Introduction
Prostate cancer (PCa) is the most prevalent
malignancy in men and the second leading cause of male
cancer-associated mortality in the United States (1). Androgens serve vital roles in the
initiation and process of PCa. Initially, PCa responds favorably to
hormone deprivation therapy; however, the majority of patients with
androgen-dependent PCa (ADPC) inevitably progresses to
castration-resistant PCa (CRPC), which has greater malignancy,
within 2 years (2). CRPC shows
poor response to currently available therapies and ultimately
progresses to become terminal. Therefore, studies of the molecular
mechanisms involved in PCa progression are of major importance and
will aid in the discovery of possible treatment strategies for
PCa.
MicroRNAs (miRNAs) are short non-coding,
single-stranded RNAs that function by regulating the protein
translation and mRNA degradation of their target genes (3). Increasing evidence has indicated that
the dysregulation of miRNAs is implicated in human carcinogenesis
and cancer progression, indicating that certain miRNAs can function
as tumor suppressor genes or oncogenes (4,5). A
series of miRNAs have been identified to be aberrantly expressed in
PCa and were suggested to have a functional contribution to PCa
tumorigenesis, including the onco-miRs miR-220/221, miR-125b and
miR-21, and the tumor suppressors miR-15a/16, miR-146a and miR-205
(6), which are associated with the
regulation of cellular differentiation, proliferation, apoptosis
and the acquisition of invasive features and/or androgen
independence.
Gene screening was performed using a miRNA chip on
ADPC and CRCP tissues, and a miRNA expression database was
constructed. In a previous study, miR-30a was identified as being
significantly downregulated in CRPC (7), with this result supported by another
study which showed miR-30a to be downregulated dramatically in CRPC
tissues compared with in ADPC and benign prostatic hyperplasia
(BPH) tissues (8). In addition,
the cyclin E2 gene (CCNE2) was predicted to be a potential target
of miR-30a by computational analysis in the present study, and had
an approximate crosscurrent of expression in the three groups of
tissues. Therefore, based on the previous findings, the aim of the
present study was to investigate the function of miR-30a in CRPC
cells and confirm whether or not CCNE2 is a direct target of
miR-30a.
Materials and methods
Tissue collection
The BPH samples were collected from trans-urethral
prostatic resection (TURP) specimens from patients treated for BPH.
The specimens were histologically confirmed not to contain any
prostate cancer cells. For the ADPC tissues, PCa samples were
obtained from patients that underwent transrectal prostatic biopsy
or radical prostatectomy, and who had not received any previous
treatment. Patients were diagnosed with CRPC based on the continual
increase in serum prostate-specific antigen levels during maximum
androgen deprivation therapy. The CRPC patients underwent TURP due
to urinary retention. Each carcinoma specimen was histologically
examined for the presence of tumor tissue (>60%) using
hematoxylin and eosin (HE) staining. All the samples were snap
frozen in liquid nitrogen and stored at −80°C prior to further
analysis. The ethics approval was obtained from the ethics
committees at Zhongda Hospital, Southeast University (Nanjing,
China) and all samples were collected following the acquisition of
informed consent from the patients.
Cell culture
The CRPC cell lines, DU145 and PC3, were obtained
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China) and cultured in Dulbecco's modified Eagle's
medium (GE Healthcare Life Sciences, Chalfont, UK) supplemented
with 10% fetal bovine serum (FBS; GE Healthcare Life Sciences), and
100 μg/ml streptomycin, 100 U/ml penicillin, (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) within a humidified
atmosphere containing 5% CO2 at 37°C.
