldehyde and staining with 2 ml 0.05% crystal violet
(Beijing Solarbio Science & Technology). The inner cells were
removed using a cotton swab and the number of cells, which were
below the microporous membrane, were counted using a microscope
(XL-71; Olympus Corporation, Tokyo, Japan). The average cell number
from 10 randomly selected fields (magnification, ×200) was
calculated.
Statistical analysis
Values are expressed as the mean ± standard error
and analyzed using SPSS 12.0 (SPSS, Inc., Chicago, IL, USA).
Western blot images were semi-quantitatively analyzed using ImageJ
version 2.1 software (National Institutes of Health, Bethesda, MD,
USA). The means of two samples were analyzed using the independent
sample t-test. P<0.05 was considered to indicate a statistically
significant difference between values.
Results
Complete inhibition of the Notch
signaling pathway inhibits cell proliferation and induces
morphological changes
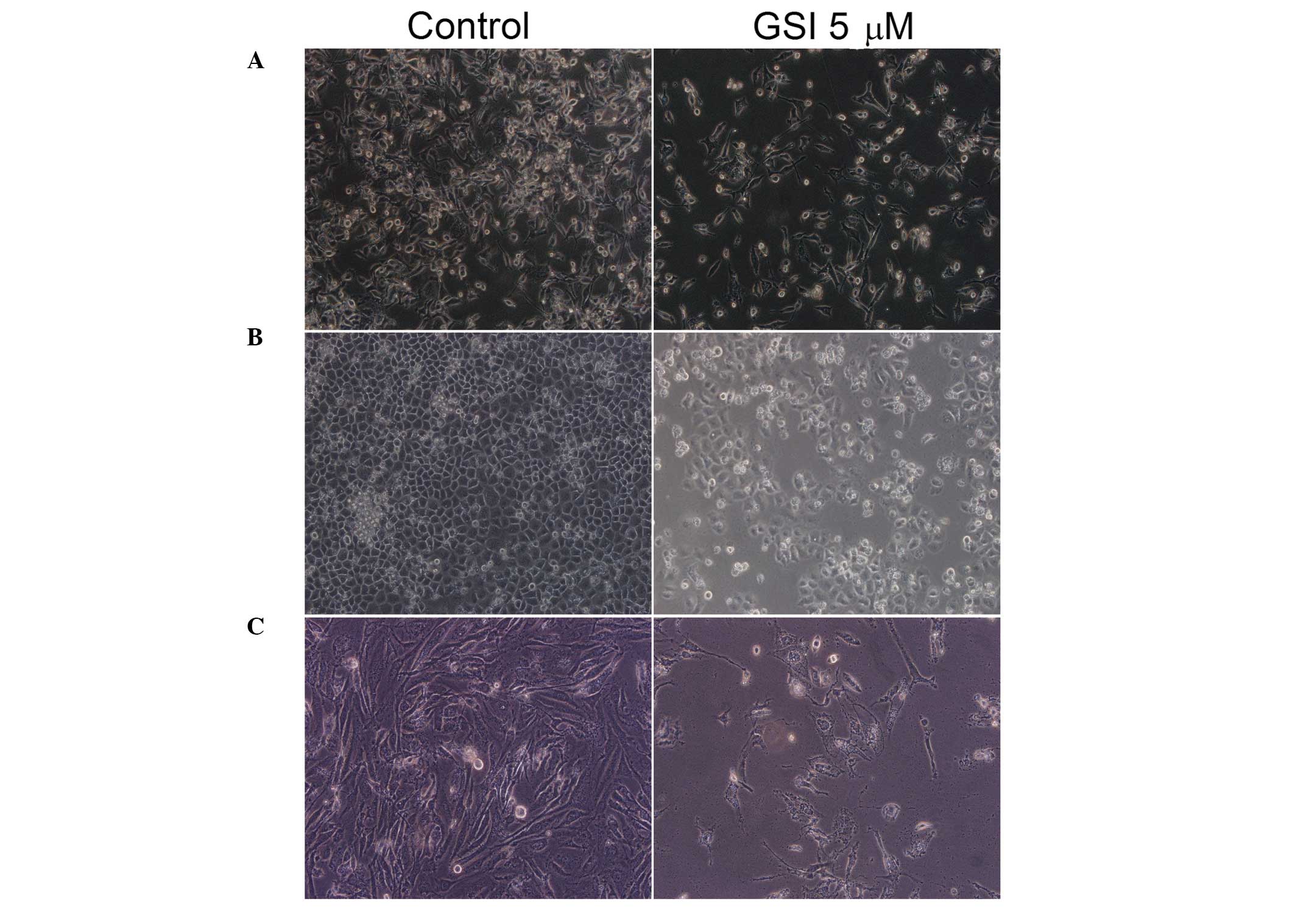
In order to determine whether the GSI caused
morphological changes in bladder cancer cells, T24, 5637 and J82
cells were examined microscopically to assess cell growth and
morphological changes following treatment with GSI for 24 h
(Fig. 1). Treatment with GSI
significantly reduced the cell density. Intercellular junction
formation was also reduced, with the majority of cells exhibiting a
shrunken appearance and underwent apoptosis.
