Introduction
Ischemic cerebrovascular disease accounts for 60–80%
of all cerebrovascular diseases (1). Compared with other forms of
cerebrovascular disease, ischemic cerebrovascular disease is
characterized by high incidence, high disability, high recurrence
and high mortality, and poses a serious threat to human health. At
present, thrombolytic therapy is the primary treatment strategy.
However, the blood reperfusion of an ischemic injury frequently
leads to further aggravation of the injury (2).
Previous studies have indicated that cerebral
ischemia-reperfusion injury (CIRI) leads to the aggravation of
injury factors, including free radicals, excitatory amino acids and
calcium overload, and additionally correlates with ischemia-induced
cerebral cell apoptosis and necrosis (1,3,4).
Apoptosis and necrosis of brain cells leads to various degrees of
dysfunction amongst patients, and results in a heavy burden being
placed upon the patient's family and society (5). Therefore, it is important to
understand the mechanisms underpinning apoptosis and necrosis in
CIRI.
Necrosis is an unprogrammed and therefore
irreversible form of cell death (6). Therefore, previous treatment
strategies of CIRI have predominantly focused upon the prevention
of apoptosis. However, clinical practice has indicated that
intervening in apoptosis is not an effective strategy for the
prevention of CIRI (1). Increasing
evidence indicates that certain forms of necrosis are regulated by
cellular signaling pathways, including the tumor necrosis factor-α
(TNF-α) receptor 1 (TNFR1)-mediated programmed necrosis pathway
(6). TNFR1 serves an important
role in inflammatory responses, cell immunity and additional
physiological and pathological processes (7). He et al (8) reported that receptor-interacting
serine/threonine-protein kinase 3 (RIP3) is a key protein in
TNF-α-induced necrosis. RIP3 belongs to the RIP family, which has a
high Ser/Thr specific protein kinase domain homology, with a
conserved kinase domain in the N-terminus and a RIP homotypic
interaction motif (RHIM) domain in the C-terminus (9). When caspase-8 (C-terminal) activation
is inhibited, RIP3 is able to bind to RIP1 through the domain,
thereby forming a cellular necrosis complex. However, when
mutations occur in 4 amino acids of the RHIM domain, the
interaction between RIP3 and RIP1 is interrupted, thereby leading
to the loss of function of RIP3 in necrosis (10–12).
Therefore, RIP3 serves an important role in the cellular death
pathways (9–13). Previous studies have demonstrated
that embryonic fibroblast cells derived from RIP3-deficient mice
exhibit resistance to TNF-α-induced cell necrosis (8). In addition, in the caerulein-induced
acute pancreatitis mouse model, RIP3 gene knockout significantly
reduces necrosis of pancreatic cells and promotes the recovery of
acute pancreatitis (14). Zhang DW
suggested that upon the stimulation of necrotic signals, RIP3
proteins act as molecular switches for TNFR1-induced apoptosis and
necrosis, and are required for necrosis (15,16).
However, the expression levels of RIP3 and TNFR1 following CIRI
remains to be investigated.
In the current study, the expression of RIP3 and
TNFR1 following CIRI were investigated using immunohistochemistry
and western blotting. Furthermore, the effects and mechanisms of
RIP3 and TNFR1 in cerebral cell necrosis were investigated. The
current study may aid in the elucidation of the various programmed
necrosis pathways involved following CIRI, and may increase
understanding of the pathogenesis of ischemic encephalopathy, thus
improving treatment strategies.
Materials and methods
Rat model of CIRI
The current study was conducted in accordance with
the guidelines for The Care and Use of Animals in Research
(17), and the protocols used were
approved by the Liaoning Medical University Animal Care and Use
Committee (Liaoning, China). Adult male Sprague-Dawley rats
(Sibeifu Co., Beijing, China; n=40) weighing 250±20 g were housed
in an environmentally controlled room at Liaoning Medical
University (Liaoning, China) (22±2°C with a 12 h/12 h light/dark
cycle). The rats were supplied with standard rat chow and water
ad libitum. Focal cerebral ischemia was induced by middle
cerebral artery occlusion (MCAO), as described previously (18) with modifications. Briefly, MCAO was
conducted using an occluding suture (diameter, 0.26 mm) for 2 h, as
follows. Rats were anesthetized with 10% chloral hydrate from
Beijing Solarbio Science & Technology Co., Ltd. (Beijing,
China; 100 g/0.3 ml) via intraperitoneal injection. The cutaneous
operational area was cleaned regularly and a 2–3 cm incision was
made in the skin of the neck. The right common carotid artery, the
external carotid artery and the internal carotid artery were
isolated. The external carotid artery was ligated, blood flow was
blocked and a 4–0 monofilament (Beijing Solarbio Science &
Technology Co., Ltd.) with a blunted tip coated with poly-L-lysine
(Beijing Solarbio Science & Technology Co., Ltd.) was inserted
into the internal carotid artery through the external carotid
artery. The suture was advanced ~18–20 mm beyond the carotid artery
bifurcation until the origin of the middle carotid artery was
blocked. Following 2 h of MCAO-evoked ischemia, the suture was
slowly drawn back to allow reperfusion. The reperfusion durations
were 6, 12, 24 and 72 h (n=6, 6, 16 and 6, respectively). The
suture was not inserted into the sham operation group (n=6). Rectal
temperature was maintained throughout the procedure at 37±0.5°C
with a temperature-regulated heating pad.
Rat groups and treatments
Rats were randomly divided into the following three
groups (n=16 rats/group): i) Sham group, S (n=6); ii) MCAO model
group received saline treatment, IR (n=6, 6, 16 and 6 for 6, 12, 24
and 72 h reperfusion, respectively); iii) MCAO model group received
the caspase inhibitor Z-VAD-FMK (zVAD; Sigma-Aldrich, St. Louis,
MO, USA) treatment 1 day prior to MCAO operation, IR+I. Rats in the
IR group received MCAO for 2 h, followed by reperfusion for 6, 12,
24 and 72 h. Rats in the S group underwent the same surgical
procedures as those in the IR group, however the suture was not
advanced beyond the internal carotid bifurcation. No additional
treatments were given in the S group and IR group. Animals in the
IR + I group received an intraperitoneal injection of the caspase
inhibitor (zVAD; 2 mg/kg) 1 day prior to MCAO.
Neurological evaluation
Rats were examined for neurological deficits
following MCAO by two investigators who were blinded to the groups,
using a 5-point neurological function score: 0, no deficit; 1,
failure to extend right forepaw fully; 2, circling to the right; 3,
falling to the right; 4, no spontaneous walking with a depressed
level of consciousness. Only rats exhibiting no or incomplete
forelimb placing with rotational asymmetry following MCAO were
included in the subsequent analysis (19).