Oligonucleotides and cell
transfection
All miRNA mimics were designed and synthesised by
Shanghai GenePharma Co., Ltd., (Shanghai, China) based on the
following sequences: hsa-miR-30a mimics,
5′-UGUAAACAUCCUCGACUGGAAG-3′; and negative control (NC),
5′-UCCAGUCGAGGAUGUUUACAUU-3′. Cell transfection was performed with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Briefly, 1×106
cells were seeded in 6-well plates at 70% confluence a day prior to
transfection. Oligonucleotides formed transfection complexes with
Lipofectamine 2000, and were added to cells and incubated for 6–8 h
prior to refreshing the medium.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA extraction was performed as previously
described (7). Analysis of mature
miR-30a expression was performed using TaqMan microRNA assay
according to the manufacturer's instructions (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Briefly, the reverse transcription
reaction was performed in a volume of 15 μl containing 5
μl total RNA, 3 μl 5X RT primer and 7 μl RT
master mix (0.15 μl 100 mM dNTPs; 1 μl MultiScribe
reverse transcriptase, 50 U/μl; 1.5 μl 10X reverse
transcription buffer; 0.19 μl RNase inhibitor, 20
U/μl; and 4.16 μl nuclease-free water). For synthesis
of cDNA, the reaction mixtures were incubated at 16°C for 30 min,
42°C for 30 min and 85°C for 5 min. The qPCR was performed with a
final volume of 20 μl containing 1 μl 20X TaqMan
MicroRNA Assays, 10 μl TaqMan Universal PCR Master Mix II
(2X), 1.33 μl RT reaction product and 7.67 μl
nuclease-free water. The relative miR-30a expression compared with
U6 was calculated by the 2−ΔΔCq method. The reaction for
miRNA detection was performed using the following conditions: 95°C
for 3 min, 40 cycles at 95°C for 12 sec and 62°C for 40 sec. The
RT-qPCR reactions were performed using a 7300 Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). All reactions
were run in triplicate.
Cell proliferation assay
Cells transfected with oligonucleotides for over 24
h were seeded into 96-well plates (3,000 cells per well). The
proliferation of the cells was determined by MTT assay. A total of
20 μl MTT (5 mg/ml) was added to each well at 24, 48 and 72
h, and the cultures were incubated for 4 h at 37°C. MTT was
carefully aspirated and the purple colored precipitates of formazan
were dissolved in 200 μl DMSO. The absorbance at 490 nm was
measured using a Model 680 automatic multi-well spectrophotometer
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). In total, 5 wells
per treatment group were measured for cell proliferation, and all
independent treatments were performed in triplicate.
Flow cytometric analysis of the cell
cycle and apoptosis
Cells were harvested 48 h post-transfection. The
cell cycle was analyzed by the propidium iodide (PI) staining
method and flow cytometry measurements according to the
manufacturer's instructions (MultiSciences Biotech Ltd., Hangzhou,
China). Apoptosis was analyzed by the annexin V-fluorescein
isothiocyanate (FITC) plus PI staining method according to the
manufacturer's instructions (Ubio Biotechnology Systems Pvt, Ltd.,
Jinan, China). The treated cells were analyzed by flow cytometry on
a FACSCalibur system (BD Biosciences, Franklin Lakes, NJ, USA). A
minimum of 20,000 cells were acquired for each sample. The
experiments were performed in triplicate.
Cell migration and invasion assays
For the migration assays, 1×105 cells in
serum-free medium were placed in the upper chamber of the Transwell
with 8 μm pore size polycarbonate membrane filters (BD
Biosciences). For the invasion assays, matrigel (BD Biosciences)
was applied to the polycarbonate membrane filters of the upper
chamber, following which 1×105 cells in serum-free
medium were seeded according to the manufacturer's protocol. To the
lower chamber, the same medium was added, containing 10% FBS and
the chamber was incubated for 24 h at 37°C. Following this, the
cells remaining on the upper membrane were removed using a
cotton-tip applicator, and the cells on the lower surface of the
membrane were fixed with methanol and stained with crystal violet.
Cells were quantified by counting five random high-powered fields.
All of the experiments were performed in triplicate.
Tumor formation assay in a nude mouse
model
Immunodeficient BALB/C nu/nu male mice (n=3; 5 weeks
old) were obtained from the Shanghai Laboratory Animal Centre
(Shanghai, China). The animal experiments were undertaken in
accordance with the National Institute of Health Guide for the Care
and Use of Laboratory Animals (9)
and were approved by the ethics committee of Zhongda Hospital,
Southeast University (Nanjing, China). PC3 cells transfected with
oligonucleotides for 48 h were harvested and the tumor formation
assay conducted as previously described (10). Tumors were formalin fixed
immediately following harvesting and paraffin embedded.
Immunohistochemical staining (IHC)
The IHC kit (NeoBioscience Technology Co., Ltd.,
Beijing, China) was used for IHC staining. The paraffin embedded
clinical specimens and xenograft tumors were processed according to
the manufacturer's instructions. Briefly, 4-μm thick
sections of the sample tissues were deparaffinized and rehydrated,
then heat-based antigen retrieval (20 min at 95°C) was conducted,
followed by endogenous peroxidase blocking with 3% hydrogen
peroxide, and nonspecific protein blocking with reagent A from the
kit (10% goat serum). Sections were incubated at 37°C for 1 h with
the following primary antibodies purchased from Abcam (Cambridge,
MA, USA): Rabbit monoclonal anti-human CCNE2 (1:250; #ab40890) and
rabbit polyclonal anti-human Ki67 (1:100; #ab66155) antibodies.