Inhibition of the Notch signaling pathway
inhibits the EMT
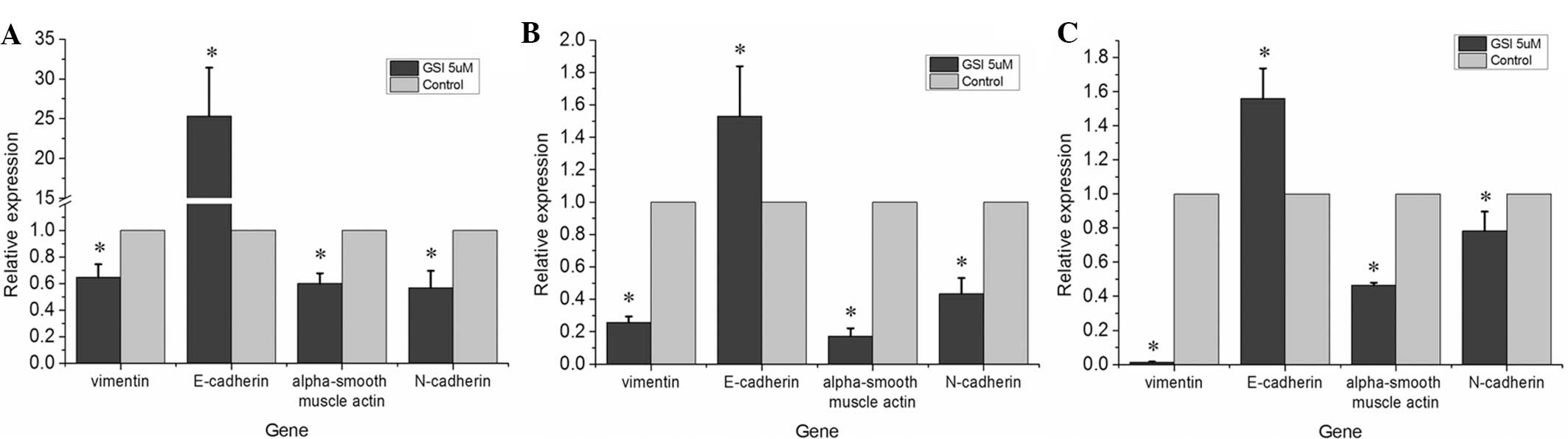
Following treatment with the GSI to inhibit the
Notch signaling pathway, the mRNA expression levels of mesenchymal
biomarkers, including N-cadherin, vimentin and α-smooth muscle
actin, were downregulated compared with those in the control group
and there was a significant upregulation of the mRNA expression of
E-cadherin (Fig. 2;
P<0.05).