Edema measurement
Rats from each group (n=5) were decapitated under
deep anesthesia with 10% chloral hydrate at 6, 12, 24 and 72 h of
reperfusion. The ipsilateral and contralateral hemispheres were
dissected and the wet weight of the tissue was measured. The
tissues were dried at 120°C for 24 h. The percentage of cerebral
water was determined as: (wet weight − dry weight)/dry weight ×
100.
Infarct measurement
Following reperfusion, rat brains (n=5 from each
group) were removed and frozen at −80°C for 5 min. Coronal slices
(2 mm) were made using a rodent brain matrix (HEAD Biotechnology
Co,. Ltd., Beijing, China). The sections were stained for 20 min at
37°C with 2% 2,3,5-triphenyltetrazolium chloride monohydrate (TTC;
Sigma-Aldrich) and were then fixed with 4% formaldehyde. The
infarct volume was calculated as previously described (20). In brief, the sections were scanned
and the infarct area in each section was calculated by subtracting
the noninfarct area of the ipsilateral side from the area of the
contralateral side using ImageJ analysis software (version 1.46;
National Institutes of Health, Bethesda, MD, USA). The infarct
areas of each section were summed and multiplied by section
thickness to give the total infarction volume.
Hematoxylin and eosin (H&E)
staining
H&E histology was conducted at 6, 12, 24 and 72
h following reperfusion. Rats were anesthetized by an
intraperitoneal injection of 10% chloral hydrate (100 g/0.3 ml) and
then perfused transcardially with saline (Beijing Solarbio Science
& Technology Co., Ltd.; 250 ml), followed by 4% formaldehyde
(Beijing Solarbio Science & Technology Co., Ltd.; 250 ml).
Brains were removed and fixed in 4% formaldehyde at 4°C for 72 h
and then dehydrated and embedded in paraffin blocks (Beijing
Solarbio Science & Technology Co., Ltd.). Coronal sections were
cut posterior to the optic chiasma, at a thickness of 3 mm. The
sections were deparaffinized and hydrated with reducing
concentrations of alcohol, stained with H&E (Beijing Solarbio
Science & Technology Co., Ltd.) and photographed using an
optical microscope (Axio Scope.A1; Zeiss AG, Oberkochen,
Germany).
Immunohistochemical staining
Rat brains were removed, post-fixed in 4%
formaldehyde overnight at 4°C and cryoprotected with 30% sucrose in
PBS. Brains were cut into coronal sections (5 µm) using a
Leica RM2135 microtome (Leica Microsystems GmbH). Sections were
obtained from each rat, and the free-floating method was used for
avidin-biotin-peroxidase labeling. Coronal sections were rinsed
with PBS and treated with 0.3% H2O2 (v/v) in
PBS for 10 min at room temperature to quench endogenous peroxidase.
Following washing with PBS, sections were incubated with a blocking
solution [10% normal goat serum (Beijing Zhongshan Jinqiao
Biological Technology Co., Ltd., Beijing, China), 0.3% Triton X-100
(Beijing Solarbio Science & Technology Co., Ltd.) in PBS] for
30 min at 37°C. The slices were incubated with polyclonal
anti-TNFR1 mouse monoclonal antibodies (1:200; cat no. sc-374185;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and polyclonal
anti-RIP3 rabbit IgG antibodies (1:200; cat no. sc-135170; Santa
Cruz Biotechnology, Inc.) for 1 h at 37°C, followed by overnight
incubation at 4°C. Following rinsing, sections were incubated with
goat anti-rabbit IgG antibodies (1:200; cat no. SAB4600015-50UL,
Sigma-Aldrich) for 1 h at 37°C. The antibodies were detected using
0.05% diaminobenzidime tetrachloride (Shanghai Kuaibo Biotechnology
Co., Ltd., Shanghai, China) and 0.03% H2O2 in
PBS (pH 7.4). Images were captured using an optical microscope
(Axio Scope.A1; Zeiss AG) at a magnification of ×40, using a
standard procedure. PBS was used as the negative control for the
primary antibody. Positive cells were counted using a CIAS-1000
cell image analysis system (CIAS-1000, Zhongguo Hengda, Ltd.,
Beijing, China). All imaging was conducted with equal interval
sections, (3 serial sections were taken and analyzed from each
animal) and within each section, 2 non-overlapping high-power
images (magnification, ×400) were obtained of the cerebral cortex.
The protein expression positive cell number was counted from each
perspective.
Western blotting
For western blot analysis, 15 mg brain tissue was
treated with 1 ml radioimmunoprecipitation assay buffer containing
50 mM Tris/HCl (pH 7.4), 150 mM NaCl 1% (v/v) NP-40, 0.1% (w/v)
sodium dodecyl sulfate (SDS), 1% (v/v) phenylmethylsulfonyl
fluoride (all purchased from Beijing Solarbio Science &
Technology Co., Ltd.), 0.3% (v/v) protease inhibitor and 0.1% (v/v)
phosphorylated proteinase inhibitor (Sigma-Aldrich). The
supernatants were then extracted from the lysates following
centrifugation at 13,000 g at 4°C for 15 min. To quantify the
relative concentration of the total proteins, a bicinchoninic
protein assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA)
was used. Subsequently, equal amounts of protein (15 µg)
were separated on an SDS-polyacrylamide gel electrophoresis gel
[10% (v/v) polyacrylamide] and transferred onto a polyvinylidene
fluoride membrane (all purchased from Merck Millipore, Darmstadt,
Germany) at 300 mA for 2 h. To block nonspecific binding, the
membrane was incubated with 8% (w/v) milk in Tris-buffered saline
Tween-20 (Beijing Solarbio Science & Technology Co., Ltd.) for
2 h at room temperature. The membranes were then incubated with
primary antibodies against β-actin mouse mAb (1:4,000; cat no.
3700), RIP3 rabbit mAb (1:1,000; cat no. 15828) and TNFR1 rabbit
mAb (1:1,000; cat no. 13377) all purchased from Cell Signaling
Technology, Inc., Danvers, MA, USA) overnight at 4°C. Subsequently,
the membranes were washed with PBS-Tween 4 times (5 min/time).
Membranes were then incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:5,000, Cell Signaling
Technology, Inc.) for 2 h at room temperature. Following
incubation, the membranes were washed 4 times (5 min/time).
