Introduction
Traumatic brain injury (TBI) is a leading cause of
mortality and disability in young individuals and represents an
important public health issue worldwide. Primary and secondary
injury cascades resulting in delayed neuronal dysfunction, synapse
loss and cell death are associated with TBI (1,2).
However, the precise mechanisms underlying the secondary injury are
not well understood, and one result of TBI is long-term cognitive
dysfunction and memory loss, which affects all patients with TBI. A
number of studies have demonstrated that TBI leads to synaptic loss
(1,3), or disrupts synaptic plasticity
(4), which contributes to
long-term behavioral disorders. However, the molecular mechanisms
by which TBI causes synapse loss in the early stages remain
unclear.
Autophagy is an evolutionarily conserved pathway
that results in the degradation of proteins and entire organelles
in cells undergoing stress (5).
Although autophagy constitutes a stress adaptation pathway that
promotes cell survival under the majority of circumstances,
increasingly studies have demonstrated that it may trigger cell
injury and death under certain pathological circumstances (6,7).
Previous studies have demonstrated that the autophagic pathway is
involved in the pathophysiological response following TBI, and
inhibition of this pathway may help to attenuate traumatic damage
and functional deficits (8–10).
Resveratrol (3,5,40-trihydroxystilbene, RV) occurs
naturally in grapes and a variety of medicinal plants, and exhibits
multiple biological activities (11,12).
In particular, RV exerts protective effects against neurological
damage in a number of animal models, including stroke, spinal cord
injury and neurodegenerative diseases (13–15).
The beneficial effects of RV in central nervous system injuries are
associated with its anti-oxidant (16), anti-inflammatory (17) and anti-apoptotic properties
(18). However, the protective
effect and mechanisms of RV treatment following TBI require further
examination.
In the current study, the effect of RV on post-TBI
brain edema, spatial cognitive function and neurological impairment
was examined in a rat model. Synaptic proteins and neuronal
autophagy were also assessed. The results may provide evidence of
RV-mediated neuroprotection in a rat model of TBI.
Materials and methods
Animals and TBI model
All experimental procedures were conducted in
accordance with the guidelines of the Chinese council on animal
protection (http://www.hebstd.gov.cn/banshi/zxyw/content_74239.htm)
and were approved by the Hebei Medical University (Shijiazhuang,
China) Committee for the use of animals in research. A total of 150
adult (age, 12–16 weeks) male Sprague-Dawley rats weighing 300–330
g were obtained from Hebei United University Experimental Animal
Center (Tangshan, China). The animals were maintained at 21–26°C
under a 12-h light/dark cycle, and water and food were provided
ad libitum prior to and following surgery or sham surgery.
The rat model of TBI was induced using a weight-drop device, as
previously described by Marmarou et al (19). Briefly, the rats were anesthetized
with 10% chloral hydrate (3 ml/kg). A midline incision was made to
expose the skull between the bregma and lambda suture lines and a
steel disc (10 mm in diameter and 3 mm thickness) was adhered to
the skull using dental acrylic. Then rats were placed on a foam
mattress underneath a weight-drop device in which a 350 g weight
falls freely through a vertical tube from 1.5 m onto the steel
disk. Following injury, the scalp was sutured. Rats were housed in
individual cages and placed on heat pads (37°C) for 24 h to
maintain normal body temperature during the recovery period. This
model is generally associated with 20% mortality within the first 5
min post-injury and no delayed mortality was observed thereafter.
The sham-operated animals were anesthetized and had the steel disk
attached to them, but they did not receive TBI.
Groups and drug administration
Rats were randomly divided into three groups: Sham
operation group (sham, n=30); TBI group (TBI, n=60); and TBI
treated with RV group (RV, n=60). RV (Sigma-Aldrich, St. Louis, MO,
USA) was freshly prepared by dissolving in 50% ethanol and diluting
in physiological saline (2%) at a concentration of 100 mg/kg, and
was administered daily via intraperitoneal injection to the rats in
the RV group for 5 days beginning immediately following TBI
(14). Both the sham and TBI
groups received equal volumes of ethanol (2%) by intraperitoneal
injection at the same time daily. Each subgroup was composed of
five rats and the rats were sacrificed at 1, 3 and 5 days following
TBI.
Evaluation of brain edema
Brain edema was evaluated by analysis of brain water
content using the wet-dry weight method, as described previously
(20). Briefly, rats were randomly
sampled from each group and anesthetized by intra-peritoneal
injection with 10% chloral hydrate (3 ml/kg; n=5). The cerebral
tissues were then removed and weighed immediately on an electric
analytic balance for the wet weight, then dried at 100°C for 24 h
to obtain the dry weight. The percentage of water in the tissues
was calculated according to the formula: % brain water = [(wet
weight - dry weight)/wet weight] × 100.
Morris water maze (MWM) test
To evaluate spatial learning and memory, rats were
tested in variation of the MWM paradigm (21). The maze consists of a water-filled
pool (180 cm diameter, 45 cm high) at 26°C and virtually divided
into 4 equivalent quadrants: North (N), west (W), south (S) and
east (E). A 2 cm submerged escape platform (diameter 12 cm, made
opaque with paint) was placed in the middle of one of the quadrants
equidistant from the sidewall and the center of the pool. All
experimental rats were trained to find the platform prior to TBI or
sham surgery. At the start of a trial, the rat was placed at one of
four fixed starting points, randomly facing a wall (designated N,
S, E and W) and allowed to swim for 60 sec or until it found the
platform. If the animal found the platform, it was allowed to
remain on it for 20 sec. If the animal failed to find the platform
within 90 sec, it was placed on the platform for 20 sec. The time
required (escape latency) to find the hidden platform with a 60 sec
limit was recorded by a video camera suspended above the maze and
interfaced with a video tracking system (HVS Imaging, Hampton, UK).