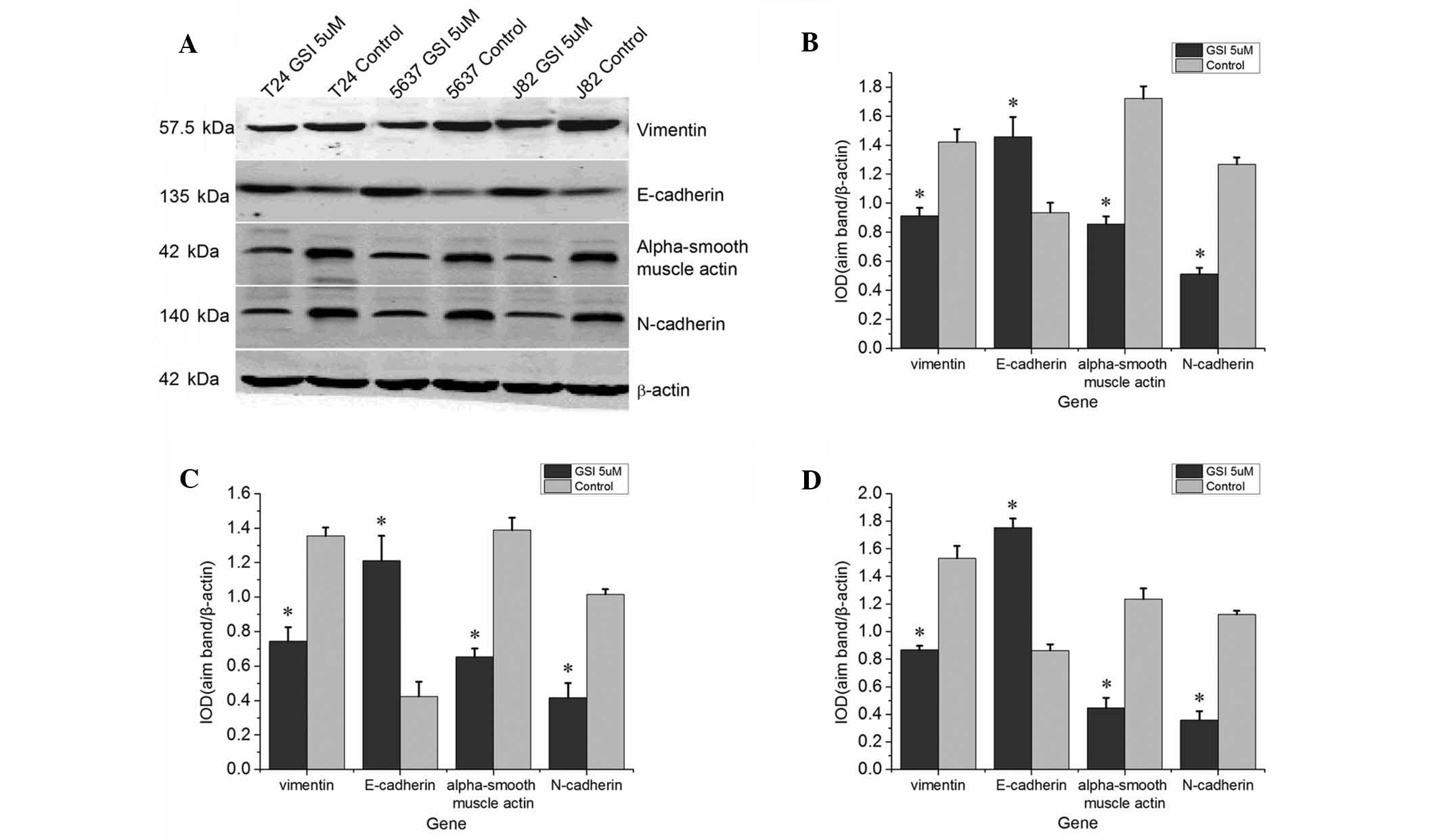
Western blot analysis demonstrated that the protein
expression levels of N-cadherin, vimentin and α-smooth muscle actin
were significantly downregulated following treatment with GSI
compared with those in the control group, whereas the protein
expression of of E-cadherin was upregulated (Fig. 3).
This observed downregulation of mesenchymal
biomarkers and upregulation of epithelial biomarkers indicated that
inhibition of Notch signaling inhibited the EMT.