This was followed by incubation with reagent B (biotinylated goat
anti rabbit IgG) for 10 min and reagent C (streptavidin-labeled
horseradish peroxidase) for 10 min at room temperature. The
reaction was visualized by DAB developer mixed from reagent D, E
and F. An Eclipse E600 microscope (Nikon Corporation, Tokyo, Japan)
was used for visualization. For the negative controls, sections
were treated following the same procedure except that they were
incubated with Tris-buffered saline (TBS) without primary
antibody.
Plasmid construction and luciferase
assays
The 3′-UTR segment of CCNE2 mRNA containing the
miR-30a binding site was amplified by PCR from human DNA extracted
from peripheral blood of the human participants. The primers used
contained the following restriction sites: Forward,
5′-CCGCTCGAGCACAAGTTACACTGCCATTC-3′ (XhoI) and reverse,
5′-CTTGCGGCCGCGCTATAGCAGCTATAGATA-3′ (NotI). The PCR product
was cloned into the XhoI and NotI restriction sites
downstream of the open reading frame of luciferase in a psiCHECK-2
Vector (Promega Corporation, Madison, WI, USA) to generate the
CCNE2 3′-UTR reporter. For the reporter assays, 1×106
cells were seeded in 24-well plates and co-transfected at 70%
confluence with 0.25 μg of CCNE2 3′UTR or control reporter
plasmid and 25 pmol miR-30a or NC mimics using Lipofectamine 2000.
Each transfection was performed in triplicate and luciferase
activity was assessed 48 h after transfection using dual luciferase
assays (Promega Corporation). Firefly luciferase activity was
normalized to Renilla luciferase activity accordingly.
Western blotting
Cells were transfected with oligonucleotides, then
harvested on ice 72 h later. Protein isolation and western blotting
analyses were conducted as previously described (10). Cells were lysed with
radioimmunoprecipitation buffer (Beyotime Institute of
Biotechnology, Haimen, China) then total protein concentrations
were determined by bicinchoninic acid assay (Beyotime Institute of
Biotechnology). Protein samples (30 μg) were separated by
10% SDS-PAGE (Beyotime Institute of Biotechnology) at 80 V for 30
min then 100 V for 1.5 h. Electrophoresed proteins were transferred
to a poly-vinylidene difluoride membrane (EMD Millipore, Billerica,
MA, USA) and subsequently blocked with 5% skimmed milk (BioSharp,
Hefei, China) at room temperature for 1 h. The membranes were
incubated with the abovementioned CCNE2 (1:1,000) antibody, or
rabbit polyclonal anti-human GAPDH antibody (1:1,000; #sc25778;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) in 5% skimmed milk
overnight at 4°C. The blots were washed 3 times with TBS (pH 7.6,
20 mM Tris-HCl, 137 mM NaCl) with 0.01% Tween 20, incubated with
horseradish peroxidase-labeled goat anti-rabbit secondary antibody
(1:3,000; #ZB2301; Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China) at 37°C for 1 h, and visualized using Immobilon
Western Chemilum HRP Substrate (EMD Millipore). Blots were exposed
to the film (5×7 inch; Carestream Health Co., Ltd., Xiamen, China)
for 5 min in X-ray film cassette (Yuehua Medical Instrument Factory
Co., Ltd., Shantou, China), and then developed and fixed (Beyotime
Institute of Biotechnology). Protein levels were determined by
normalization against GAPDH.
