Introduction
Primary gallbladder cancer (GBC) is a rare
malignancy of the digestive system ranking sixth most common,
however it is the most common malignancy of the biliary tract,
accounting for 80–95% of biliary tract cancers. In 2008, it
accounted for an estimate of 1.1% of all newly diagnosed cancer
cases, ranking 16th amongst all types of tumors; furthermore, it
accounted for 1.4% of all cancer-associated mortalities, ranking
9th amongst all cancers (1). Due
to its high degree of malignancy and few effective therapeutic
options, the prognosis of GBC patients is currently poor. In recent
years, the incidence of GBC has increased in northern India,
Pakistan and Korea (1,2). As GBC has no specific clinical
symptoms at the early stage, the majority of patients are diagnosed
at the advanced stage, at which palliative treatment is the only
option. Therefore, the five-year survival rate for GBC is only ~10%
(3). While chemotherapy is the
major treatment option for patients at advanced cancer stages, GBC
is resistant to standard cytotoxic drugs, and no effective
chemotherapeutic options are available at present (4). Hence, it is required to discover
novel drugs or means of sensitizing GBC cells to chemotherapeutics,
and to explore novel therapeutic strategies and improved
diagnostics.
In 1972 Chinese scientists extracted an effective
antimalarial component from Artemisia annua L, named
artemisinin (qinghaosu). Artemisinin belongs to the new
sesquiterpene lactone drugs and contains an endoperoxide moiety,
and its derivatives include dihydroartemisinin, artemether,
arteether and artesunate (5,6).
Traditional antimalarial drugs such as quinoline have been replaced
with artemisinin due to its lower toxicity and higher antimalarial
activities, and artemisinin has become the preferred drug of the
World Health Organization for treating Plasmodium falciparum
infection and cerebral malaria. Studies have revealed that besides
its anti-parasitic effects, artemisinin possessed further
pharmacological activities, including anti-inflammatory, antitumor
and immunomodulatory effects. The molecular mechanisms of the
anticancer effects of artemisinin are complex. It has been
demonstrated that artemisinin inhibits tumor proliferation,
angiogenesis, invasion and metastasis, induces cell cycle arrest
and apoptosis, reverses multidrug resistance and sensitizes cancer
cells to chemotherapy (7). A
number of studies have shown that artemisinin and its derivatives
inhibit the growth of colon cancer (8), ovarian cancer (9), lymph node metastasis of lung cancer
cells (10), breast cancer
(11), hepatocellular carcinoma
(12) and prostate cancer
(13) in vivo or in
vitro, while its efficacy against gallbladder cancer cells has
not been reported. Therefore, the present study was performed to
examine the effects of artemisinin on the proliferation, cell cycle
and apoptosis of the gallbladder cancer cell lines GBC-SD and NOZ
and to explore the underlying molecular mechanisms.
Materials and methods
Cell culture
The GBC-SD and NOZ gallbladder cancer cell lines
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China) and the Japanese Collection of Research
Bioresources Cell Bank (Osaka, Japan). Cell lines were maintained
at 37°C in Dulbecco's modified Eagle's medium (DMEM; Gibco, Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA), 1 mM
non-essential amino acids (Sigma-Aldrich) and 1%
penicillin/streptomycin (Sigma-Aldrich) at 37°C in a humidified
atmosphere containing 5% CO2. Cells were divided and
subcultured upon reaching 80% confluence and passaged with 0.25%
trypsin (Gibco).
Cell proliferation assay
Cells cultured in the presence or absence of
artemisinin were subjected to a viability assay using the WST-1
cell proliferation reagent (Roche, Mannheim, Germany). In brief,
5×103 cells were seeded into each well of a 96-well
plate and incubated for attachment overnight. Artemisinin was added
to the wells resulting in the following concentrations: 0, 2.5, 5,
10, 20, 40, 80 and 160 µM, and cells were incubated for 48
h. The wells were subsequently washed once with 100 µl
phosphate-buffered saline (PBS). DMEM (100 µl) containing
10% FBS was placed in the wells, followed by 10 µl WST-1
diluted 1:10 in culture medium. Following incubation for 2 h, the
optical density (OD) at 450 nm was measured using a Model 550
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell survival rates were calculated using the following equation:
(ODExperimental group)/(ODControl group) ×
100%.
Xenograft study
To evaluate the effects of artemisinin on
gallbladder tumors in vivo, GBC-SD and NOZ-derived xenograft
mouse models were used. All animal procedures were approved by the
Ethical Commission of the First Affiliated Hospital of Bengbu
Medical College (Bengbu, China). Male BALB/c nude mice (n=24; age,
5 weeks; weight, 250–300 g) were obtained from Shanghai SLAC
Laboratory Animal Co., Ltd. (Shanghai, China) and raised under
specific pathogen-free conditions. They were maintained in
conditions of 21°C and 50% humidity, under a 12-h light/dark cycle.
Mice had free access to food and water. After one week,
1×107 GBC-SD or NOZ cells were subcutaneously injected
into the right flank of each mouse (n=12 in each group) following
anesthesia with isoflurane (Merck Millipore, Darmstadt, Germany).
Tumors had formed in all mice after two weeks. When tumors reached
a volume of ~0.5 cm3, the mice were orally administered
artemisinin (100 mg/kg per day; dissolved in drinking water and
delivered by oral gavage) or a control over 30 days. The tumor
dimensions were measured every five days using a CD-6 CS caliper
(Mitutoyo Corporation, Kawasaki, Japan), and the tumor volume was
calculated according to the following modified ellipsoidal formula:
Tumor volume = 1/2(length × width2). On day 30 of
artemisinin administration, mice were anesthetized with isoflurane
prior to sacrifice by cervical dislocation, and tumors were
weighed.