Inhibition of the Notch signaling pathway
reduces the drug resistance and invasiveness of bladder cancer
cells
Following treatment with the GSI (5 μM) for
48 h, the T24, 5637 and J82 bladder cancer cells were cultured with
different concentrations of the bladder cancer therapeutic
mitoxantrone and the cell viability was detected using an MTT
assay. The results revealed that the GSI-treated cells exhibited an
increased sensitivity to mitoxantrone-induced cytotoxicity compared
with that of the control (untreated) cells (Fig. 4; P<0.05).
Similarly, a Transwell assay demonstrated that the
GSI-treated cells were more sensitive to mitoxantrone compared with
the untreated cells, as indicated by decreased invasiveness.
Treatment with GSI reduced the number of cells which passed through
the Transwell membrane, which indicated a deficit in the invasive
capacity of those cells which lacked Notch signaling (Fig. 5). Therefore, the present study
demonstrated that inhibiting the Notch signaling pathway inhibited
the invasion of bladder cancer cells.
Discussion
Studies have demonstrated that the morbidity of
bladder cancer has been increasing in China. Bladder cancer is the
most common malignancy of the urinary system, 90% of which are
transitional cell carcinoma of the urinary system (5). The proliferation and metastasis of
bladder cancer cells involves various transient changes, which
involve sequential regulation of a number of different genes and
signaling pathways (5,10,11).
The Notch signaling pathway and the EMT have been demonstrated to
be involved in the proliferation and metastasis of the majority
human cancer types (12–15). However, the role of Notch signaling
and the EMT in bladder cancer cells remains to be elucidated.
The Notch signaling pathway, a conserved signaling
pathway, controls cell proliferation, survival, apoptosis,
differentiation and the development and function of various organs
(16,17). It has been previously reported that
the activation of the Notch signaling pathway predominantly depends
upon the activity of GSI (14).
GSI, as an effective inhibitor of all Notch receptors, has been
used clinically to treat several types of carcinoma (18,19).
The present study analyzed the effect of the GSI on the T24, 5637
and J82 bladder cancer cells. A previous study has indicated that
Notch 4 activates mouse mammary tumor virus and causes mammary
tumors in mice (20). Notch 1 and
Notch 2 cooperate with the EIA to transform epithelial cells in
common rodent models (21,22). Previous studies have demonstrated
that Notch facilitates cell cycle progression and inhibits cell
apoptosis (23). Shi et al
(8) revealed that the expression
of Notch 1 is decreased in non-invasive bladder cancer, while it is
highly expressed in invasive bladder cancer. The present study
demonstrated a significant inhibition of cell proliferation upon
inhibition of the Notch signaling pathway, suggesting that the
Notch signaling pathway may trigger carcinogenesis in bladder
cancer, particularly those associated with a high expression of
Notch 1.
Accumulating evidence has indicated that there is a
correlation between the EMT and metastasis, since the EMT can equip
cancer cells with an increased migratory ability (24). Grego-Bessa et al (25) demonstrated that inducing the EMT
caused a downregulation of expression levels of epithelial markers,
including E-cadherin, α-catenin and β-catenin, and an upregulation
of mesenchymal biomarkers, including N-cadherin, vimentin,
fibronectin, matrix metalloproteinase and α-smooth muscle actin.
Following complete inhibition of the Notch signaling pathway, T24,
5637 and J82 bladder cancer cells exhibited increased expression of
E-cadherin and down-regulation of the expression levels of
N-cadherin, vimentin and α-smooth muscle actin, as confirmed by
RT-qPCR and western blot analysis. In addition, the invasiveness of
bladder cancer cells following inhibition of the Notch signaling
pathway was significantly reduced. Consequently, these data
suggested that inhibiting the Notch signaling pathway inhibited the
EMT in bladder cancer cells, thereby reducing cell invasiveness. A
previous study also demonstrated that the Slug molecule was one of
the vital components in the process of EMT via the Notch signaling
pathway (25). Upregulation of
Slug activated the promoter of E-cadherin, resulting in EMT
(26,27). However, whether the EMT is
dependent upon the expression of Slug in bladder cancer remains to
be elucidated.