Proteins were detected using enhanced chemiluminescence (EMD
Millipore, Billerica, MA, USA) according to the manufacturer's
instructions, and the relative protein levels were determined. To
measure the alterations in protein expression, target proteins were
normalized to β-actin.
Statistical analysis
All values are presented as the mean ± standard
deviation. Data were analyzed for significance using the
nonparametric Mann-Whitney U-test or analysis of variance using
SPSS software, version 13.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
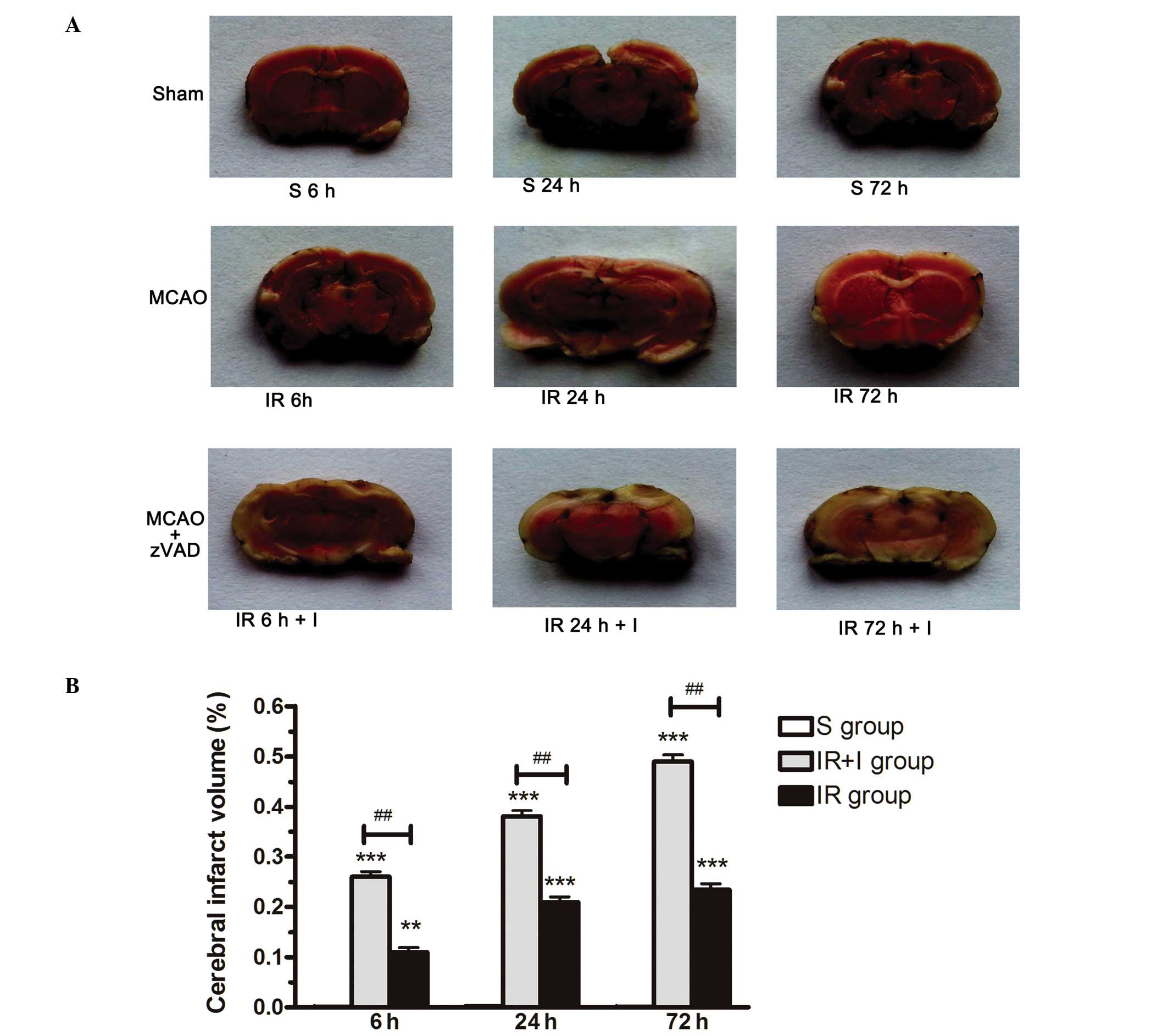
zVAD increases the volume of cerebral
infarction post-reperfusion
Infarct volume as a measure of stroke severity was
first determined in the three rat groups (S group, IR group and IR
+ I group). In the IR group, the volume of the infarct region was
significantly increased compared with the S group at 6, 24 and 72
h, as assessed by TTC staining (Fig.
1A and B). To investigate whether zVAD enhances the infarct
volume resulting from MCAO, 2 mg/kg zVAD or saline was administered
intraperitoneally 10 min prior to the initiation of MCAO (or sham
MCAO). Administration of zVAD increased the infarct volume in rat
brains by 0.15, 0.18 and 0.25% respectively at 6, 24 and 72 h
post-reperfusion (P<0.05; Fig.
1). These results demonstrated the pro-necrotic effect of zVAD
against CIRI.
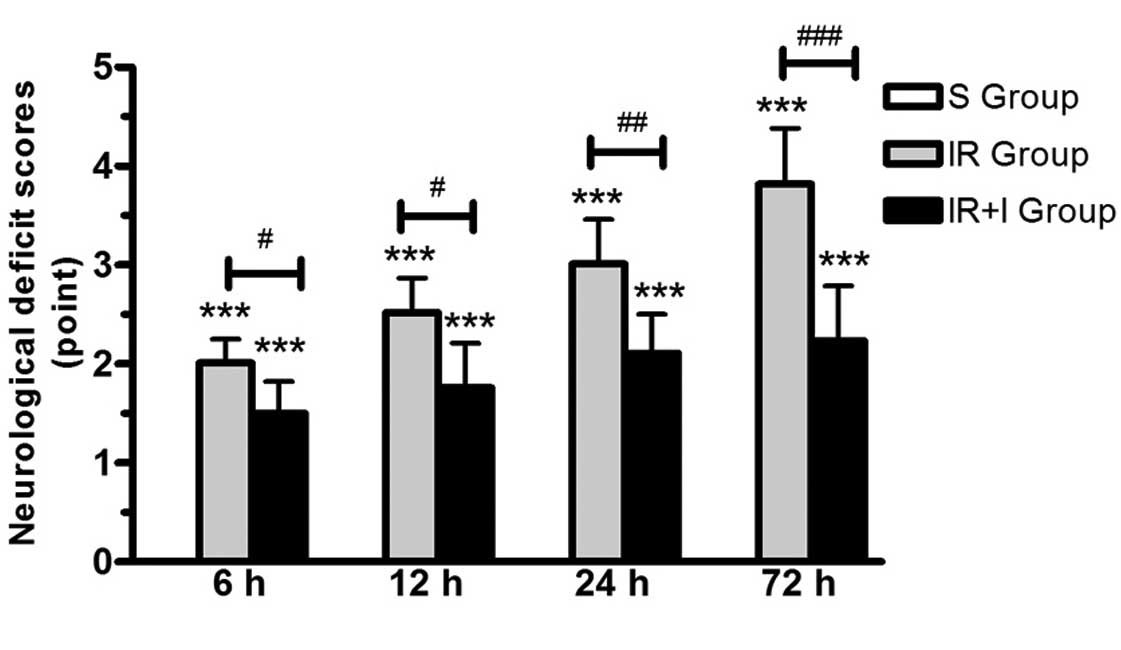
zVAD enhances neurological deficit scores
post-reperfusion
In the S group, no rats exhibited symptoms of
neurological deficits, while there were clear neurological deficits
in the IR group. In addition, the neurological function was
significantly enhanced in the IR + I group rats at 24 and 48 h
post-reperfusion compared with the sham group (P<0.05; Fig. 2).