Western blot analysis
Western blotting was used to determine the levels of
certain proteins. GBC-SD and NOZ cells were treated with
20µM artemisinin for 24 h. Total protein was extracted using
the Cell Lysis Buffer for Western and IP (Beyotime Institute of
Biotechnology, Haimen, China) according to the manufacturer's
protocols and the protein concentration of the extract was
determined using the Bradford Protein assay (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Equal amounts (25 µg) of protein
samples were separated using 12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (Beyotime Institute of
Biotechnology) and transferred onto a 0.2-µm polyvinylidene
difluoride membrane (EMD Millipore, Billerica, MA, USA). The
membrane was blocked in 5% non-fat milk. Membranes were probed with
primary antibodies overnight at 4°C. The primary antibodies used
were as follows: Polyclonal rabbit anti-phosphorylated
extracellular signal-regulated kinase 1/2 (p-ERK1/2; 1:200; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA; cat. no. sc-23759);
monoclonal mouse anti-p16 (1:100; Santa Cruz Biotechnology, Inc.;
cat. no. sc-377412); mouse monoclonal anti-β-actin (1:200; Santa
Cruz Biotechnology, Inc.; cat. no. sc-47778); mouse monoclonal
anti-cyclin D kinase (CDK)4 (1:200; Santa Cruz Biotechnology, Inc.;
cat. no. sc-23896); mouse monoclonal anti-cyclin D1 (1:200; Santa
Cruz Biotechnology, Inc.; cat. no. sc-20044); polyclonal rabbit
anti-caspase-3 (1:200; Santa Cruz Biotechnology, Inc.; cat. no.
sc-7148); polyclonal rabbit anti-poly(adenosine diphosphate ribose)
polymerase (PARP)-1 (1:200; Santa Cruz Biotechnology, Inc.; cat.
no. sc-7150); mouse monoclonal anti-cytochrome c (1:200;
Santa Cruz Biotechnology, Inc.; cat. no. sc-65396); and mouse
monoclonal anti-cycloxygenase IV (1:100; Santa Cruz Biotechnology,
Inc.; cat. no. sc-376731). The membranes were then washed three
times in Tris-buffered saline containing Tween 20 (TBST), incubated
with goat anti-mouse (1:2,000; Santa Cruz Biotechnology, Inc.; cat.
no. sc-2005) or anti-rabbit horseradish peroxidase antibody
(1:5,000; Santa Cruz Biotechnology, Inc.; cat. no. sc-2004) for 1 h
at room temperature and then washed three times in TBST. Protein
signals were visualized using an enhanced chemiluminescence
solution (ECL Plus; GE Healthcare, Little Chalfont, UK) and blots
were exposed to X-Omat LS film (Eastman Kodak Co., Rochester, NY,
USA). Cyclooxygenase IV and β-actin were used as the cytoplasmic
and mitochondrial marker, respectively.
Cell cycle analysis
To determine the effect of artemisinin on the cell
cycle of gallbladder cancer cells, GBC-SD and NOZ cells were seeded
in a six-well plate and incubated overnight for attachment.
Artemisinin (20 µM) was added to the wells of the plates for
24 h. After two washes with PBS, the cells were fixed in 70%
pre-cooled ethanol for 12 h. The cells were then treated with RNase
A (50 µg/ml; Sigma-Aldrich) and stained with propidium
iodide (PI; EMD Millipore, Billerica, MA, USA) according to the
manufacturer's instructions. Samples were analyzed on a FACSCalibur
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and their
DNA content was analyzed using ModFit LT software (version 3.1;
Verity Software House, Topsham, ME, USA).
Apoptosis assay
Annexin V-fluorescein isothiocyanate (FITC)/PI
staining (BD Biosciences) was used for apoptosis analysis. In
brief, cells were treated with artemisinin (20 µM) for 24 h,
harvested, rinsed with cold PBS and resuspended in Annexin V-FITC
binding buffer at a final density of 1×105 cells/ml.
Annexin V-FITC (5 µl) and 50 µg/ml PI (10 µl)
were added to 195 µl of the cell suspension. The cell
suspension was gently vortexed and then incubated for 15 min at
room temperature protected from light. Subsequently, samples were
analyzed using a FACSCalibur flow cytometer within 1 h.
Mitochondrial membrane potential (Δψm)
assay
Loss of Δψm is a fundamental early event in the
apoptotic process. The reagent
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimid-azolcarbocyanine
iodide (JC-1) is one of the most specific agents for measuring
changes in Δψm. The high Δψm of normal cells loaded with JC-1
allows for the formation of J-aggregates, which exhibit red
fluorescence. Upon loss of Δψm, the J-aggregates dissociate into
monomers, leading to a shift in fluorescence wavelength from red to
green. Therefore, JC-1 was used to assess changes in Δψm in the
present study. This assay was performed with the JC-1 mitochondrial
membrane potential detection kit (Biotium, Inc., Hayward, CA, USA).
Cell suspension containing 5×105 cells was centrifuged
at 500 × g for 5 min at room temperature and the cells were
resuspended in 0.5 ml JC-1 working solution at 37°C for 15 min.
Cells were then rinsed in 2 ml 1X JC-1 staining buffer twice and
resuspended in the same buffer prior to analysis by flow cytometry.
The fluorescence intensity of JC-1 was measured by flow cytometry
(FACSCalibur) with an excitation wavelength of 490 nm and an
emission wavelength of 530 nm for JC-1 monomers, and an excitation
wavelength of 525 nm and an emission wavelength of 590 nm for JC-1
aggregates.
Measurement of intracellular reactive
oxygen species (ROS)
A 2′,7′-dichlorofluorescein diacetate (DCFH-DA)
assay was used to measure intracellular ROS production. DCFH-DA
diffuses passively through the cellular membrane and is hydrolyzed
by intracellular esterase to the non-fluorescent DCFH. In the
presence of ROS, DCFH can be oxidized to the highly fluorescent
DCF. After incubation with artemisinin (20 µM) for 24 h,
cells were treated with DCFH-DA (10 µM in serum-free medium;
Sigma-Aldrich) at 37°C for 30 min. The fluorescence intensity of
DCF was measured using a fluorescence microplate reader with an
excitation wavelength of 488 nm and an emission wavelength of 525
nm.
Statistical analysis
All statistical analyses were performed by using
SPSS 14.0 (SPSS, Inc., Chicago, IL, USA). Values are expressed as
the mean ± standard deviation. For comparison of the means of two
groups, the independent samples t-test was used. For comparison of
the magnitude of changes among three or more groups, one-way
analysis of variance was used. All experiments of the present study
were performed independently at least three times.