It has been reported that the EMT correlates with
drug resistance in cancer, including drug resistance to paclitaxel,
vinblastine, oxaliplatin, gemcitabine and epidermal growth factor
receptor-targeted agents (28).
Shah et al (29)
demonstrated that gemcitabine-resistant pancreatic carcinoma cells
exhibited the EMT phenotype, alongside downregulation of epithelial
markers and upregulation of mesenchymal markers. In addition,
activation of the Notch signaling pathway was shown to partially
reverse the EMT phenotype (29,30).
These data suggested that the activation of Notch signaling may
trigger the EMT in cancer cells and, therefore, cause drug
resistance. The present study revealed that the inhibition of the
Notch signaling pathway in bladder cancer cells caused a higher
sensitivity to the anti-cancer drug mitoxantrone as compared with
that of the control group. The results indicated that regulation of
the EMT by the Notch signaling pathway may be associated with drug
resistance in bladder cancer, and that Notch inhibition may be
utilized to counteract drug resistance.
In conclusion, treatment with the GSI demonstrated
that inhibiting the Notch signaling pathway inhibited the EMT of
bladder cancer cells. Inhibiting the Notch signaling pathway
triggered reversion of EMT-associated biomarker levels and reduced
the invasiveness and drug resistance of bladder cancer cells. These
data provided further evidence that there is a clear association
between the Notch signaling pathway and the EMT. In further studies
it is be necessary to identify the key molecule that regulates the
EMT through the Notch signaling pathway, so that the precise
signaling regulation network for EMT and the Notch signaling
pathway can be determined. Finally, it is necessary to investigate
novel molecular mechanisms underlying the genesis, metastasis and
drug-resistance of bladder cancer in order to develop novel
treatment strategies.
Acknowledgments
This study was supported by the National Natural
Science Foundation of P.R. China (no. 81160272), the Natural
Science Foundation of Jiangxi (no. 800GZY0039), the Jiangxi
Province Science Foundation for Youths (no. 2010JX02761) and the
Science and Technology Development Fund of Macau, China Special
Administrative Region (no. 064/2012/A).
References
|
1
|
Ploeg M, Aben KK and Kiemeney LA: The
present and future burden of urinary bladder cancer in the world.
World J Urol. 27:289–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Floor S, van Staveren WC, Larsimont D,
Dumont JE and Maenhaut C: Cancer cells in epithelial-to-mesenchymal
transition and tumor-propagating-cancer stem cells: Distinct,
overlapping or same populations. Oncogene. 30:4609–4621. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gomes LR, Terra LF, Sogayar MC and
Labriola L: Epithelial-mesenchymal transition: Implications in
cancer progression and metastasis. Curr Pharm Biotechnol.
12:1881–1890. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yao D, Dai C and Peng S: Mechanism of the
mesenchymal-epithelial transition and its relationship with
metastatic tumor formation. Mol Cancer Res. 9:1608–1620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A,
Siefker-Radtke A and Dinney C: Role of epithelial-to-mesenchymal
transition (EMT) in drug sensitivity and metastasis in bladder
cancer. Cancer Metastasis Rev. 28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi TP, Ma X, Fu B, Li H, Ai X, Xu H, Ju
Z, Wang C, Zhang G, Wang BJ and Zhang X: Expression of Notch
signaling in bladder cancer and its relationship with patients’
prognosis. Chin J Exp Surg. 9:1305–1307. 2010.In Chinese.
|
|
9
|
Wang Z, Li Y, Kong D and Sarkar FH: The
role of Notch signaling pathway in epithelial-mesenchymal
transition (EMT) during development and tumor aggressiveness. Curr
Drug Targets. 11:745–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mills RD, Turner WH, Fleischmann A,
Markwalder R, Thalmann GN and Studer UE: Pelvic lymph node
metastases from bladder cancer: Outcome in 83 patients after
radical cystectomy and pelvic lymphadenectomy. J Urol. 166:19–23.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamai T, Tsujii T, Arai K, Takagi K, Asami
H, Ito Y and Oshima H: Significant association of Rho/ROCK pathway
with invasion and metastasis of bladder cancer. Clin Cancer Res.