The test was conducted at 3–5 days following TBI or sham surgery,
and each rat was tested for four trials per day for three
consecutive days. The average escape latency of the total of the
four trials was calculated.
Modified neurological severity score
Neurological deficits were evaluated using the
Neurologic Severity Score (NSS) on a 10-point scale, as previously
described by Chen et al (22) by a researcher blinded to treatment.
One point is awarded for failure to perform a particular task, and
a score of ten reflects maximal impairment, thus, a normal rat
scores zero. Post-injury, the NSS was evaluated at days 1–5.
Western blot analysis
Western blotting was performed as previously
described (23). At scheduled time
points, rats were anesthetized with 10% chloral hydrate and
decapitated. The brains were quickly removed and the hippocampal
tissues were dissected on ice. Total proteins were extracted and
the protein concentration was determined using a bicinchoninic acid
kit (Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China). Equal amounts of protein (50 mg) was subjected to 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Beyotime
Institute of Biotechnology, Haimen, China). Separated proteins on
the gel were transferred onto polyvinylidene fluoride membranes
(Roche Diagnostics GmbH, Mannheim, Germany) by a transfer apparatus
at 200 mA for 50 min. The membrane was then blocked with 5%
fat-free dry milk for 2 h at room temperature. Subsequently, blots
were incubated with the following primary antibodies overnight at
4°C: Polyclonal rabbit anti-microtubule-associated protein light
chain 3 (LC3) [cat. no. PD014; Molecular & Biological
Laboratories Co., Ltd., Nagoya, Japan (dilution, 1:1,000)],
polyclonal rabbit anti-Beclin1 [cat. no. JM-3663-100; Molecular
& Biological Laboratories Co., Ltd. (dilution, 1:1,000)],
polyclonal rabbit anti-p62 [cat. no. ARH4210; Antibody Revolution,
Inc. San Diego, CA, USA (dilution, 1:1,000)], monoclonal rabbit
anti-postsynaptic density protein 95 (PSD95) [cat. no. ab76108;
Abcam, Cambridge, UK (dilution,1:500)], monoclonal rabbit
anti-synaptophysin (SYN) [cat. no. ARH4007; Antibody Revolution,
Inc. (dilution, 1:500)], polyclonal rabbit anti-β-actin [cat. no.
AF7018; Affinity Biologicals, Inc., Cincinnati, OH, USA (dilution,
1:1,000)]. The membranes were then incubated with horseradish
peroxidase-conjugated anti-rabbit IgG [cat. no. sc-2004; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA (dilution, 1:5,000)] for 2 h
at room temperature, prior to development using an enhanced
chemiluminescence detection system [MultiSciences (LIANKE) Biotech
Co., Ltd., Hangzhou, China] and the densitometric signals were
quantified using an imaging program (Image Lab 4.1; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The immunoreactive bands
were normalized to rabbit anti-β-actin polyclonal antibody
(dilution, 1:1,000). Results were analyzed using the National
Institutes of Health ImageJ 1.41 software (Bethesda, MD, USA).
Immunofluorescence analyses
Rats were perfused transcardially with saline under
deep anesthesia [10% chloral hydrate (3 ml/kg) by intraperitoneal
injection]. The brain tissues were the fixed using 4% formaldehyde
for 24 h, transferred to a 30% sucrose solution [0.1 M
phosphate-buffered saline (PBS), pH 7.4] until they sank to the
bottom, following which they were embedded in optimal cutting
temperature compound. The brains were cut into 15 µm-thick
sections coronally from the anterior to posterior hippocampus
(bregma −2.0 to −3.5 mm) using a cryostat. Frozen sections were
treated with 0.4% Triton X-100 for 30 min, and blocked in normal
donkey serum for 1 h. For double labeling, the frozen sections were
incubated with a mixture of polyclonal rabbit anti-LC3 (1:100) and
monoclonal mouse anti-NeuN [cat. no. MAB377; EMD Millipore,
Billerica, MA, USA (dilution, 1:100)] antibodies overnight at 4°C.
The following day, the sections were incubated with a mixture of
donkey anti-rabbit IgG-fluorescein isothiocyanate (FITC) and donkey
anti-mouse IgG-FITC [cat. nos. sc-2090 and sc-2099, respectively;
Santa Cruz Biotechnology, Inc. (dilution, 1:1,000)] for 2 h at 37°C
in the dark. All cell nuclei were counterstained by
4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a
laser scanning confocal microscope (Olympus Fluoview™ FV1000;
Olympus Corporation, Tokyo Japan). Primary antibodies were replaced
with PBS in the negative control group.
Statistical analysis
Statistical analysis was performed using SPSS
software, version 16.0 (SPSS, Chicago, IL, USA). Data are presented
as the mean ± standard deviation. Statistical analysis was
performed using one-way analysis of variance followed by the
Student-Newman-Keuls post hoc multiple comparisons test. P<0.05
was considered to indicate a statistically significant
difference.