Results
Artemisinin inhibits the proliferation of
GBC cells in vitro and in vivo
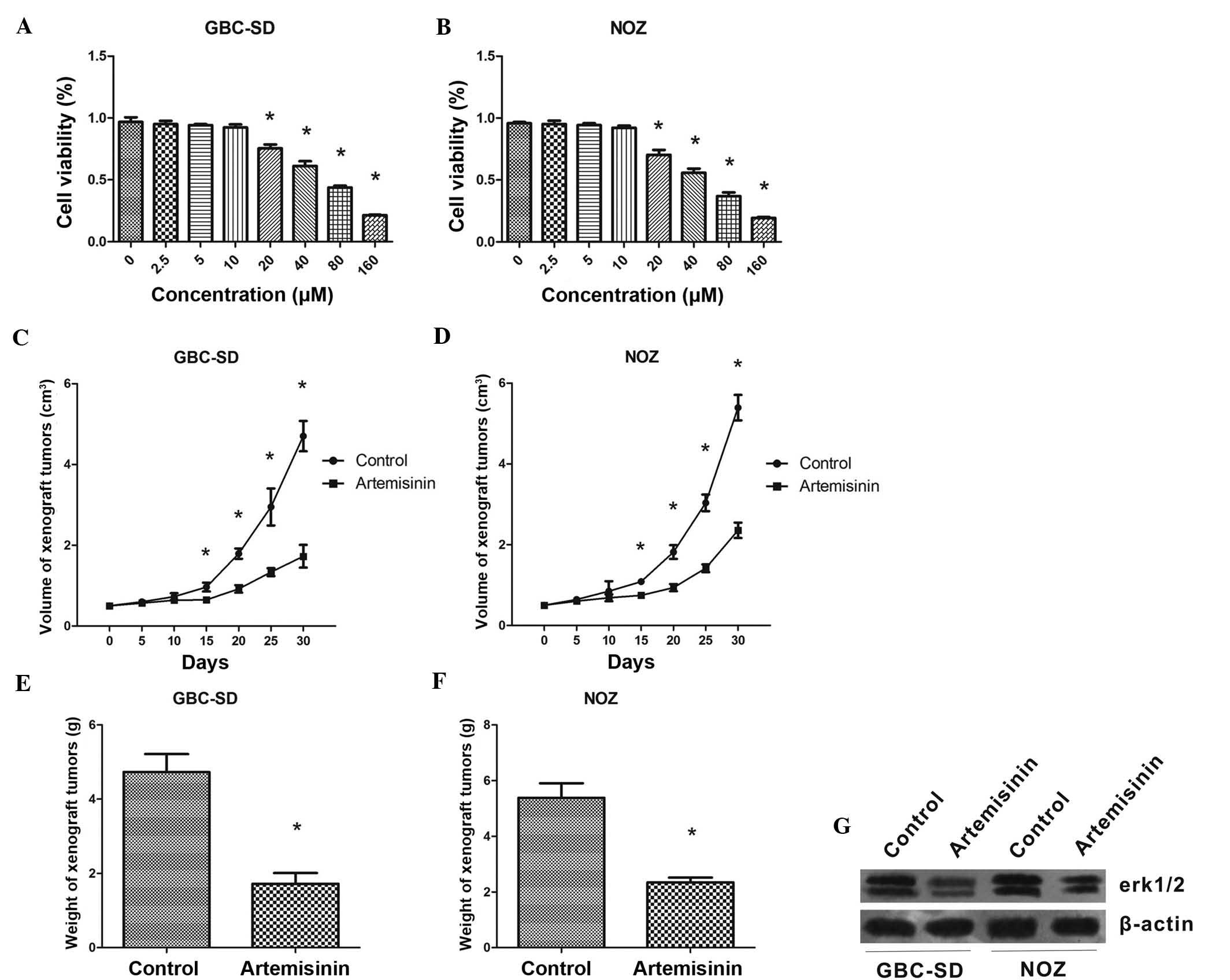
First, the effects of artemisinin on the
proliferation of GBC cells were assessed in vitro. A WST-1
assay showed that at concentrations of 20-160 µM,
artemisinin concentration-dependently inhibited the proliferation
of GBS-SD and NOZ cells (P<0.05) (Fig. 1A and B). The IC50 values
of artemisinin were 49.14±1.69 and 58.60±1.77 µM for GBS-SD
and NOZ cells, respectively.
In the xenograft experiment, artemisinin showed a
significant inhibitory effect on GBC cell-derived tumors in
vivo. From treatment day 15 onwards, mice treated with
artemisinin began to show a significantly reduced tumor volume
compared with that of the control mice (P<0.05) (Fig. 1C and D). The weight of tumors grown
in mice treated with artemisinin was also significantly lower than
that in the control animals (P<0.05) (Fig. 1E and F).
In addition, western blot analysis showed that after
24 h of treatment with artemisinin, the protein levels of p-ERK1/2
were obviously decreased in GBC-SD and NOZ cells. (Fig. 1G).