9:2632–2641. 2003.PubMed/NCBI
|
|
12
|
Ohashi S, Natsuizaka M, Naganuma S, Kagawa
S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D,
Gimotty PA, et al: A NOTCH3-mediated squamous cell differentiation
program limits expansion of EMT-competent cells that express the
ZEB transcription factors. Cancer Res. 71:6836–6847. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Espinoza I and Miele L: Notch inhibitors
for cancer treatment. Pharmacol Ther. 139:95–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishida T, Hijioka H, Kume K, Miyawaki A
and Nakamura N: Notch signaling induces EMT in OSCC cell lines in a
hypoxic environment. Oncol Lett. 6:1201–1206. 2013.PubMed/NCBI
|
|
15
|
Li Y, Ma J, Qian X, Wu Q, Xia J, Miele L,
Sarkar FH and Wang Z: Regulation of EMT by Notch signaling pathway
in tumor progression. Curr Cancer Drug Targets. 13:957–962. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schwanbeck R, Martini S, Bernoth K and
Just U: The Notch signaling pathway: Molecular basis of cell
context dependency. Eur J Cell Biol. 90:572–581. 2011. View Article : Google Scholar
|
|
17
|
Borggrefe T and Oswald F: The Notch
signaling pathway: Transcriptional regulation at Notch target
genes. Cell Mol Life Sci. 66:1631–1646. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Capobianco AJ, Zagouras P, Blaumueller CM,
Artavanis-Tsakonas S and Bishop JM: Neoplastic transformation by
truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol Cell Biol.
17:6265–6273. 1997.PubMed/NCBI
|
|
19
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
The importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shun XD and Wang HZ: Research progress in
bladder cancer markers and its detection met hods. J Mod Urol.
17:319–321. 2012.
|
|
21
|
Gallahan D, Kozak C and Callahan R: A new
common integration region (int-3) for mouse mammary tumor virus on
mouse chromosome 17. J Virol. 61:218–220. 1987.PubMed/NCBI
|
|
22
|
Joshi I, Minter LM, Telfer J, Demarest RM,
Capobianco AJ, Aster JC, Sicinski P, Fauq A, Golde TE and Osborne
BA: Notch signaling mediates G1/S cell-cycle progression in T cells
via cyclin D3 and its dependent kinases. Blood. 113:1689–1698.
2009. View Article : Google Scholar :
|
|
23
|
Shi TP, Xu H, Wei JF, Ai X, Ma X, Wang BJ,
Ju ZH, Zhang GX, Wang C, Wu ZQ, Zhang X, et al: Association of low
expression of notch-1 and jagged-1 in human papillary bladder
cancer and shorter survival. J Urol. 180:361–366. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meng F and Wu G: The rejuvenated scenario
of epithelial-mesenchymal transition (EMT) and cancer metastasis.
Cancer Metastasis Rev. 31:455–467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grego-Bessa J, Díez J, Timmerman L and de
la Pompa JL: Notch and epithelial-mesenchyme transition in
development and tumor progression: Another turn of the screw. Cell
Cycle. 3:718–721. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leong KG, Niessen K, Kulic I, Raouf A,
Eaves C, Pollet I and Karsan A: Jagged1-mediated Notch activation
induces epithelial-to-mesenchymal transition through Slug-induced
repression of E-cadherin. J Exp Med. 204:2935–2948. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niessen K, Fu Y, Chang L, Hoodless PA,
McFadden D and Karsan A: Slug is a direct Notch target required for
initiation of cardiac cushion cellularization. J Cell Biol.
182:315–325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sabbah M, Emami S, Redeuilh G, Julien S,
Prevost G, Zimber A, Ouelaa R, Bracke M, De Wever O and Gespach C:
Molecular signature and therapeutic perspective of the
epithelial-to-mesenchymal transitions in epithelial cancers. Drug
Resist Updat. 11:123–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shah AN, Summy JM, Zhang J, Park SI,
Parikh NU and Gallick GE: Development and characterization of
gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol.
14:3629–3637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|