Results
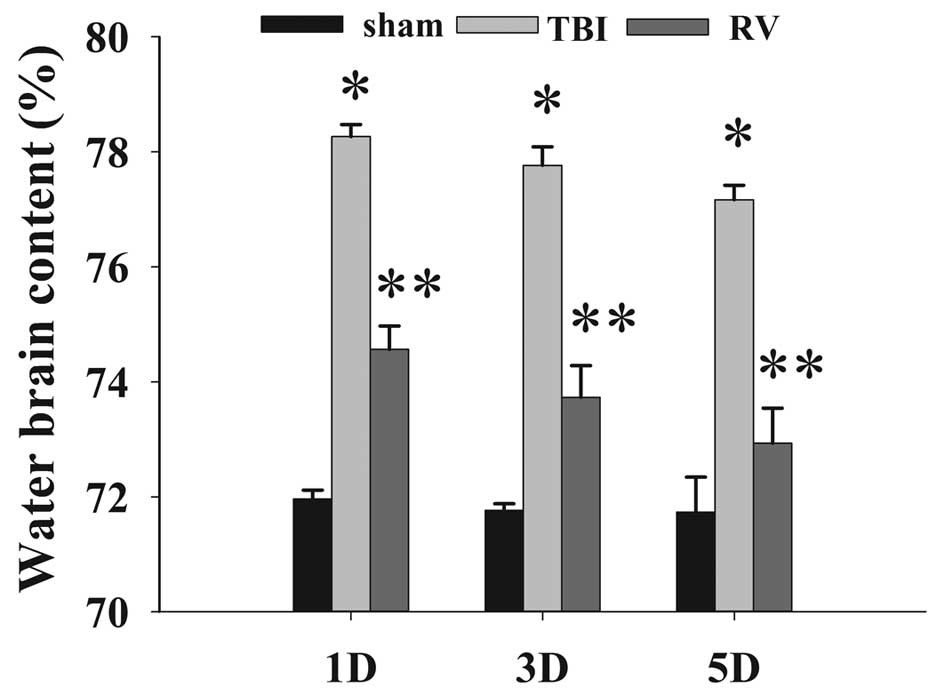
RV treatment attenuates brain edema
The wet-dry weight method was used to evaluate brain
edema. As shown in Fig. 1, TBI
induced a significant increase in brain edema at 1, 3 and 5 days in
the TBI group compared with the sham control. RV administered
post-injury significantly reduced brain edema following TBI.
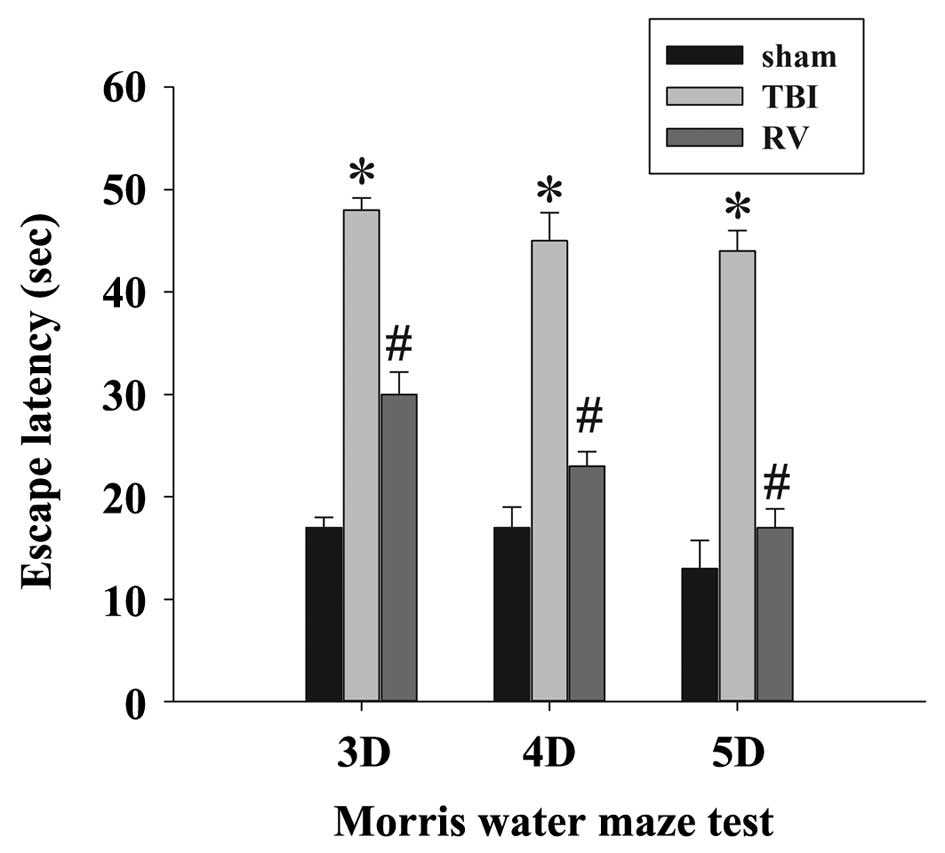
RV treatment improves learning and memory
ability and motor deficits
To determine the neuroprotective effects of RV
against TBI-induced brain damage, the effects of RV pretreatment on
the learning and memory ability (Fig.
2) and motor deficits (Fig. 3)
were examined using the MWM and NSS score following TBI,
respectively. As expected, TBI resulted in a significant deficit in
spatial learning compared with the sham group, and the
administration of RV significantly reduced the escape latency at 3,
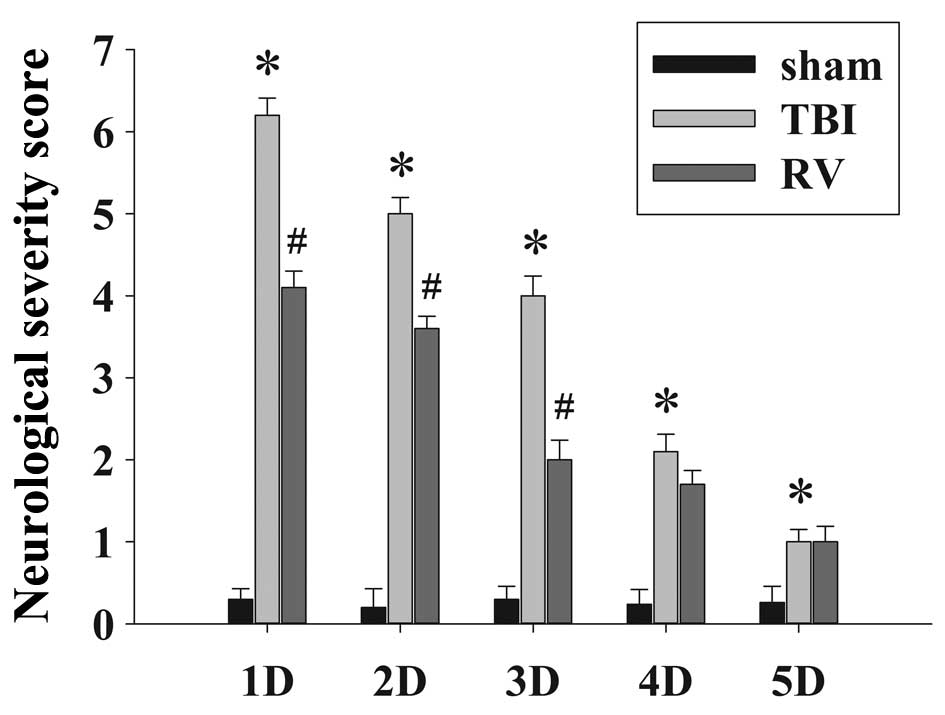
4 and 5 days compared with the TBI group. In addition, the NSS of
the rats in the TBI group were observed to significantly increase
in comparison with the sham group at 1–4 days, and RV treatment
significantly improved the motor function recovery of the TBI rats
at 1–3 days following injury.