Artemisinin induces G1-phase arrest in
GBC-SD and NOZ cells via downregulation of CDK4 and cyclin D1 and
upregulation of p16
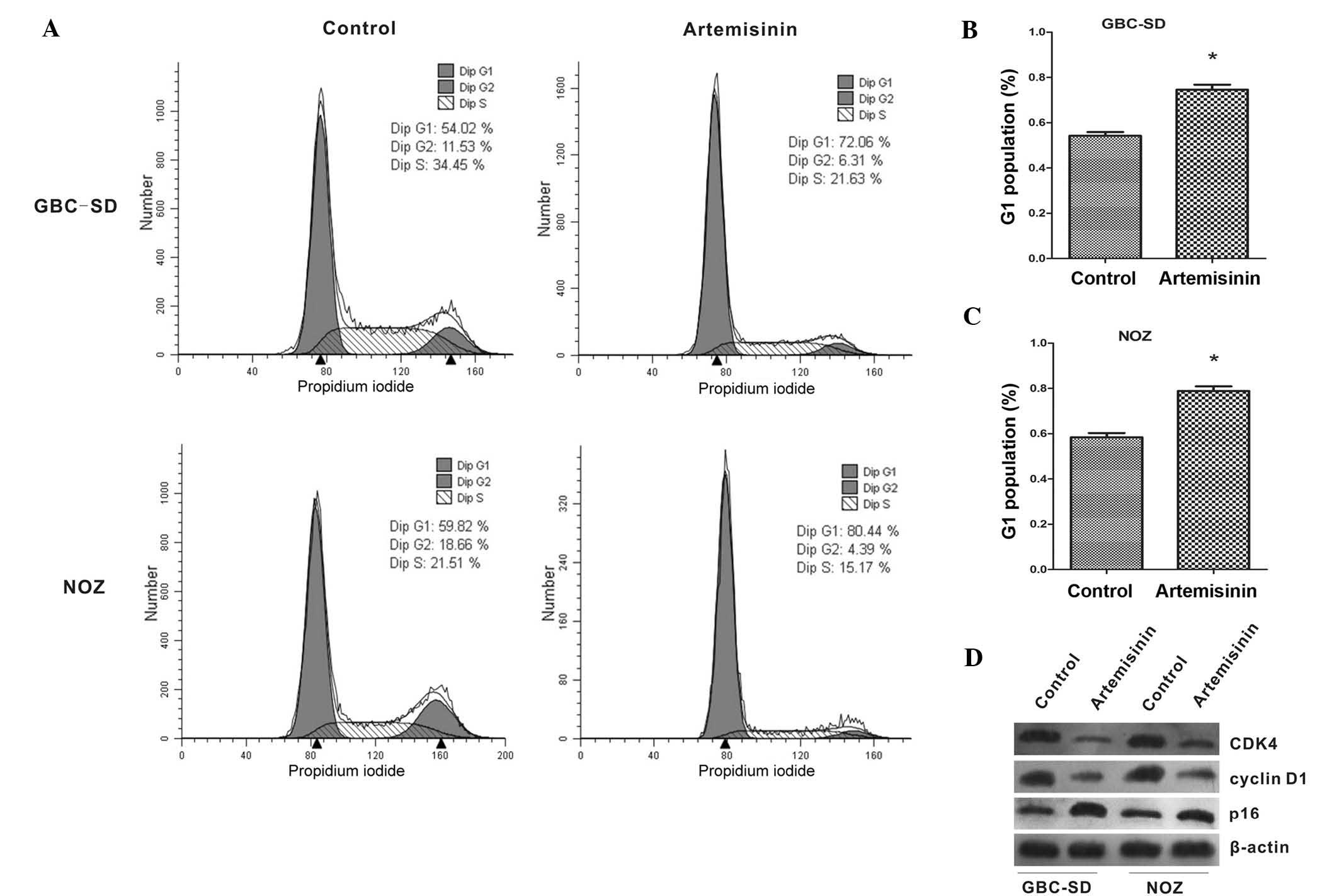
The effects of artemisinin on the cell cycle of
gallbladder cancer cells were further examined. As shown in
Fig. 2A–C, the proportion of cells
in G1 phase was significantly increased after treatment with
artemisinin when compared with that of the controls (P<0.05).
Furthermore, the expression of cell cycle-associated proteins was
analyzed by western blot. After treatment with artemisinin, the
expression of CDK4 and cyclin D1 was significantly decreased, while
p16 showed a marked increase (Fig.
2D).
Artemisinin induces apoptosis of GBC-SD
and NOZ cells through activation of caspase-3
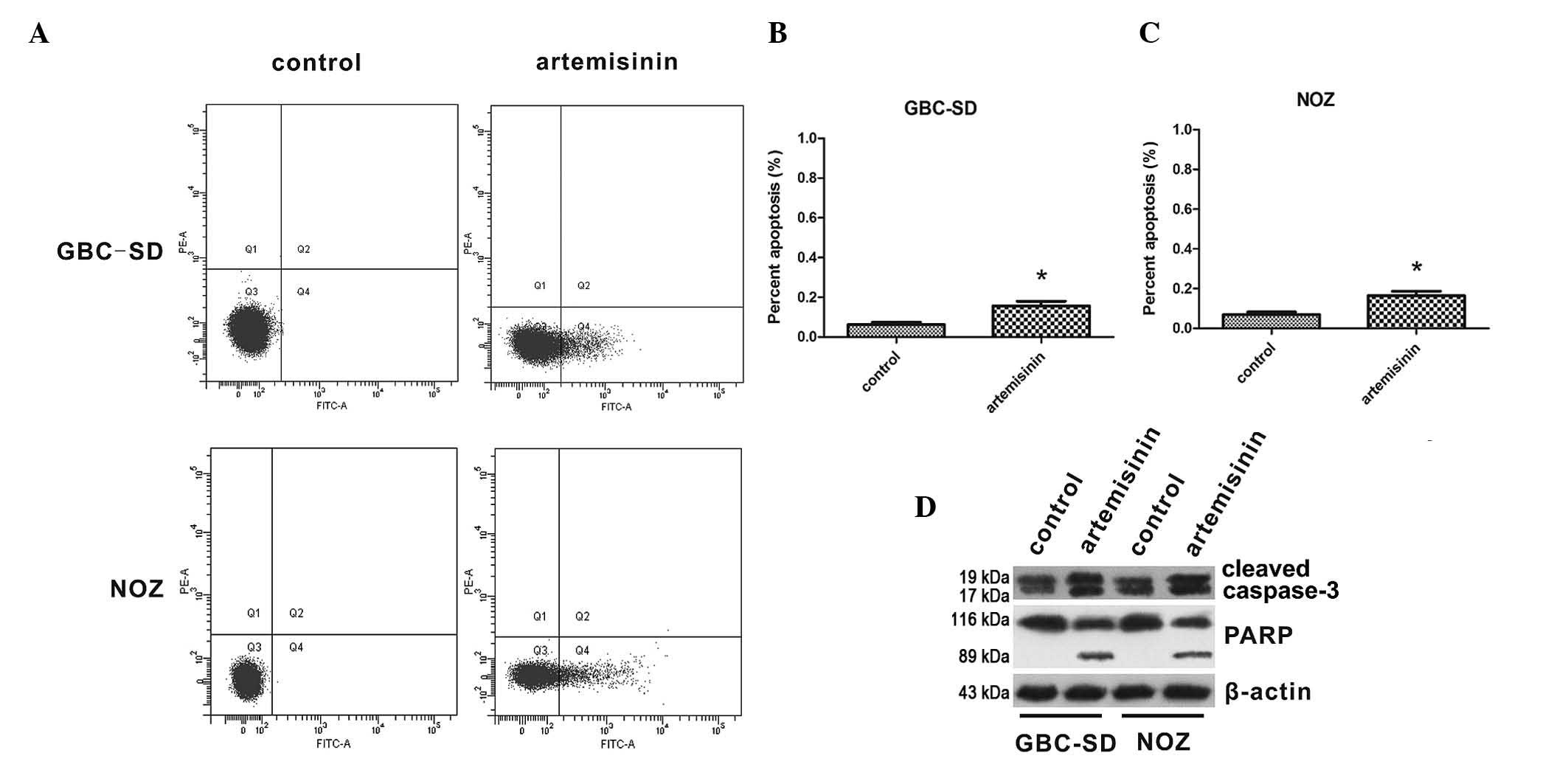
The effects of artemisinin on apoptosis of GBC cells
were then assessed. The apoptotic rates of GBC-SD and NOZ cells are
shown in Fig. 3A–C. The proportion
of apoptotic cells was significantly increased following treatment
with 20 µM artemisinin for 24 h compared to that in the
control group (P<0.05). Furthermore, the effect of artemisinin
on the levels of cleaved caspase 3 and cleaved PARP were detected,
indicating that caspase 3 and PARP were activated by artemisinin in
GBC-SD and NOZ cells (Fig.