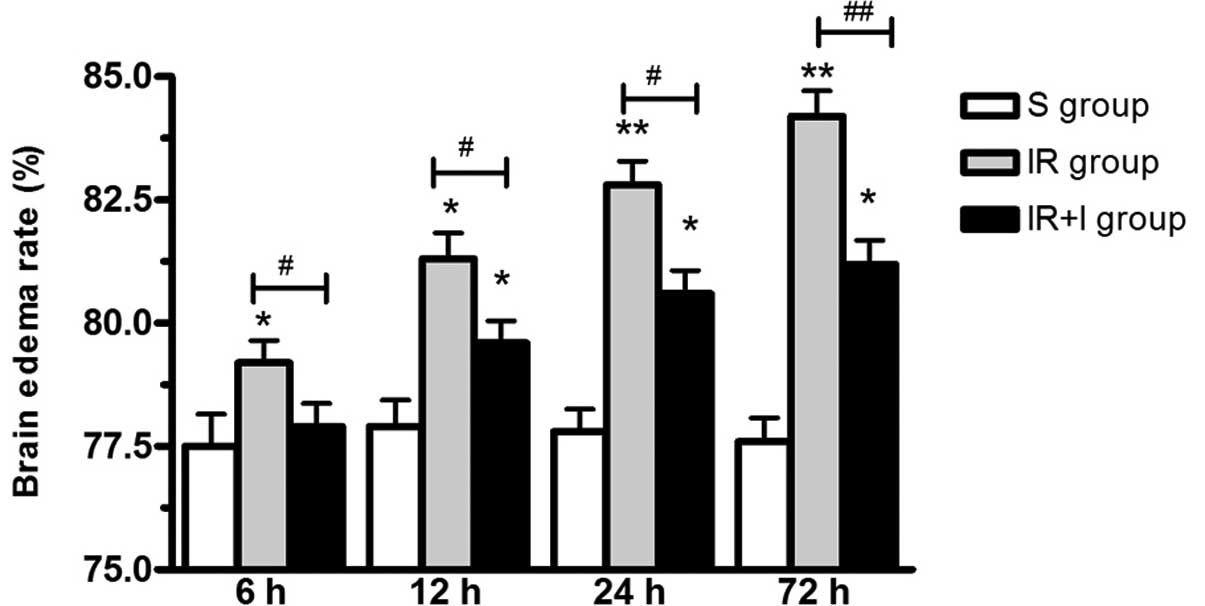
zVAD increases brain edema
post-reperfusion
As presented in Fig.
3, brain edema was significantly increased post-reperfusion in
the IR group. To investigate the function of zVAD, it was
administered intravenously 10 min prior to the initiation of MCAO.
The water content of the ischemic brains was significantly
increased at 6, 12, 24 and 72 h post-reperfusion (P<0.05;
Fig. 3).
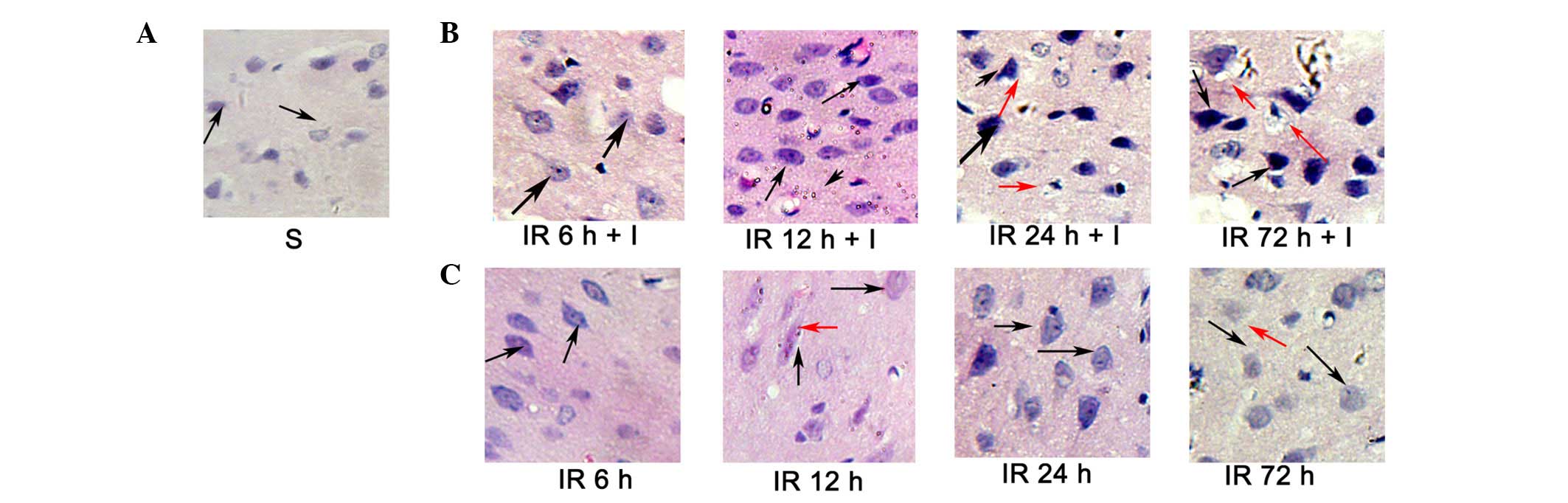
Evaluation of cerebral histology by
H&E
H&E staining (Fig.