Bioinformatic and statistical
analysis
The miRNA target predicting algorithms miRDB
(www.mirdb.org/miRDB/) and TargetScan
(www.targetscan.org/) were used to
predict miRNAs targeting CCNE2 and the binding regions. Data in the
present study was obtained from at least three independent
experiments and presented as the mean ± standard error. The
correlation between the expression of miR-30a and CCNE2 was
examined by Spearman correlation analysis. Group means were
compared by Student's t-test. Expression of miR-30a and
CCNE2 in three groups were analyzed by analysis of variance
followed by Tukey's multiple comparisons test. All statistical
analyses were performed using SPSS software, version 16.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of miR-30a and CCNE2 and the
correlation between miR-30a and CCNE2 in the three patient
groups
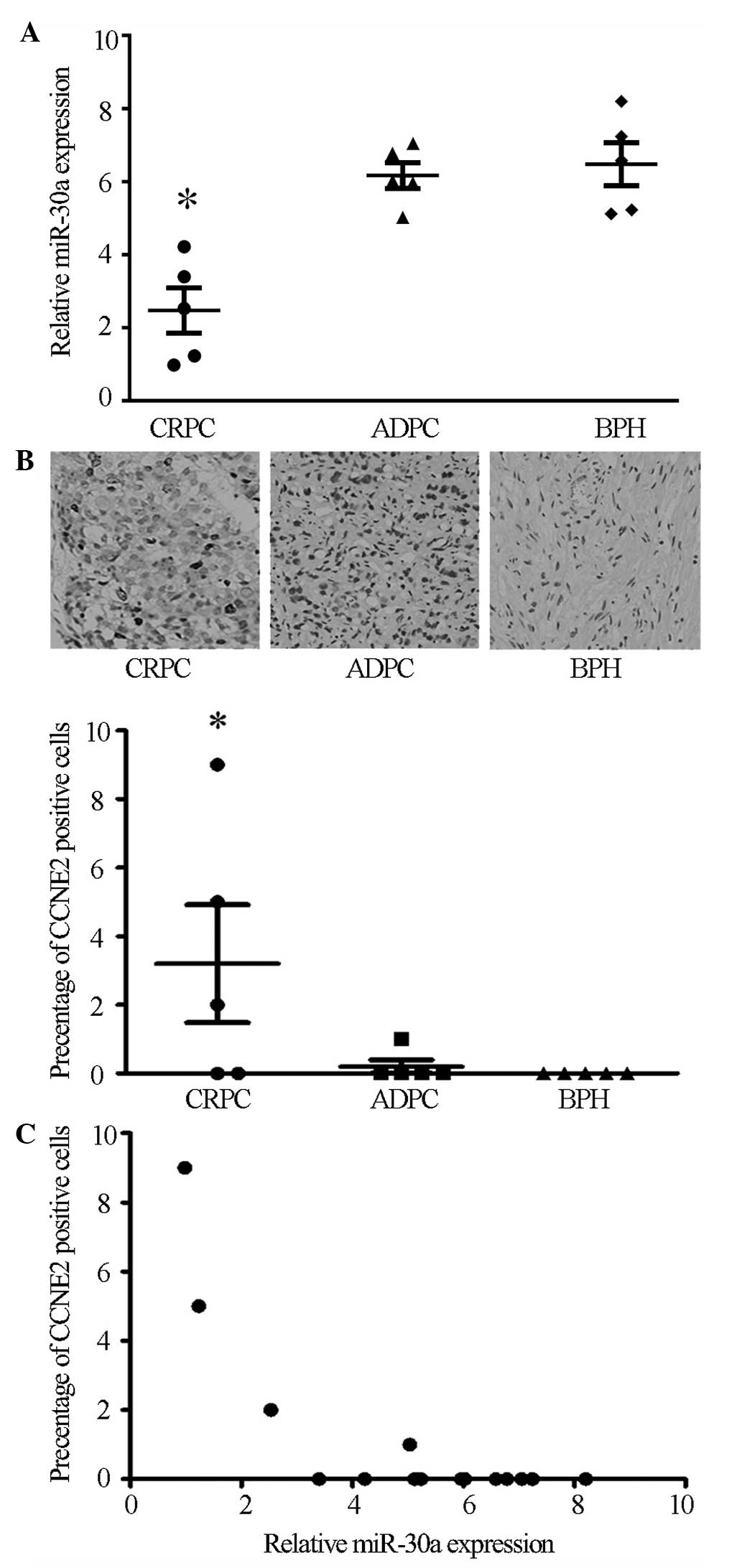
Microarray data from a previous study indicated that
miR-30a expression in the CRPC tissues was lower compared with the
ADPC tissues (7). To verify these
microarray results, the current study assessed the miR-30a
expression levels in 5 BPH, 5 ADPC and 5 CRPC tissues by RT-qPCR.
Compared with the ADPC and BPH tissues, miR-30a expression was
markedly downregulated in CRPC tissues (P<0.05), while no
significant difference was observed between the ADPC and BPH
tissues (Fig. 1A).
Immunohistochemistry was conducted to detect CCNE2 protein
expression in the BPH, ADPC and CRPC tissues. The results showed
that CCNE2 was expressed strongly in 3 of 5 CRPC tissues
(P<0.05), while no significant expression was observed in ADPC
or BPH tissues (Fig. 1B). In
agreement with miR-30a inhibition of CCNE2 in cultured cells, the
expression levels of miR-30a negatively correlated with CCNE2
expression in the prostate samples (P<0.05, r=−0.72; Fig. 1C). Taken together, these data
suggest a potential interaction between miR-30a and CCNE2 in the
progression of ADPC to CRPC.