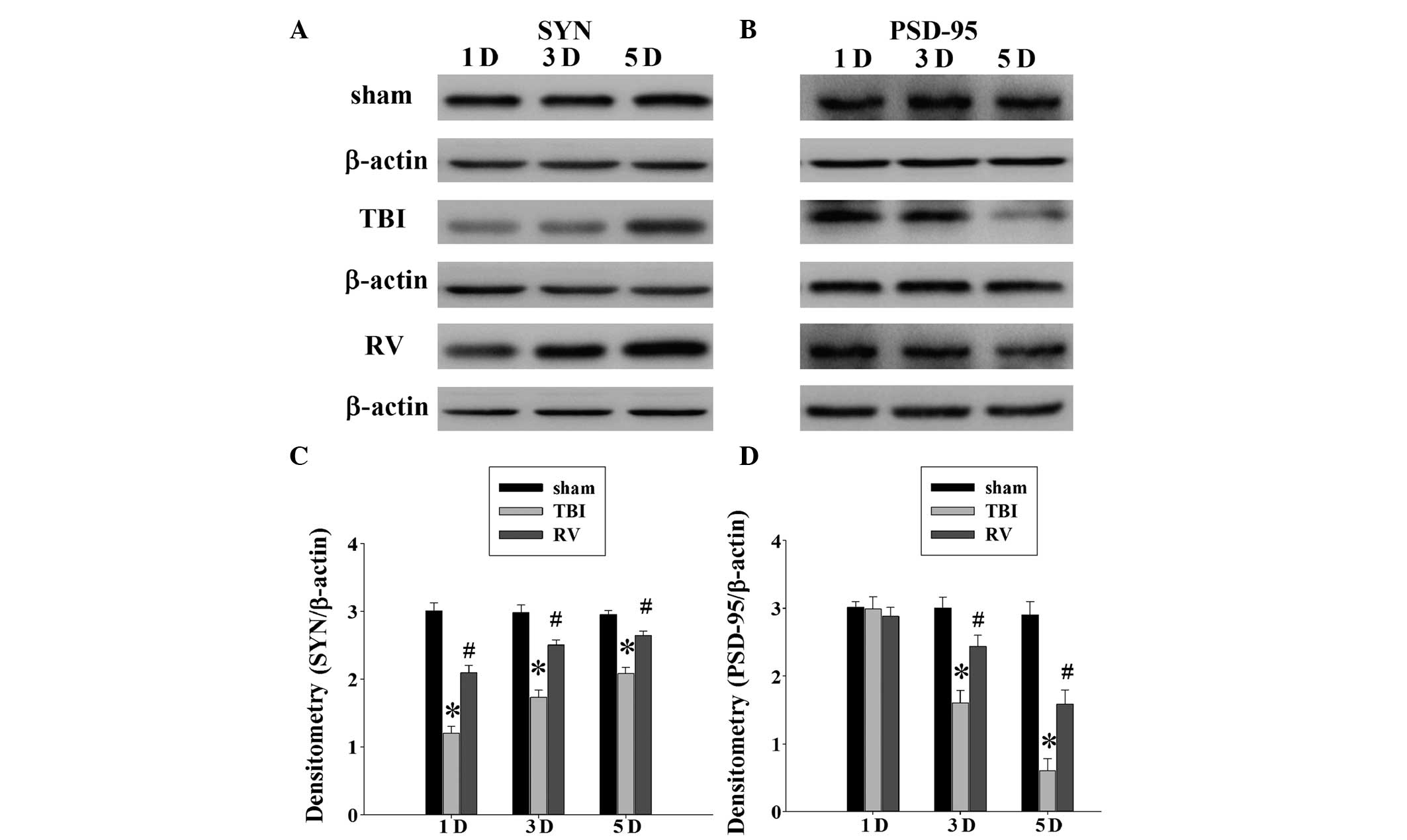
RV treatment attenuates synaptic protein
loss in the hippocampus
A number of studies have reported the significant
loss of synapses in the days following brain injury, including in
the brain regions connected to the site of initial injury, such as
the hippocampus (1,4,24).
Considering RV was able to improve spatial learning, it was
examined whether RV treatment could affect synaptic alterations in
the rat hippocampus following TBI. The expression of two synaptic
proteins was evaluated in the rat hippocampus using western blot
analysis at 1, 3 and 5 days following TBI. As demonstrated in
Fig. 4A, there was a reduction in
the expression levels of synaptophysin in the TBI group compared
with the sham group at 1, 3 and 5 days. The reduction in the levels
of synaptophysin indicated a loss of synapses in the TBI rats. The
rats treated with RV exhibited significantly greater levels
compared with the TBI group. In addition, Fig. 4B indicates that the levels of
PSD-95 were reduced at 3 and 5 days following injury, with the RV
treated rats significantly improved compared with the TBI
group.