4) was conducted to investigate the histopathological
alterations occurring following focal ischemia. Normal cells in the
contralateral hemisphere had round, lightly stained nuclei
(Fig. 4A), whereas dying cells in
the ipsilateral hemisphere had pyknotic nuclei (black arrows;
Fig. 4B and C). In addition, dying
cells exhibited signs of vacuolization (red arrows, Fig. 4B and C) in the core ischemic zone
and the ischemic penumbra at 24 and 72 h following reperfusion.
However, the pyknotic nuclei were significantly increased in the IR
+ I group rats at 6, 12, 24 and 72 h post-reperfusion compared with
the saline group (P<0.05; Fig.
4B).
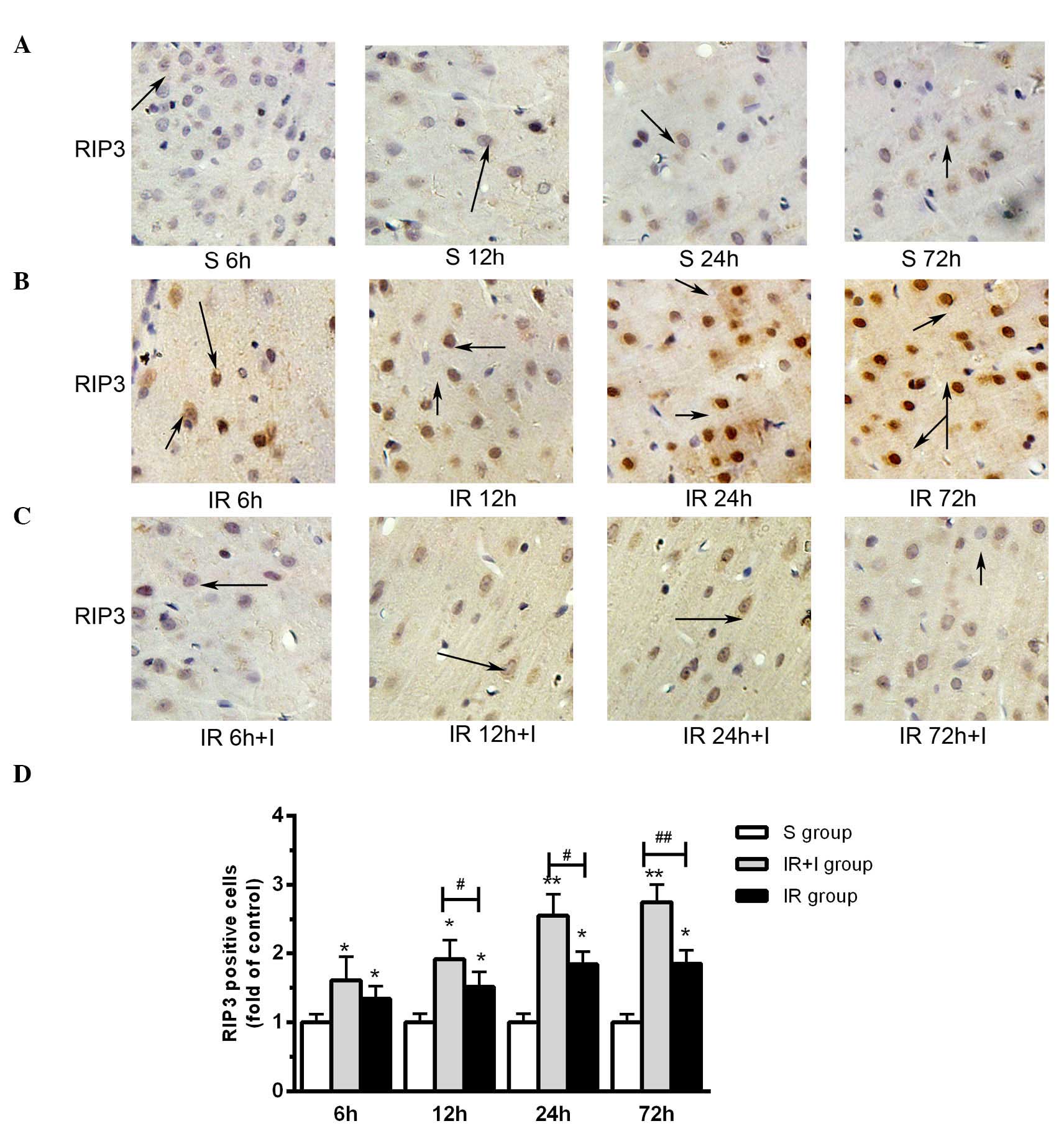
zVAD upregulates the expression levels of
RIP3 in the rat brain cortex
It has been previously reported that the expression
levels of RIP3 were increased in the cortex following reperfusion
and 2 h ischemia (20), which was
in accordance with the findings of the present study (Fig. 5A). Compared with the saline groups,
pretreatment with zVAD significantly increased the protein
expression level of RIP3 in the IR + I group (Fig. 5B). As presented in Fig. 5C, enhanced RIP3 immunostaining was
observed in cells scattered throughout the ischemic cortex 6, 12,
24 and 72 h post-reperfusion. Quantification of RIP3 positive cells
in the three groups are displayed in Fig. 5D.