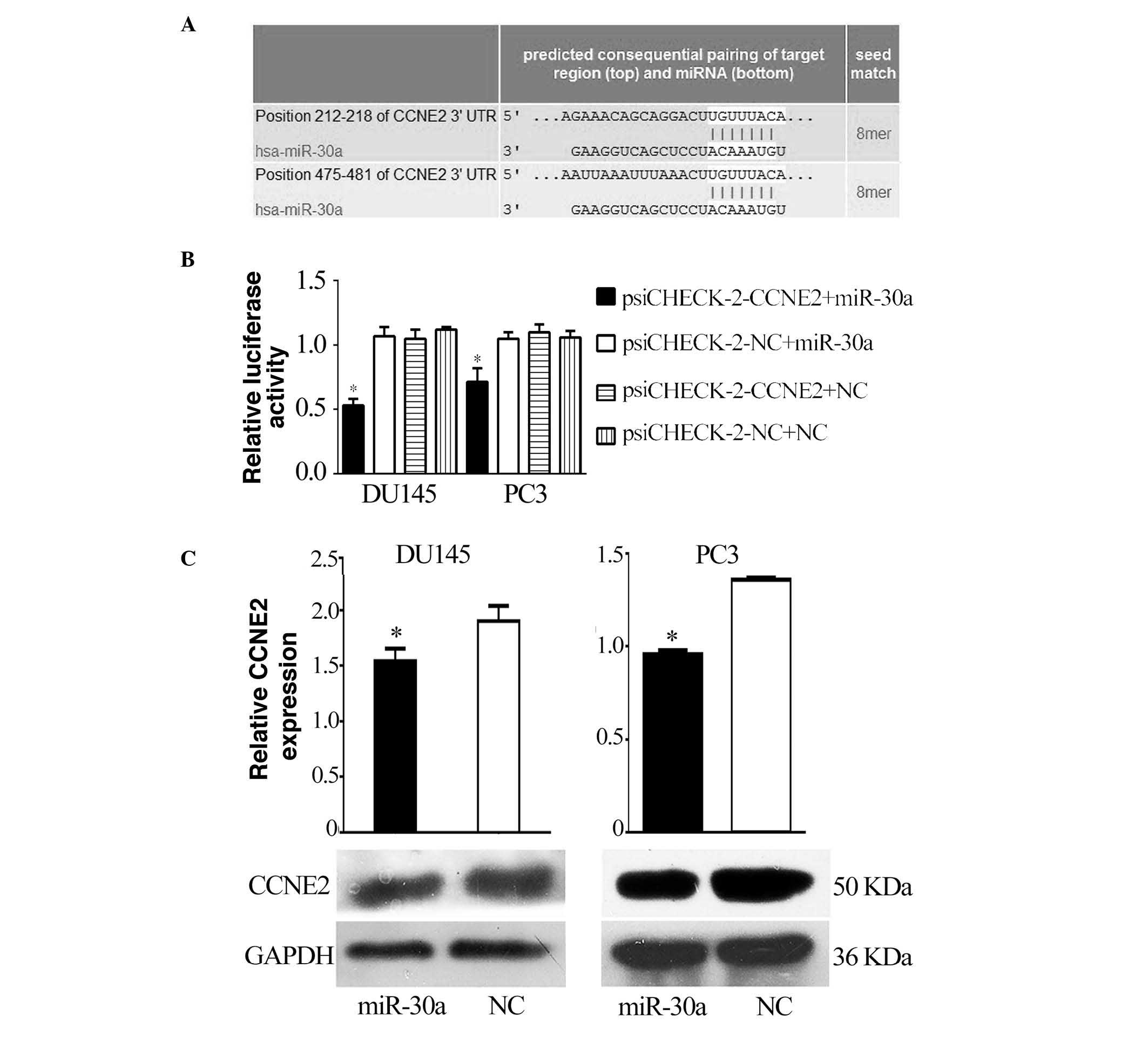
CCNE2 is a direct target of miR-30a
A bioinformatic search (TargetScan, miRDB) was
performed for putative targets of miR-30a, and miR-30a was
identified as being able to bind to target sequences located in
nucleotides 212–218 and 475–481 of the 3′-UTR of CCNE2 mRNA
(Fig. 2A). To ascertain the direct
miRNA-target interaction, the CCNE2 3′-UTR was cloned into a
luciferase reporter cloning site in a psiCHECK-2 dual luciferase
vector. With increasing miR-30a levels, the luciferase activities
were markedly reduced (P<0.05; Fig.