RV treatment suppresses neuronal
autophagy in the hippocampus
To determine how autophagic activity is altered
following TBI, LC3 expression in hippocampal neurons at 24 h
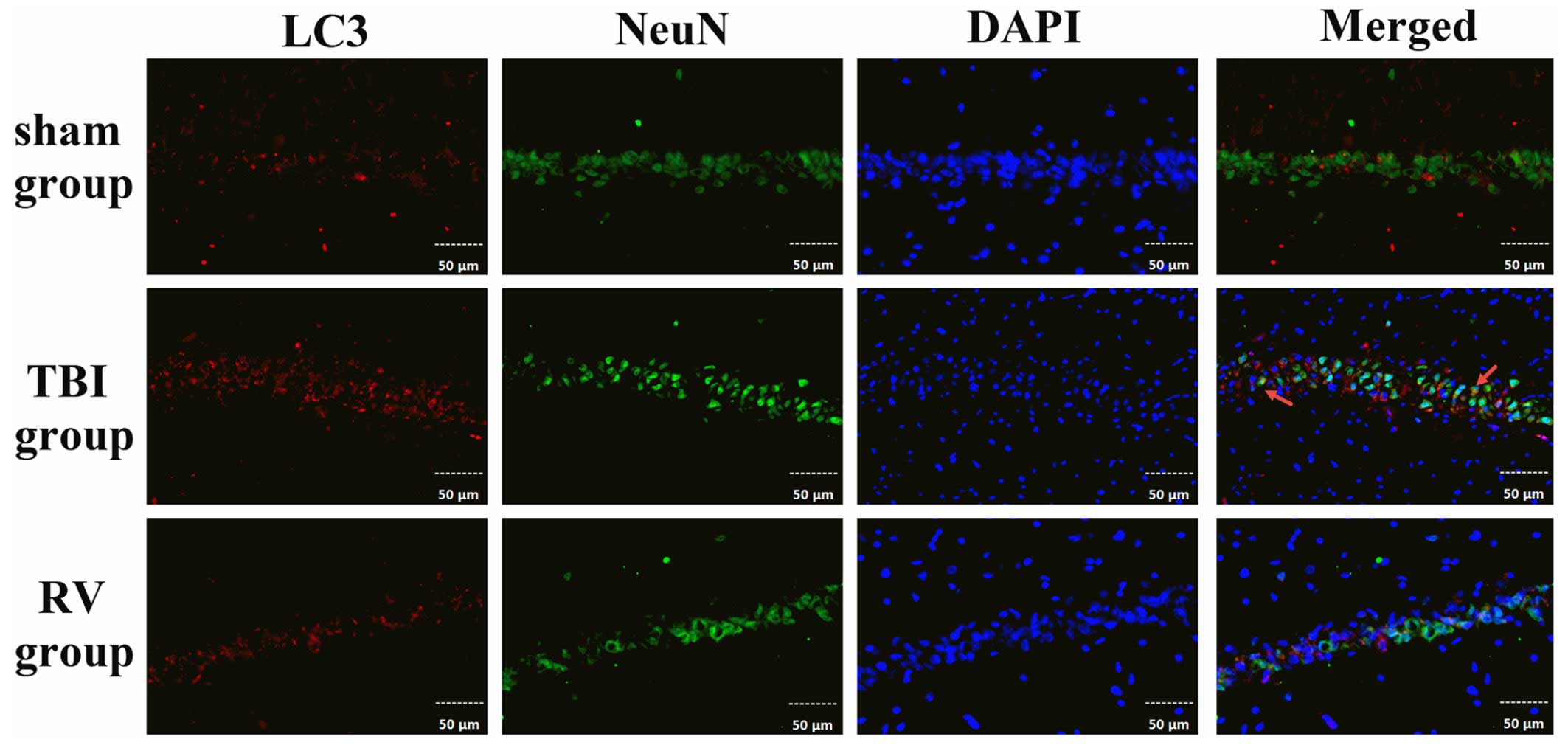
following TBI was detected using immunofluorescence. As shown in
Fig. 5, LC3 staining is indicated
in red, neurons stained with NeuN are in green, and DAPI staining
of nuclei is in blue. The images were merged, and the
co-localization of NeuN and LC3 was observed in the
immunofluorescent staining at 24 h. The results indicated that
alterations in LC3 proteins had occurred in neurons in the
hippocampal region following TBI. In order to confirm the ability
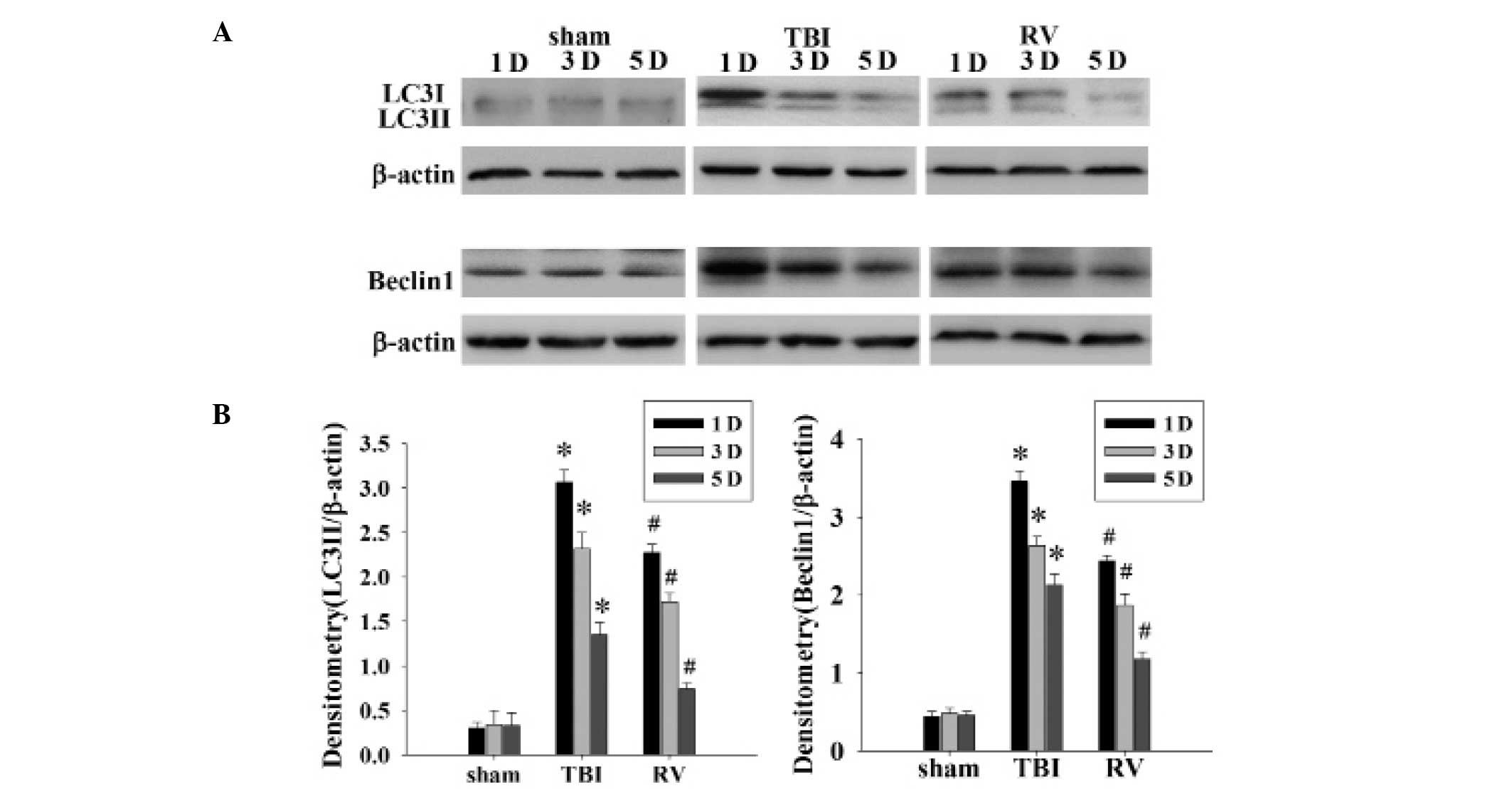
of RV to inhibit autophagy, the protein levels of LC3-II (ratio of
LC3-II to LC3-I) and Beclin1 were determined by western blot
analysis. The protein expression levels of LC3-II and Beclin1 in
the hippocampus were significantly upregulated at 1, 3 and 5 days
following TBI, with the highest level observed at 1 day (Fig. 6A). As the densitometry analysis
indicates in Fig. 6B, treatment
with RV significantly reduced the relative protein expression of
LC3-II and Beclin1 at 1, 3 and 5 days in the hippocampus compared
with the TBI group.
Discussion
Traumatic brain injury is characterized by neuronal
damage and commonly, secondary cell death, leading to neurological
dysfunction. The loss of neurons in the hippocampus contributes to
the impairment of learning and memory following TBI. Thus,
impairment of cognitive function has long been thought to be the
result of rapid cell death following TBI in humans. As a
therapeutic strategy, use of safe nutritional supplements may be
promising for reducing brain damage and staving off long-term
cognitive deficits as observed in instances of TBI. With respect to
the latter, the nutritional supplement, RV, is a promising therapy
to treat secondary brain injury following TBI. RV has been
previously reported to provide neuroprotection against secondary
brain injury, through its antioxidant and anti-inflammatory
activity (16,25), reducing neuronal death and glial
activation (26), reducing
hippocampal degeneration and improving cognitive performance