3D).
Artemisinin induces Δψm collapse of
GBC-SD and NOZ cells via cytochrome c release
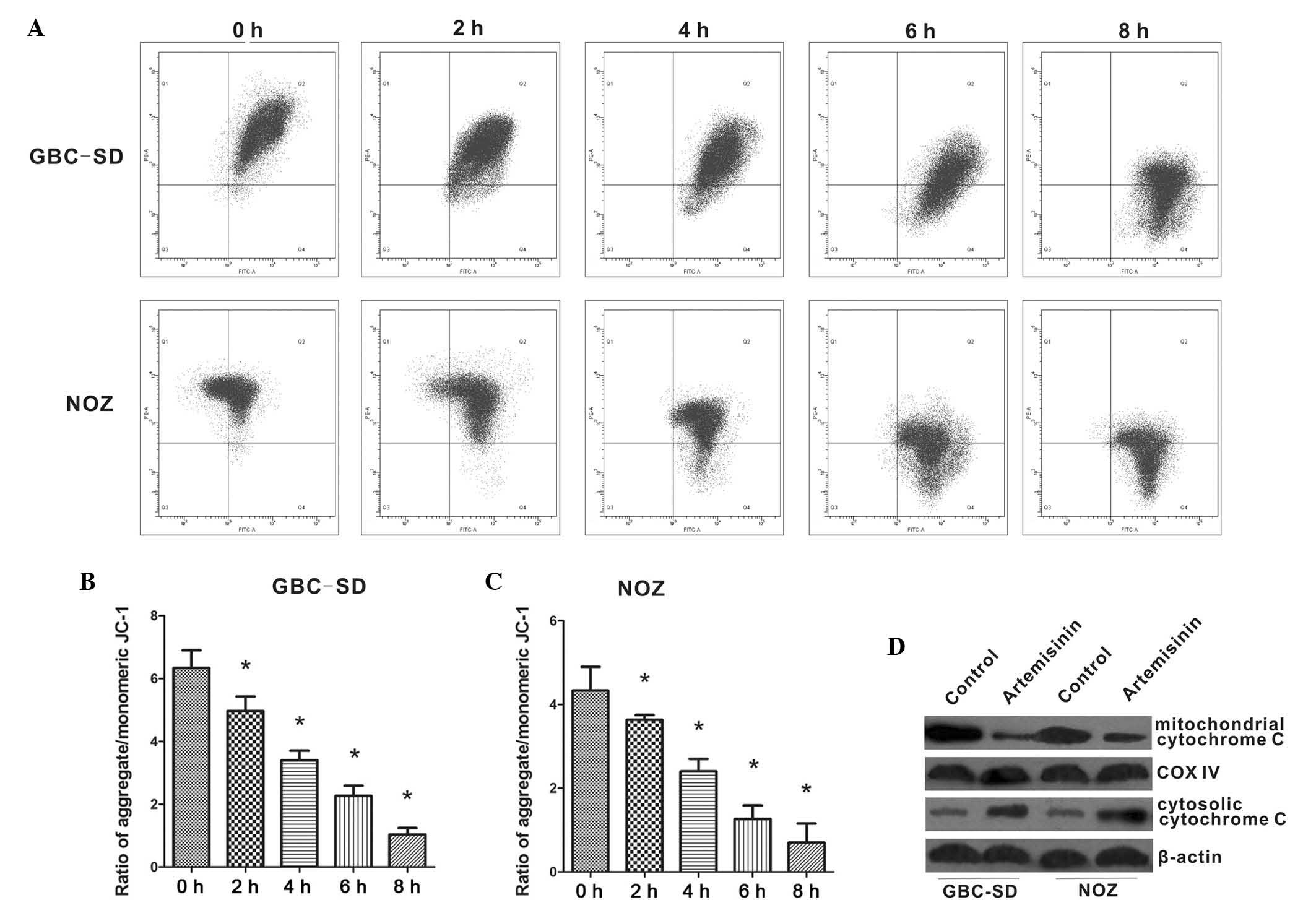
Furthermore, the present study examined the effects
of artemisinin on mitochondrial functions of GBC-SD and NOZ cells.