2B). Furthermore, western blotting indicated that the levels of
CCNE2 were reduced by treatment with miR-30a mimics (P<0.05;
Fig. 2C).
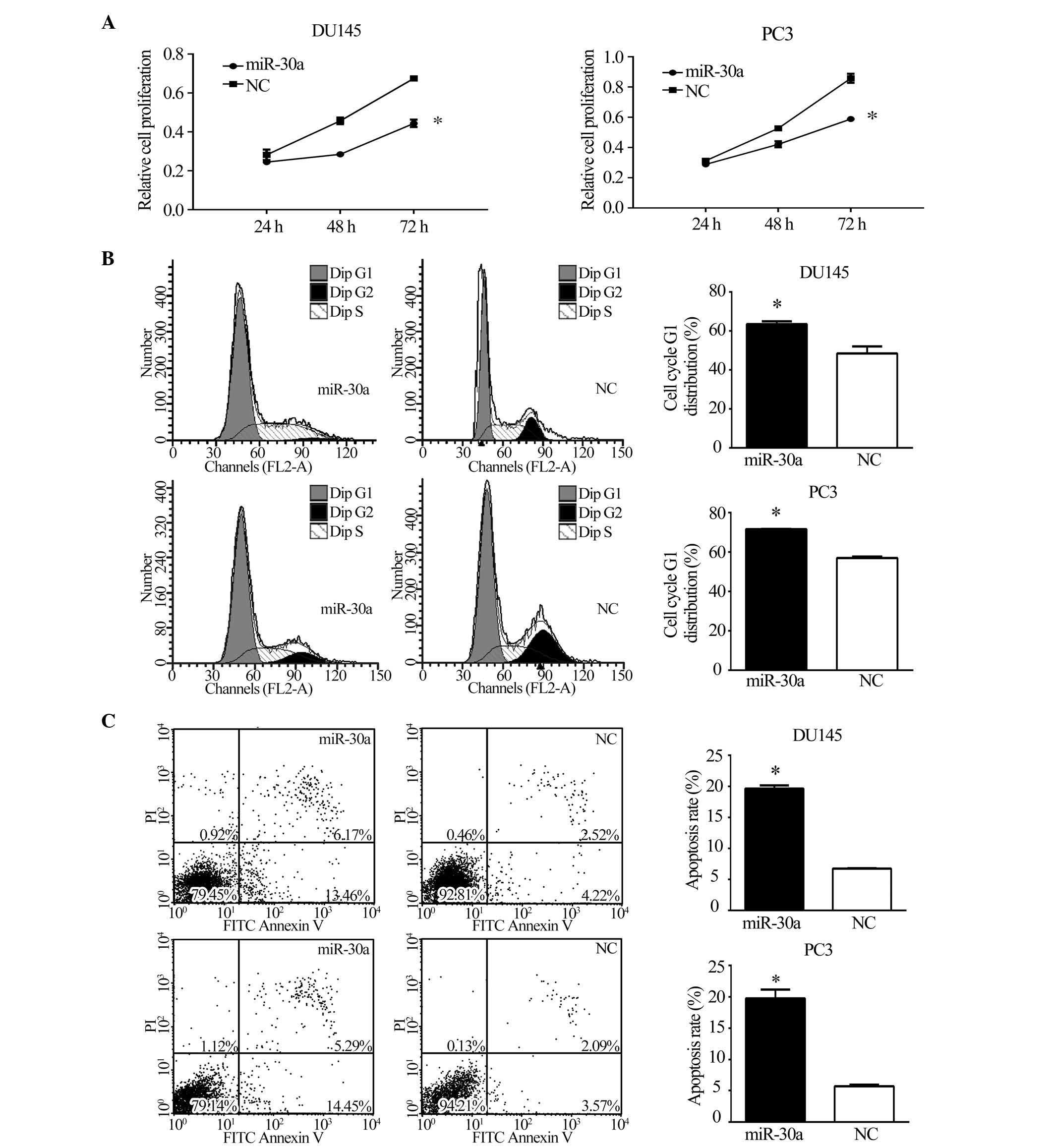
miR-30a suppresses tumorigenicity and
metastasis in vitro
To investigate the function of miR-30a in CRPC,
miR-30a expression was restored in CRPC cell lines. DU145 and PC3
cells were transfected with miR-30a or NC, following which
functional assays were conducted. The MTT assay, as shown in
Fig. 3A, demonstrated that the
restoration of miR-30a expression significantly inhibited the
growth of CRPC cells at 48 and 72 h (P<0.05). To assess the role
of miR-30a in cell cycle progression, miR-30a expression was
restored in DU145 and PC3 cells. Compared with NC transfectants,
flow cytometric analysis of PI-stained cells transfected with
miR-30a demonstrated a G1 accumulation 48 h following
transfection (P<0.05; Fig. 3B).