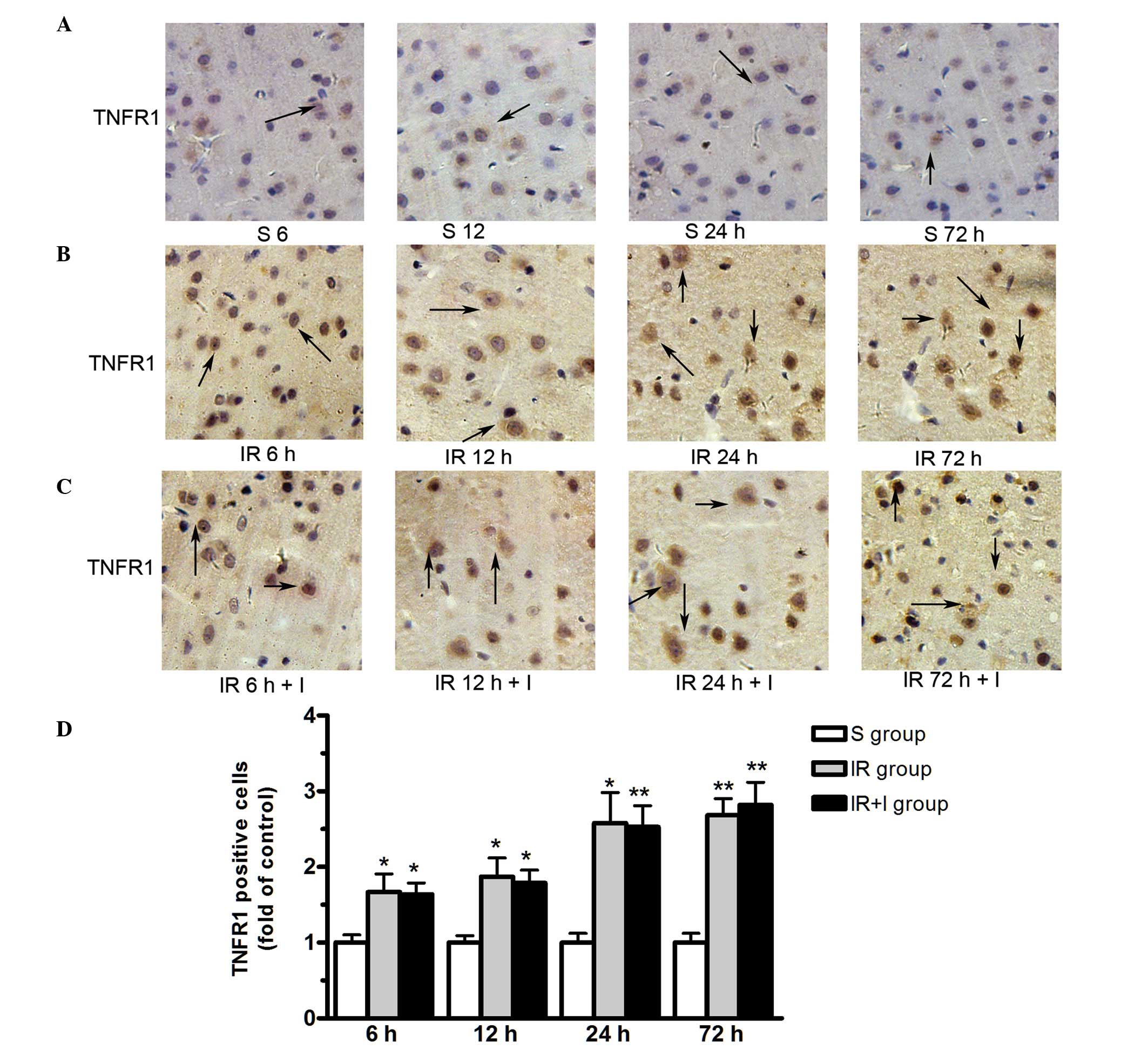
zVAD does not alter TNFR1 protein
expression levels in the rat brain cortex
As presented in Fig.
6, TNFR1 protein was expressed in the plasma membrane. The
expression level of TNFR1 were significantly upregulated in the
ischemic hemisphere at 6, 12, 24 and 72 h post-reperfusion.
However, zVAD does not alter the TNFR1 protein expression levels in
the rat brain cortex (Fig. 6).
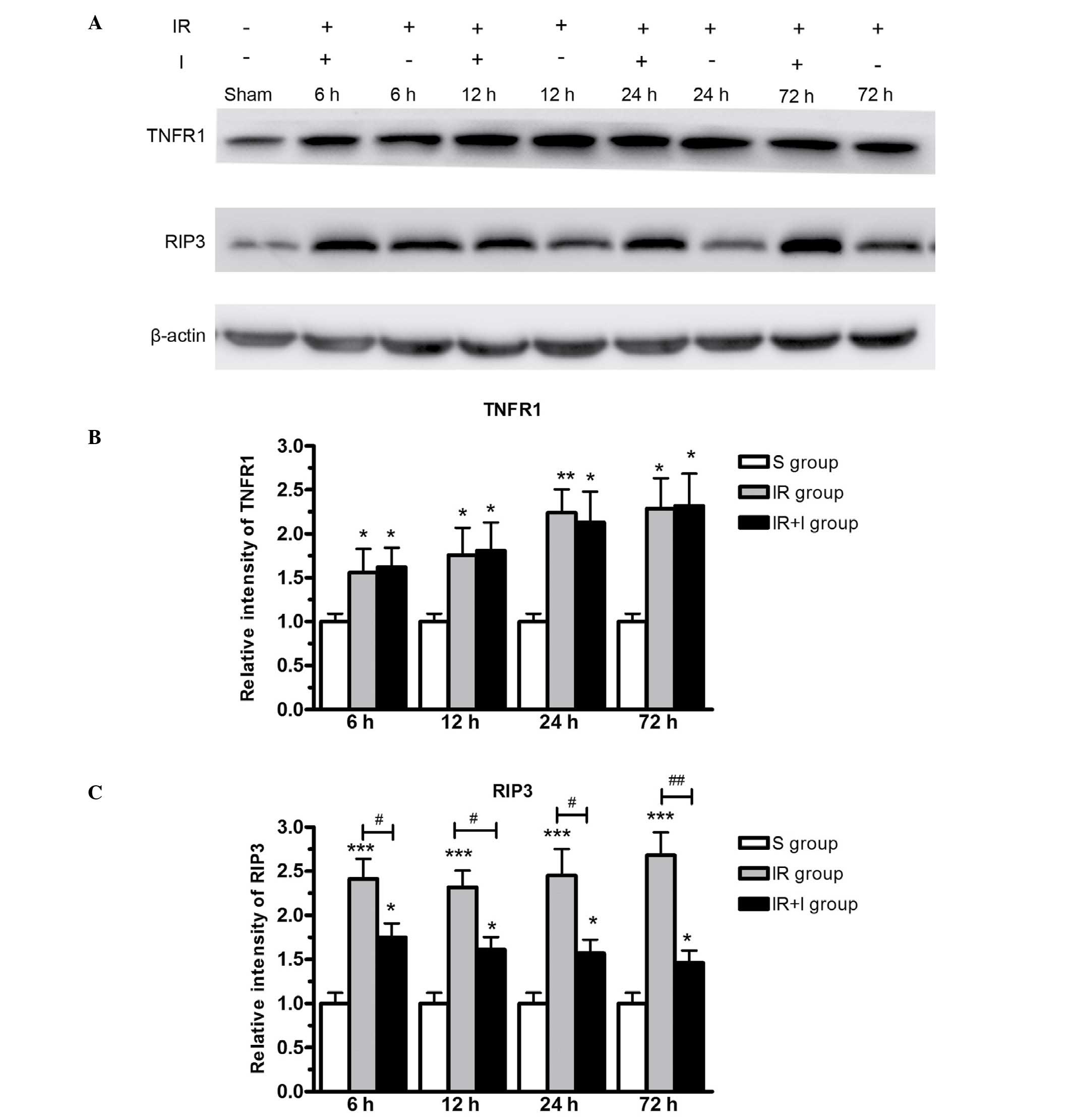
Protein expression levels of RIP3 and
TNFR1 in the lesion boundary zone of the ipsilateral
hemisphere
To further investigate the effect of zVAD on the
expression of RIP3 and TNFR1, western blotting was used to measure
target protein levels in the lesion boundary zone in the three rat
groups. The western blotting data (Fig. 7) supported the immunohistochemical
data (Figs. 5 and 6), in that the protein expression levels
of RIP3 and TNFR1 were significantly greater in the IR group
compared with the S group (P<0.05). RIP3 was expressed at
greater levels in the IR + I group, while no significant
alterations were observed in TNFR1 levels (Fig. 7).