As shown in Fig. 4A–C, the
red/green fluorescence ratio was high in untreated GBC-SD and NOZ
cells. However, the addition of 20 µM artemisinin caused a
reduction of the red/green fluorescence ratio, which reflected the
collapse of the Δψm in artemisinin-treated cells. Immunoblotting of
cell extracts showed decreased mitochondrial cytochrome c in
cells treated with 20 µM artemisinin, while cytoplasmic
cytochrome c was significantly increased (Fig. 4D), suggesting that artemisinin
promotes cytochrome c release from mitochondria to
cytoplasm.
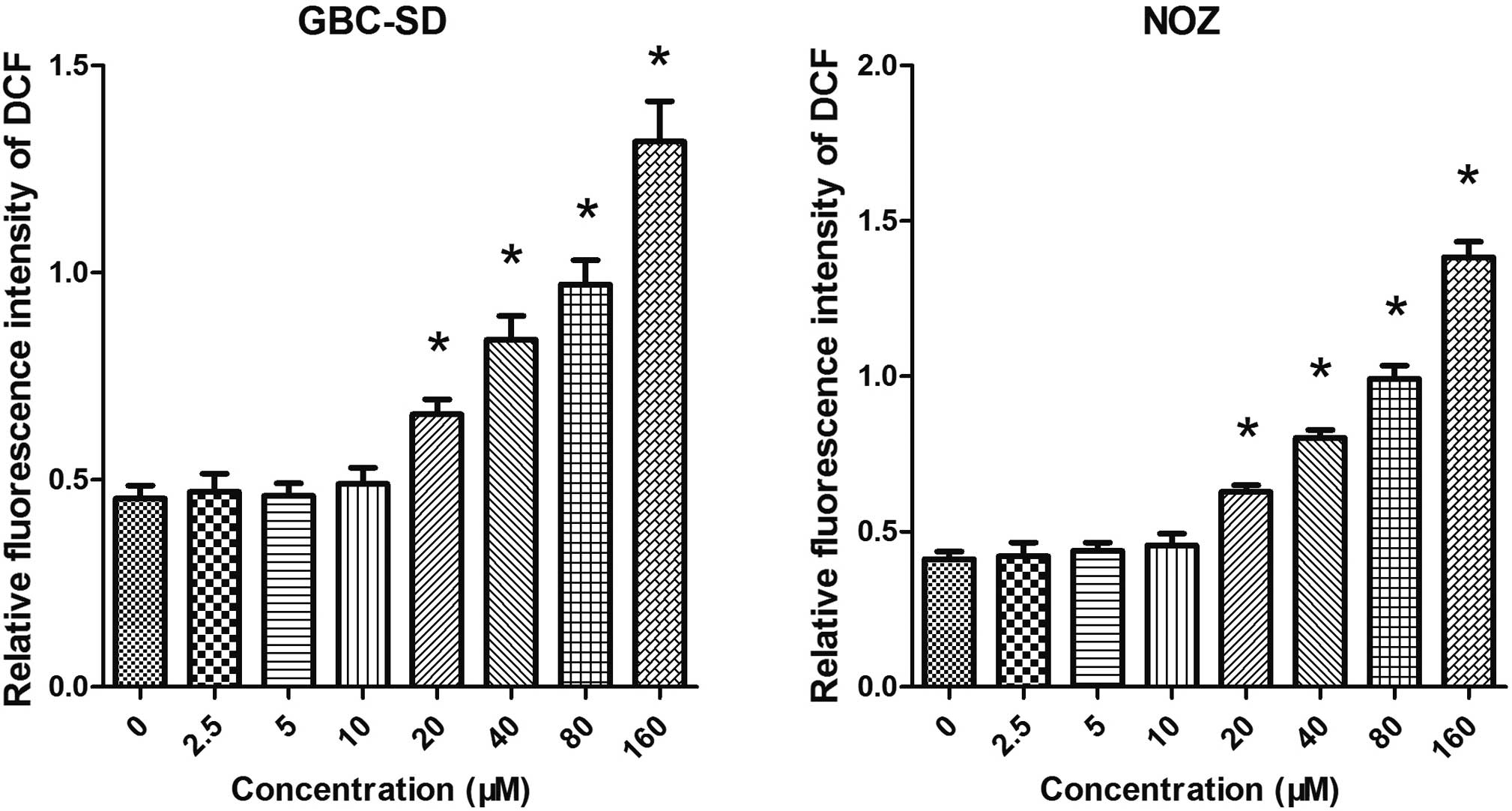
Artemisinin induces the generation of
ROS
As mitochondrial functions are tightly associated
with ROS generation, the present study further examined the effect
of artemisinin on the generation of ROS in GBC-SD and NOZ cells. As
shown in Fig. 5, cells treated
with artemisinin exhibited a significantly higher ROS-associated
fluorescence intensity. It was therefore indicated that artemisinin
can promote the generation of ROS.
Discussion
The five-year survival rate of GBC patients is
currently low due to insufficient diagnostic tools and inefficient
treatment strategies as well as the occurrence of drug resistance.
Therefore, it is urgently required to explore novel and effective
therapeutic options for patients with gallbladder cancer.
The aim of the present study was to assess the
potency of artemisinin, a pharmaceutically approved drug currently
used for the treatment of malaria and which has been demonstrated
to have inhibitory effects on a variety of cancer types, against
GBC and to explore the underlying molecular mechanisms. It was
demonstrated that artemisinin inhibited GBC cell proliferation
in vitro and in vivo, inhibited the cell cycle and
induced apoptosis, likely through the generation of ROS.
Studies have shown that artemisinin inhibits the
proliferation of numerous tumor cell types (8–13).
The present study was the first to reveal that artemisinin also
inhibited the proliferation of the GBC cell lines GBC-SD and NOZ.
An in vitro cell viability demonstrated that and artemisinin
(20–160 µM) exerted its growth inhibitory effects in a
concentration-dependent manner. The calculated IC50
values for GBC-SD and NOZ cells were 49.14±1.69 and 58.60±1.77
µM, respectively. In addition, artemisinin showed
significant inhibitory activity against GBC cell-derived tumors in
a murine xenograft model.