(27). However, the specific
mechanisms by which RV exerts its neuroprotective activity remain
unclear.
In the present study, the neuroprotective potential
of RV was investigated in a rat model of TBI. Rats treated with RV
(100 mg/kg/day, up to 5 days) exhibited reduced posttraumatic brain
edema and improved learning and memory ability and motor deficits.
These results are similar to previous studies, which reported that
RV treatment resulted in neuroprotection against a variety of
neurologic disorders, in particular, acute brain injury (27). In addition, the current study
demonstrates that RV was observed to protect synaptic proteins in
the hippocampus. TBI leads to a decline in both synaptophysin and
PSD-95, and treatment with RV showed significantly greater proteins
levels compared with the TBI group, with protein levels peaking at
5 days. This is the first report, to the best of our knowledge,
that RV can protect key synaptic proteins following TBI.
Synaptophysin is a 38-kDa calcium-binding
glycoprotein found in the membranes of presynaptic vesicles in
neurons and is involved in vesicular trafficking, docking,
synaptogenensis, synaptic reorganization and vesicular fusion with
the synaptic plasma membrane (28). It has been reported to be expressed
in presynaptic vesicles of the brain, spinal cord and retina, in
addition to at neuromuscular junctions. Furthermore, synaptophysin
has been widely used as a marker protein to quantify the number of
synapses during neuroanatomical remodeling or following injury
(29,30). In the current study, synaptophysin
was used to measure synapse loss following TBI. TBI was observed to
result in synapse loss or damage, and may contribute to the
observed behavioral, cognitive and neurological deficits. These
results are supported by previous reports that TBI causes
significant synapse loss in the hippocampal CA1 region at 2 days
post-injury by 60% (1). In
addition, Ding et al (31)
quantified the synaptic loss following closed TBI in rats, and
observed that a diffuse brain injury achieved by the use of the
Marmarou model resulted in synaptic damage and reduced
synaptophysin expression at both the transcriptional and
translational levels (31).