Discussion
As a fundamental cellular process, programmed cell
death widely regulates cell proliferation and homeostasis. Previous
studies have indicated two important cellular death pathways,
including apoptosis and programmed necrosis (21). To the best of our knowledge, the
role of necrosis in CIRI remains to be fully elucidated. In the
current study, RIP3-mediated cell necrosis was enhanced by caspase
blockade using zVAD, and the function of zVAD was observed to be
independent of TNFR1 signaling. The current study is the first to
demonstrate the effect of zVAD in rat brains using a model of
transient MCAO. In addition to enhanced structural injury, a
reduction in neurological outcomes was observed, furthermore zVAD
increased infarct size and brain edema following reperfusion.
Over 80% of stroke cases result from ischemic
stroke, which is characterized by high morbidity and mortality
(22). Following a period of
ischemia, reperfusion damage may occur when blood flow resumes in
the brain (23), and reperfusion
serves an important role in the pathology of cerebral ischemia
(24). Numerous studies report the
correlation between certain pathologies and CIRI, including brain
edema, inflammation, apoptosis and necrosis (25–27).
Apoptosis is an important cellular process in the
maintenance of homeostasis in all tissues. Caspases belong to the
cysteine protease family and serve key roles in the initiation of
apoptosis (28). Caspase-8 is
considered to be an initiator caspase and therefore is critically
involved in apoptosis (29). Upon
the stimulation of TNF-like cytokines, death receptors are
triggered and the caspase cascade is activated, leading to
apoptotic cell death (30).
Previous studies have indicated that the inhibition of caspase
activity by specific caspase inhibitors, such as zVAD, is able to
lead to apotosis-independent cell death, namely programmed necrosis
(necroptosis) (31–33). Necroptosis has been demonstrated to
be regulated by two key kinases, RIP1 and RIP3 (29).
RIP1 and RIP3 are suggested to serve key roles in
TNF-induced cell necrosis (8,11,16).
Previous studies have indicated that the binding of TNF-like death
cytokines to TNFR1 triggers the formation of a large protein
complex, including TNFR1-associated death domain protein, TNF
receptor-associated factor 2 and RIP1 (34). It has been reported that necrosis
is initiated by RIP1 and RIP3 following the activation of death
receptors (35). RIP3 belongs to
the RIP family, which has homologs to RIP1, RIP2 and RIP4 (36,37).
However, the C-terminal domain of RIP3 is significantly different
from other RIPs (36,37). Transient overexpression of RIP3 in
certain cells has been demonstrated to induce cell death (36).
Following the induction of necrosis, RIP3 is
recruited to RIP1 and constitutes a necroptosis-inducing complex.
It has been suggested that RIP3 functions as a molecular switch for
necrosis (14). Previous studies
have demonstrated that knockout of RIP3 is able to promote cell
survival even in the lethal phenotype of caspase-8 deficient mice
(12,38). In addition, studies have reported
that caspase-8 significantly inhibits RIP3-RIP1-kinase-dependent
necroptosis in line with the activation of death receptor (8,16).
At the molecular level, caspase-8 is observed to proteolytically
cleave and inactivate RIP1 and RIP3, thereby controlling
RIP-3-mediated necrosis. In the present study, the expression of
TNFR1 and RIP3 was measured in rat brains subjected to MCAO. This
demonstrated that TNFR1 and RIP3 were significantly upregulated,
suggesting that inflammation-induced cell necrosis occurs following
CIRI. However, pre-administration with zVAD significantly increased
the protein level of RIP3, with no effect on TNFR1, accompanied by
enhanced cell necrosis. These results suggest that zVAD mediates
caspase-independent cell death. Thus, the inhibition of caspase-8
using zVAD may sensitize cerebral cells to RIP-mediated necroptotic
cell death following IR. Taken together, RIP3-mediated cell
necrosis may be enhanced by caspase blockade using zVAD, and the
function of zVAD is indicated to be independent of TNFR1 signaling
following IR.
Acknowledgments
The current study was supported by the Natural
Science Foundation of Liaoning Province (grant no. 2014022008).
References
|
1
|
Cerqueira NF, Hussni CA and Yoshida WB:
Pathophysiology of mesenteric ischemia/reperfusion: A review. Acta
Cir Bras. 20:336–343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takahashi H, Xia P, Cui J, Talantova M,
Bodhinathan K, Li W, Holland EA, Tong G, Piña Crespo J, Zhang D, et
al: Pharmacologically targeted NMDA receptor antagonism by
NitroMemantine for cerebrovascular disease. Sci Rep. 5:147812015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu M, Chen X, Gu Y, Peng T, Yang D, Chang
RC, So KF, Liu K and Shen J: Baicalin can scavenge peroxynitrite
and ameliorate endogenous peroxynitrite-mediated neurotoxicity in
cerebral ischemia-reperfusion injury. J Ethnopharmacol.
150:116–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang J, Wang P, Li S, Wang S, Li Y, Liang
N and Wang M: Mdivi-1 prevents apoptosis induced by
ischemia-reperfusion injury in primary hippocampal cells via
inhibition of reactive oxygen species-activated mitochondrial
pathway. J Stroke Cerebrovasc Dis. 23:1491–1499. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Herreweghe F, Festjens N, Declercq W
and Vandenabeele P: Tumor necrosis factor-mediated cell death: To
break or to burst, that's the question. Cell Mol Life Sci.