The PI3K/Akt/ERK1/2 signaling pathway is critical in
cell proliferation and apoptosis (14). ERK1/2 is highly expressed or
constitutively activated in a variety of tumor types, including
gallbladder cancer (15,16). Inhibition of ERK1/2 expression or
activity can suppress tumor cell proliferation (17). The present study revealed that
cells treated with artemisinin reduced the expression of p-ERK1/2,
indicating that the inhibition of cell proliferation by artemisinin
may by dependent on the suppressed activation of the ERK1/2
signaling pathway.
It has been indicated that artemisinin and its
derivatives may inhibit tumor cell proliferation through blocking
the cell cycle. Studies have shown that the drugs can induce cell
cycle arrest at the different phases, such as G1-phase arrest in
Ishikawa endometrial carcinoma (18), MCF7 breast cancer (19), A431 skin cancer (20) and LNCaP prostate cancer (21) cells, or G2/M phase arrest in A549
non-small cell lung cancer (22),
H69 small cell lung cancer, HCT116 colon cancer and U251 glioma
cells (23). These data suggested
that the cell cycle regulatory effect of artemisinin and its
derivatives is cell type specific, possibly due to differences in
the regulation of certain key cell cycle regulators. However, the
effect of artemisinin on the cell cycle of GBC cell lines has not
been previously reported. The present study revealed that
artemisinin induced G1-phase arrest in GBC-SD and NOZ cells. To
examination of the underlying molecular mechanisms, the levels of
certain cell cycle regulatory proteins were assessed. As is known,
the transformation from G1 phase to S phase is a key step during
cell cycle progression, and the G1/S checkpoint is regulated by
multiple proteins. CDKs, the key regulators of cell cycle
progression, can be activated during a specific cell cycle phase
and subsequently form complexes with cyclins such as the
CDK4/6-cyclin D1 complex, which triggers downstream events that
promote cell cycle progression. The cyclin-dependent kinase
inhibitor protein p16 can competitively bind with CDK4/6 to inhibit
its activation and block cell cycle progression by triggering G1
arrest (24). The present study
revealed that artemisinin treatment upregulated the expression of
p16 protein while downregulating CDK4 and cyclin D1 expression,
suggesting that artemisinin may induce G1-phase arrest by
regulating the p16/CDK4/cyclin D1 pathway.
Besides induction of cell cycle arrest, artemisinin
can also inhibit tumor cell proliferation via induction of
apoptosis (25–27). Studies have shown that artemisinin
can activate the p53-dependent and p53-independent apoptotic
pathway (28–30), and that active oxygen has a crucial
role in artemisinin-induced apoptosis (31). When reacting with iron, artemisinin
produces large amounts of free radicals (32). Increases in intracellular ROS lead
to the opening of the mitochondrial permeability transition pore
(MPT), thus increasing the mitochondrial membrane permeability,
resulting in a reduction of the Δψm. ROS can trigger and accelerate
the opening of the MPT, which in turn promotes the generation of
ROS (33,34). This feedback mechanism leads to a
self-amplifying effect of ROS; thus, the opening of the MPT
triggers an irreversible decrease of Δψm, leading to cell
apoptosis. The present study found that artemisinin can induce the
generation of ROS in GBC-SD and NOZ cells, along with the collapse
of Δψm and activation of caspase-3, a key executioner of apoptosis.
These results demonstrated that artemisinin exerts its anti-cancer
effects by activating the ROS-mediated mitochondrial apoptotic
pathway.
In conclusion, the present study showed that
artemisinin can inhibit the proliferation of GBC-SD and NOZ cells
by generating ROS. It also triggers G1-phase arrest through
upregulation of p16 protein expression and downregulation of CDK4
and cyclin D1 expression, as well as downregulation of ERK1/2. The
ROS generated by artemisinin activate mitochondrial-mediated
apoptosis. All of these findings suggested that artemisinin may be
used as a novel drug for gallbladder cancer, which is facilitated
by its previous approval for pharmaceutical use.
Acknowledgments
This study was supported by the Key Program of
Bengbu Medical College (no. Bykf12A05) and the Key Program of the
Foundation for Youth Talents in colleges and universities of Anhui
province (no. 2013SQRL052ZD).
References
|
1
|
Hundal R and Shaffer EA: Gallbladder
cancer: Epidemiology and outcome. Clin Epidemiol. 6:99–109.
2014.PubMed/NCBI
|
|
2
|
Eslick GD: Epidemiology of gallbladder
cancer. Gastroenterol Clin North Am. 39:307–330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Misra S, Chaturvedi A, Misra NC and Sharma
ID: Carcinoma of the gallbladder. Lancet Oncol. 4:167–176. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caldow Pilgrim CH, Groeschl RT, Quebbeman
EJ and Gamblin TC: Recent advances in systemic therapies and
radiotherapy for gallbladder cancer. Surg Oncol. 22:61–67. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miller LH and Su X: Artemisinin: Discovery
from the Chinese herbal garden. Cell. 146:855–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu YX, Wu W, Liang YJ, Jie ZL, Wang H,
Wang W and Huang YX: New uses for old drugs: The tale of
artemisinin derivatives in the elimination of Schistosomiasis
japonica in China. Molecules. 19:15058–15074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ho WE, Peh HY, Chan TK and Wong WS:
Artemisinins: Pharmacological actions beyond antimalarial.
Pharmacol Ther. 142:126–139. 2014. View Article : Google Scholar
|
|
8
|
Lu JJ, Chen SM, Zhang XW, Ding J and Meng
LH: The anticancer activity of dihydroartemisinin is associated
with induction of iron-dependent endoplasmic reticulum stress in
colorectal carcinoma HCT116 cells. Invest New Drugs. 29:1276–1283.
2011. View Article : Google Scholar
|
|
9
|
Chen T, Li M, Zhang R and Wang H:
Dihydroartemisinin induces apoptosis and sensitizes human ovarian
cancer cells to carboplatin therapy. J Cell Mol Med. 13:1358–1370.
2009. View Article : Google Scholar
|
|
10
|
Wang J, Zhang B, Guo Y, Li G, Xie Q, Zhu
B, Gao J and Chen Z: Artemisinin inhibits tumor lymphangiogenesis
by suppression of vascular endothelial growth factor C.