PSD-95, a scaffolding protein that is abundantly expressed within
excitatory synapses, has been implicated in various important roles
in the regulation of ion-channel function, neuronal
differentiation, synaptogenesis, synaptic plasticity, and the
processes of learning and memory (32,33).
Western blotting of PSD-95 was performed to determine the effect of
TBI on the number of synapses in the hippocampus. In the present
study, the PSD-95 levels were markedly reduced in the hippocampus
at 3 and 5 days following TBI. A previous study indicated that
PSD-95 follows a time-dependent reduction following TBI in rats.
Following controlled cortical impact, using to model moderate TBI
in rats, a marked reduction in PSD-95 levels in the hippocampus was
observed at day 3–7 following TBI (34). The loss of PSD-95 has been directly
correlated with a reduction in cognitive function in rats,
suggesting a possible cellular mechanism that may, at least in
part, explain the neurological deficits observed weeks and even
months following the initial trauma (35). The present study observed that RV
was able to increase the levels of both pre- and post-synaptic
proteins in the hippocampus, which may explain why RV improved
learning and memory ability, and may indicate it to be a promising
treatment. Further studies are required to clarify the
physiological consequences of the hippocampal protection.
Furthermore, it is worth noting that the present
study observed that following injury, administration of RV was able
to suppress neuronal autophagy in the rat hippocampus. Accumulating
evidence suggests that the autophagic pathway is involved in the
pathophysiological response to TBI, and that the inhibition of this
pathway may help attenuate traumatic damage and functional outcome
deficits (8–10,23,36).
Lin et al (18) reported
that RV can increase cell survival by suppressing glycogen synthase
kinase-3-mediated autophagy and apoptosis using in vivo and
in vitro TBI models. Another previous study demonstrated
that the combination of rapamycin and RV blocked rapamycin-induced
autophagy, however, promoted apoptosis in tuberous sclerosis
complex 2-deficient cells (37).
In the present study, the results are in agreement with previous
studies, and therefore it may be hypothesized that the
neuroprotective effects of RV on TBI may be associated with the
attenuation of neuronal autophagy, which is a contributing factor
to neuronal death.
In conclusion, the results of the current study
demonstrated that the post-injury administration of RV was able to
attenuate brain edema and enhance cognitive functional recovery. In
addition, RV treatment resulted in a marked elevation of synaptic
proteins and suppressed neuronal autophagy induced following TBI in
rats. It remains to be investigated whether RV is able to provide
significant neuroprotection when administration is delayed
following injury. It is additionally unclear whether the current
dosage and route of administration of RV provides the maximal
neuroprotective benefit. Therefore, further investigations are
required to improve the understanding of the neuroprotective
effects of RV in TBI. The results of the present study suggest that
RV is a potential therapeutic compound and may provide novel
clinical efficacy for the treatment of TBI.
Acknowledgments
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (grant no.
H2014105079).
Abbreviations:
|
RV
|
resveratrol
|
|
TBI
|
traumatic brain injury
|
|
SYN
|
synaptophysin
|
|
PSD95
|
post synaptic density 95
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
NeuN
|
neuron-specific nuclear protein
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
NSS
|
neurologic severity score
|
References
|
1
|
Scheff SW, Price DA, Hicks RR, Baldwin SA,
Robinson S and Brackney C: Synaptogenesis in the hippocampal CA1
field following traumatic brain injury. J Neurotrauma. 22:719–732.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baldwin SA, Gibson T, Callihan CT,
Sullivan PG, Palmer E and Scheff SW: Neuronal cell loss in the CA3
subfield of the hippocampus following cortical contusion utilizing
the optical dissector method for cell counting. J Neurotrauma.
14:385–398. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rafols JA, Morgan R, Kallakuri S and
Kreipke CW: Extent of nerve cell injury in Marmarou's model
compared to other brain trauma models. Neurol Res. 29:348–355.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semchenko VV, Bogolepov NN, Stepanov SS,
Maksimishin SV and Khizhnyak AS: Synaptic plasticity of the
neocortex of white rats with diffuse-focal brain injuries. Neurosci
Behav Physiol. 36:613–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pozuelo-Rubio M: 14-3-3ζ binds class III
phosphatidylinositol-3-kinase and inhibits autophagy. Autophagy.
7:240–242. 2011. View Article : Google Scholar
|
|
6
|
Bursch W, Hochegger K, Torok L, Marian B,
Ellinger A and Hermann RS: Autophagic and apoptotic types of
programmed cell death exhibit different fates of cytoskeletal
filaments. J Cell Sci. 113:1189–1198. 2000.PubMed/NCBI
|
|
7
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao
HJ, Dai DK, Shen YW, Xu HF, Ni H, et al: Autophagy is involved in
traumatic brain injury-induced cell death and contributes to
functional outcome deficits in mice. Neuroscience. 184:54–63. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cordaro M, Impellizzeri D, Paterniti I,
Bruschetta G, Siracusa R, De Stefano D, Cuzzocrea S and Esposito E:
Neuroprotective effects of Co-ultraPEALut on secondary inflammatory
process and autophagy involved in traumatic brain injury. J
Neurotrauma. 2015.
|
|
10
|
Bao HJ, Zhang L, Han WC and Dai DK:
Apelin-13 attenuates traumatic brain injury-induced damage by
suppressing autophagy. Neurochem Res. 40:89–97. 2015. View Article : Google Scholar
|
|
11
|
Cucciolla V, Borriello A, Oliva A,
Galletti P, Zappia V and Della Ragione F: Resveratrol: From basic
science to the clinic. Cell Cycle. 6:2495–2510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Delmas D, Jannin B and Latruffe N:
Resveratrol: Preventing properties against vascular alterations and
ageing. Mol Nutr Food Res. 49:377–395. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakata Y, Zhuang H, Kwansa H, Koehler RC
and Doré S: Resveratrol protects against experimental stroke:
Putative neuroprotective role of heme oxygenase 1. Exp Neurol.