Annexin V-FITC/PI stained cells transfected with miR-30a showed a
higher rate of apoptosis (P<0.05; Fig. 3C). These results indicate that
miR-30a induces cell cycle arrest and apoptosis in CRPC cells.
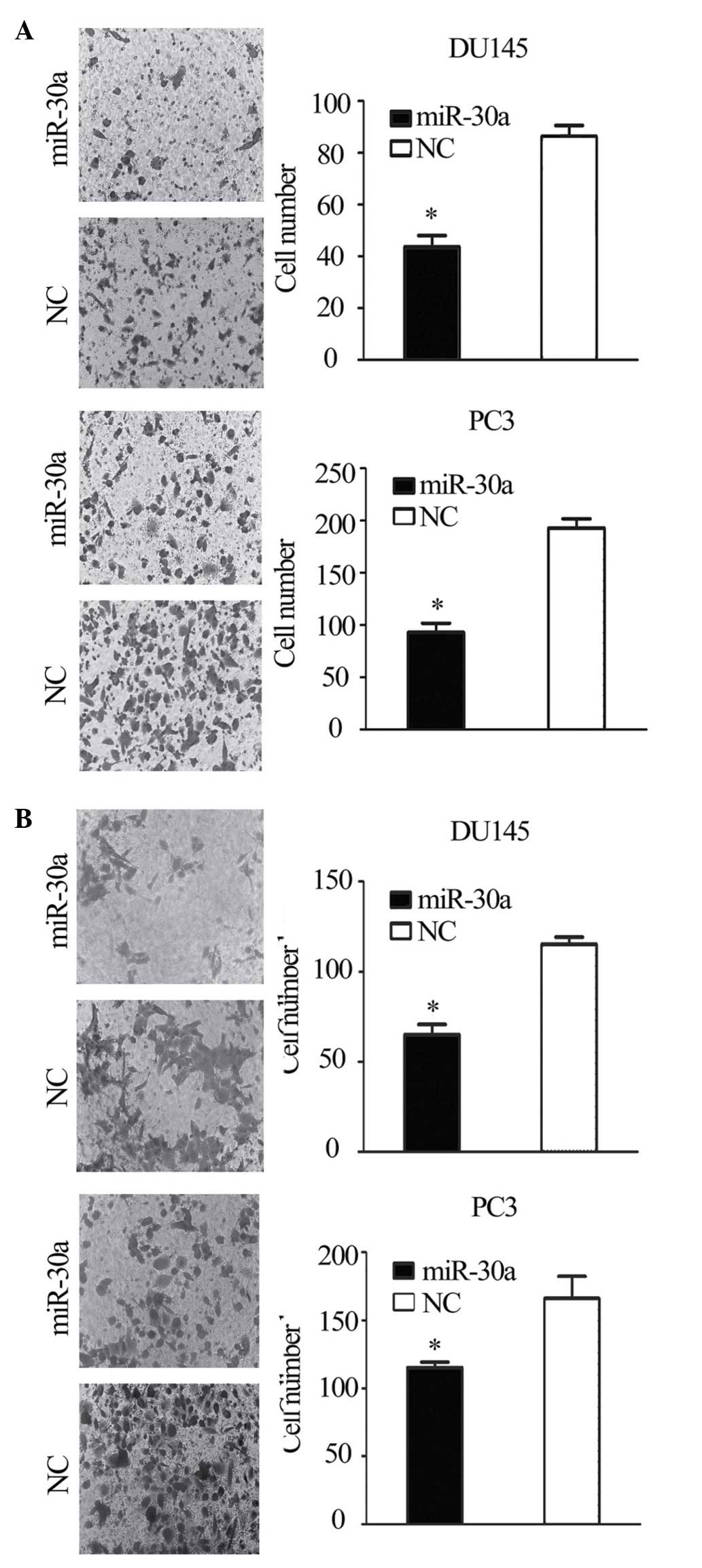
Transwell migration and invasion assays showed that the migration
and invasion of miR-30a-transfected cells was reduced compared with
the NC transfectants (P<0.05; Fig.
4A and B). This suggests that the restoration of miR-30a
expression suppresses the tumorigenicity and metastasis of CRPC
cells in vitro.