Pharmacology. 82:148–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sundar SN, Marconett CN, Doan VB,
Willoughby JA Sr and Firestone GL: Artemisinin selectively
decreases functional levels of estrogen receptor-alpha and ablates
estrogen-induced proliferation in human breast cancer cells.
Carcinogenesis. 29:2252–2258. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weifeng T, Feng S, Xiangji L, Changqing S,
Zhiquan Q, Huazhong Z, Peining Y, Yong Y, Mengchao W, Xiaoqing J
and Wan-Yee L: Artemisinin inhibits in vitro and in vivo invasion
and metastasis of human hepatocellular carcinoma cells.
Phytomedicine. 18:158–162. 2011. View Article : Google Scholar
|
|
13
|
Morrissey C, Gallis B, Solazzi JW, Kim BJ,
Gulati R, Vakar-Lopez F, Goodlett DR, Vessella RL and Sasaki T:
Effect of artemisinin derivatives on apoptosis and cell cycle in
prostate cancer cells. Anticancer Drugs. 21:423–432. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lei YY, Wang WJ, Mei JH and Wang CL:
Mitogen-activated protein kinase signal transduction in solid
tumors. Asian Pac J Cancer Prev. 15:8539–8548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Q and Yang Z: Expression of
phospho-ERK1/2 and PI3-K in benign and malignant gallbladder
lesions and its clinical and pathological correlations. J Exp Clin
Cancer Res. 28:652009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jinawath A, Akiyama Y, Yuasa Y and
Pairojkul C: Expression of phosphorylated ERK1/2 and homeodomain
protein CDX2 in cholangiocarcinoma. J Cancer Res Clin Oncol.
132:805–810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
18
|
Tran KQ, Tin AS and Firestone GL:
Artemisinin triggers a G1 cell cycle arrest of human Ishikawa
endometrial cancer cells and inhibits cyclin-dependent kinase-4
promoter activity and expression by disrupting nuclear factor-kB
transcriptional signaling. Anticancer Drugs. 25:270–281. 2014.
View Article : Google Scholar :
|
|
19
|
Tin AS, Sundar SN, Tran KQ, Park AH,
Poindexter KM and Firestone GL: Antiproliferative effects of
artemisinin on human breast cancer cells requires the downregulated
expression of the E2F1 transcription factor and loss of E2F1-target
cell cycle genes. Anticancer Drugs. 23:370–379. 2012. View Article : Google Scholar
|
|
20
|
Jiang Z, Chai J, Chuang HH, Li S, Wang T,
Cheng Y, Chen W and Zhou D: Artesunate induces G0/G1 cell cycle
arrest and iron-mediated mitochondrial apoptosis in A431 human
epidermoid carcinoma cells. Anticancer Drugs. 23:606–613. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Willoughby JA Sr, Sundar SN, Cheung M, Tin
AS, Modiano J and Firestone GL: Artemisinin blocks prostate cancer
growth and cell cycle progression by disrupting Sp1 interactions
with the cyclin-dependent kinase-4 (CDK4) promoter and inhibiting
CDK4 gene expression. J Biol Chem. 284:2203–2213. 2009. View Article : Google Scholar :
|
|
22
|
Zhao Y, Jiang W, Li B, Yao Q, Dong J, Cen
Y, Pan X, Li J, Zheng J, Pang X and Zhou H: Artesunate enhances
radiosensitivity of human non-small cell lung cancer A549 cells via
increasing no production to induce cell cycle arrest at G2/M phase.
Int Immunopharmacol. 11:2039–2046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steinbrück L, Pereira G and Efferth T:
Effects of artesunate on cytokinesis and G2/M cell cycle
progression of tumour cells and budding yeast. Cancer Genomics
Proteomics. 7:337–346. 2010.
|
|
24
|
Semczuk A and Jakowicki JA: Alterations of
pRb1-cyclin D1-cdk4/6-p16 (INK4A) pathway in endometrial
carcinogenesis. Cancer Lett. 203:1–12. 2004. View Article : Google Scholar
|
|
25
|
Singh NP and Lai HC: Artemisinin induces
apoptosis in human cancer cells. Anticancer Res. 24:2277–2280.
2004.PubMed/NCBI
|
|
26
|
Nam W, Tak J, Ryu JK, Jung M, Yook JI, Kim
HJ and Cha IH: Effects of artemisinin and its derivatives on growth
inhibition and apoptosis of oral cancer cells. Head Neck.
29:335–340. 2007. View Article : Google Scholar
|
|
27
|
Hou J, Wang D, Zhang R and Wang H:
Experimental therapy of hepatoma with artemisinin and its
derivatives: In vitro and in vivo activity, chemosensitization and
mechanisms of action. Clin Cancer Res. 14:5519–5530. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamachika E, Habte T and Oda D:
Artemisinin: An alternative treatment for oral squamous cell
carcinoma. Anticancer Res. 24:2153–2160. 2004.PubMed/NCBI
|
|
29
|
Disbrow GL, Baege AC, Kierpiec KA, Yuan H,
Centeno JA, Thibodeaux CA, Hartmann D and Schlegel R:
Dihydroartemisinin is cytotoxic to papillomavirus-expressing
epithelial cells in vitro and in vivo. Cancer Res. 65:10854–10861.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Efferth T: Mechanistic perspectives for
1,2,4-trioxanes in anticancer therapy. Drug Resist Updat. 8:85–97.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Hu W, Zhang JL, Wu XH and Zhou HJ:
Dihydroartemisinin induces autophagy and inhibits the growth of
iron-loaded human myeloid leukemia K562 cells via ROS toxicity.
FEBS Open Bio. 2:103–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haynes RK, Cheu KW, N'Da D, Coghi P and
Monti D: Considerations on the mechanism of action of artemisinin
antimalarials: Part 1-the 'carbon radical' and 'heme' hypotheses.
Infect Disord Drug Targets. 13:217–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
34
|
Oh SH and Lim SC: A rapid and transient
ROS generation by cadmium triggers apoptosis via caspase-dependent
pathway in HepG2 cells and this is inhibited through
N-acetylcysteine-mediated catalase upregulation. Toxicol Appl
Pharmacol. 212:212–223. 2006. View Article : Google Scholar
|