224:325–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Shi Z, Fan L, Zhang C, Wang K and
Wang B: Resveratrol improves neuron protection and functional
recovery in rat model of spinal cord injury. Brain Res.
1374:100–109. 2011. View Article : Google Scholar
|
|
15
|
Saiko P, Szakmary A, Jaeger W and Szekeres
T: Resveratrol and its analogs: Defense against cancer, coronary
disease and neurodegenerative maladies or just a fad? Mutat Res.
658:68–94. 2008. View Article : Google Scholar
|
|
16
|
Ates O, Cayli S, Altinoz E, Gurses I,
Yucel N, Sener M, Kocak A and Yologlu S: Neuroprotection by
resveratrol against traumatic brain injury in rats. Mol Cell
Biochem. 294:137–144. 2007. View Article : Google Scholar
|
|
17
|
Gatson JW, Liu MM, Abdelfattah K,
Wigginton JG, Smith S, Wolf S and Minei JP: Resveratrol decreases
inflammation in the brain of mice with mild traumatic brain injury.
J Trauma Acute Care Surg. 74:470–474. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin CJ, Chen TH, Yang LY and Shih CM:
Resveratrol protects astrocytes against traumatic brain injury
through inhibiting apoptotic and autophagic cell death. Cell Death
Dis. 5:e11472014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats. Part I: Pathophysiology and biomechanics. J
Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang J, Liu J, Zhou C, Alexander JS, Nanda
A, Granger DN and Zhang JH: MMP-9 deficiency enhances
collagenase-induced intracerebral hemorrhage and brain injury in
mutant mice. J Cereb Blood Flow Metab. 24:1133–1145. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hui-guo L, Kui L, Yan-ning Z and Yong-jian
X: Apocynin attenuate spatial learning deficits and oxidative
responses to intermittent hypoxia. Sleep Med. 11:205–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Constantini S, Trembovler V,
Weinstock M and Shohami E: An experimental model of closed head
injury in mice: Pathophysiology, histopathology and cognitive
deficits. J Neurotrauma. 13:557–568. 1996.PubMed/NCBI
|
|
23
|
Cui C, Cui Y, Gao J, Sun L, Wang Y, Wang
K, Li R, Tian Y, Song S and Cui J: Neuroprotective effect of
ceftriaxone in a rat model of traumatic brain injury. Neurol Sci.
35:695–700. 2014. View Article : Google Scholar
|
|
24
|
Gao X, Deng P, Xu ZC and Chen J: Moderate
traumatic brain injury causes acute dendritic and synaptic
degeneration in the hippocampal dentate gyrus. PLoS One.
6:e245662011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singleton RH, Yan HQ, Fellows-Mayle W and
Dixon CE: Resveratrol attenuates behavioral impairments and reduces
cortical and hippocampal loss in a rat controlled cortical impact
model of traumatic brain injury. J Neurotrauma. 27:1091–1099. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Q, Xu J, Rottinghaus GE, Simonyi A,
Lubahn D, Sun GY and Sun AY: Resveratrol protects against global
cerebral ischemic injury in gerbils. Brain Res. 958:439–447. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sönmez U, Sönmez A, Erbil G, Tekmen I and
Baykara B: Neuroprotective effects of resveratrol against traumatic
brain injury in immature rats. Neurosci Lett. 420:133–137. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Südhof TC: The synaptic vesicle cycle: A
cascade of protein-protein interactions. Nature. 375:645–653. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brock TO and O'Callaghan JP: Quantitative
changes in the synaptic vesicle proteins synapsin I and p38 and the
astrocyte-specific protein glial fibrillary acidic protein are
associated with chemical-induced injury to the rat central nervous
system. J Neurosci. 7:931–942. 1987.PubMed/NCBI
|
|
30
|
Meng H, Walker N, Su Y and Qiao X:
Stargazin mutation impairs cerebellar synaptogenesis, synaptic
maturation and synaptic protein distribution. Brain Res.
1124:197–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ding JY, Kreipke CW, Schafer P, Schafer S,
Speirs SL and Rafols JA: Synapse loss regulated by matrix
metalloproteinases in traumatic brain injury is associated with
hypoxia inducible factor-1alpha expression. Brain Res.
1268:125–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen P, Gu Z, Liu W and Yan Z: Glycogen
synthase kinase 3 regulates N-methyl-D-aspartate receptor channel
trafficking and function in cortical neurons. Mol Pharmacol.
72:40–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ehrlich I, Klein M, Rumpel S and Malinow
R: PSD-95 is required for activity-driven synapse stabilization.
Proc Natl Acad Sci USA. 104:4176–4181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ansari MA, Roberts KN and Scheff SW: A
time course of contusion-induced oxidative stress and synaptic
proteins in cortex in a rat model of TBI. J Neurotrauma.
25:513–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wakade C, Sukumari-Ramesh S, Laird MD,
Dhandapani KM and Vender JR: Delayed reduction in hippocampal
post-synaptic density protein-95 expression temporally correlates
with cognitive dysfunction following controlled cortical impact in
mice. J Neurosurg. 113:1195–1201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jing CH, Wang L, Liu PP, Wu C, Ruan D and
Chen G: Autophagy activation is associated with neuroprotection
against apoptosis via a mitochondrial pathway in a rat model of
subarachnoid hemorrhage. Neuroscience. 213:144–153. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alayev A, Sun Y, Snyder RB, Berger SM, Yu
JJ and Holz MK: Resveratrol prevents rapamycin-induced upregulation
of autophagy and selectively induces apoptosis in TSC2-deficient
cells. Cell Cycle. 13:371–382. 2014. View
Article : Google Scholar :
|