67:1567–1579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xanthoulea S, Pasparakis M, Kousteni S,
Brakebusch C, Wallach D, Bauer J, Lassmann H and Kollias G: Tumor
necrosis factor (TNF) receptor shedding controls thresholds of
innate immune activation that balance opposing TNF functions in
infectious and inflammatory diseases. J Exp Med. 200:367–376. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-α. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, McQuade T, Siemer AB, Napetschnig J,
Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et
al: The RIP1/RIP3 necrosome forms a functional amyloid signaling
complex required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu W, Liu P and Li J: Necroptosis: An
emerging form of programmed cell death. Crit Rev Oncol Hematol.
82:249–258. 2012. View Article : Google Scholar
|
|
11
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaiser WJ, Upton JW, Long AB,
Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T and
Mocarski ES: RIP3 mediates the embryonic lethality of
caspase-8-deficient mice. Nature. 471:368–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vandenabeele P, Declercq W, Van Herreweghe
F and Vanden Berghe T: The role of the kinases RIP1 and RIP3 in
TNF-induced necrosis. Sci Signal. 3:re42010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moriwaki K and Chan FK: RIP3: A molecular
switch for necrosis and inflammation. Genes Dev. 27:1640–1649.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang DW, Zheng M, Zhao J, Li YY, Huang Z,
Li Z and Han J: Multiple death pathways in TNF-treated fibroblasts:
RIP3- and RIP1-dependent and independent routes. Cell Res.
21:368–371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
National Research Council (USA) Institute
for Laboratory Animal Research: Guide for the Care and Use of
Laboratory Animals. 8th edition. National Academies Press (USA);
Washington, DC: 1996
|
|
18
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thored P, Wood J, Arvidsson A, Cammenga J,
Kokaia Z and Lindvall O: Long-term neuroblast migration along blood
vessels in an area with transient angiogenesis and increased
vascularization after stroke. Stroke. 38:3032–3039. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Swanson RA, Morton MT, Tsao-Wu G, Savalos
RA, Davidson C and Sharp FR: A semiautomated method for measuring
brain infarct volume. J Cereb Blood Flow Metab. 10:290–293. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SJ and Li J: Caspase blockade induces
RIP3-mediated programmed necrosis in Toll-like receptor-activated
microglia. Cell Death Dis. 4:e7162013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Durai Pandian J, Padma V, Vijaya P, Sylaja
PN and Murthy JM: Stroke and thrombolysis in developing countries.
Int J Stroke. 2:17–26. 2007. View Article : Google Scholar
|
|
23
|
Xu D, Du W, Zhao L, Davey AK and Wang J:
The neuroprotective effects of isosteviol against focal cerebral
ischemia injury induced by middle cerebral artery occlusion in
rats. Planta Med. 74:816–821. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Budohoski KP, Guilfoyle M, Helmy A,
Huuskonen T, Czosnyka M, Kirollos R, Menon DK, Pickard JD and
Kirkpatrick PJ: The pathophysiology and treatment of delayed
cerebral ischaemia following subarachnoid haemorrhage. J Neurol
Neurosurg Psychiatry. 85:1343–1353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu H, Zhang Y, Sun H, Chen S and Wang F:
Effects of acupuncture at GV20 and ST36 on the expression of matrix
metalloproteinase 2, aquaporin 4, and aquaporin 9 in rats subjected
to cerebral ischemia/reperfusion injury. PLoS One. 9:e974882014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen HJ, Shen YC, Shiao YJ, Liou KT, Hsu
WH, Hsieh PH, Lee CY, Chen YR and Lin YL: Multiplex Brain Proteomic
Analysis Revealed the Molecular Therapeutic Effects of Buyang
Huanwu Decoction on Cerebral Ischemic Stroke Mice. PLoS One.
10:e01408232015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Tan C, Liu Y, Liu X, Wang X, Gui Y,
Qin L, Deng F, Yu Z, Hu C and Chen L: Resveratrol ameliorates
oxidative stress and inhibits aquaporin 4 expression following rat
cerebral ischemia-reperfusion injury. Mol Med Rep. 12:7756–7762.
2015.PubMed/NCBI
|
|
28
|
Saini MK, Sanyal SN and Vaiphei K:
Piroxicam and C-phycocyanin mediated apoptosis in
1,2-dimethylhydrazine dihydrochloride induced colon carcinogenesis:
Exploring the mitochondrial pathway. Nutr Cancer. 64:409–418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Günther C, Martini E, Wittkopf N, Amann K,
Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF
and Becker C: Caspase-8 regulates TNF-α-induced epithelial
necroptosis and terminal ileitis. Nature. 477:335–339. 2011.
View Article : Google Scholar
|
|
30
|
Bamias G, Jia LG and Cominelli F: The
tumor necrosis factor-like cytokine 1A/death receptor 3 cytokine
system in intestinal inflammation. Curr Opin Gastroenterol.
29:597–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma J, Endres M and Moskowitz MA:
Synergistic effects of caspase inhibitors and MK-801 in brain
injury after transient focal cerebral ischaemia in mice. Br J
Pharmacol. 124:756–762. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Madsen PM, Clausen BH, Degn M, Thyssen S,
Kristensen LK, Svensson M, Ditzel N, Finsen B, Deierborg T,
Brambilla R and Lambertsen KL: Genetic ablation of soluble tumor
necrosis factor with preservation of membrane tumor necrosis factor
is associated with neuroprotection after focal cerebral ischemia. J
Cereb Blood Flow Metab. Oct 19–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Y, Wang J, Song X, Wei R, He F, Peng G
and Luo B: Protective mechanisms of CA074-me (other than
cathepsin-B inhibition) against programmed necrosis induced by
global cerebral ischemia/reperfusion injury in rats. Brain Res
Bull. 120:97–105. 2016. View Article : Google Scholar
|
|
34
|
Moulin M, Anderton H, Voss AK, Thomas T,
Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J and Vaux
DL: IAPs limit activation of RIP kinases by TNF receptor 1 during
development. EMBO J. 31:1679–1691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
36
|
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB,
Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV and Mak TW:
Requirement for Casper (c-FLIP) in regulation of death
receptor-induced apoptosis and embryonic development. Immunity.
12:633–642. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boise LH, Minn AJ, Noel PJ, June CH,
Accavitti MA, Lindsten T and Thompson CB: CD28 costimulation can
promote T cell survival by enhancing the expression of Bcl-XL.
Immunity. 3:87–98. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oberst A, Dillon CP, Weinlich R, McCormick
LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS and Green DR:
Catalytic activity of the caspase-8-FLIP(L) complex inhibits
RIPK3-dependent necrosis. Nature. 471:363–367. 2011. View Article : Google Scholar : PubMed/NCBI
|