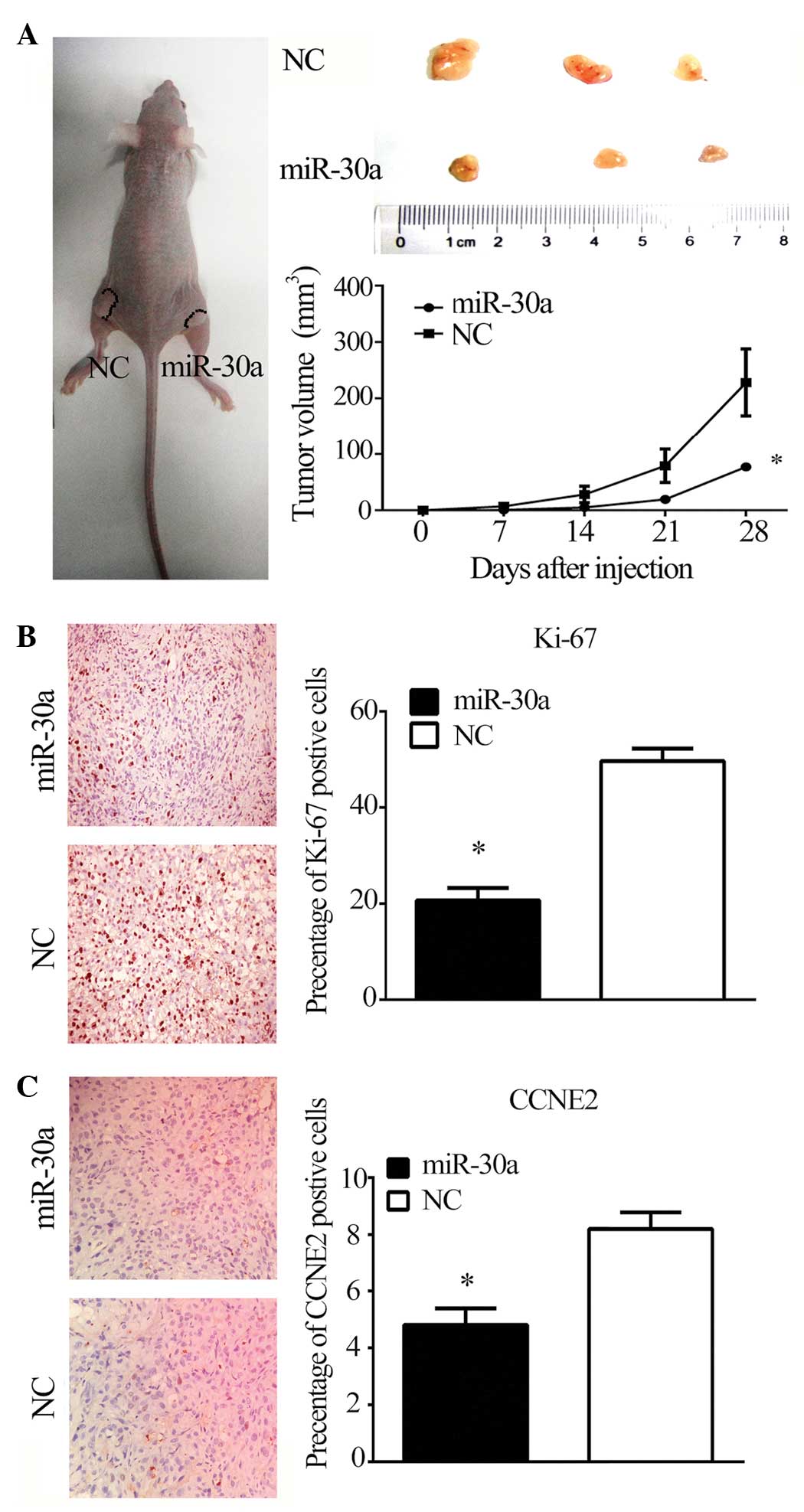
miR-30a suppresses tumor growth in
vivo
To investigate the effect of miR-30a on
tumorigenicity and tumor progression in vivo, miR-30a and
NC-transfected PC3 cells were subcutaneously injected into either
flank of nude mice. As predicted, miR-30a reduced the volume of
tumors formed from PC3 cells (P<0.05; Fig. 5A). Furthermore, miR-30a reduced
Ki-67 and CCNE2 staining in tumor xenografts (P<0.05; Fig. 5B and C), suggesting that the
miR-30a/CCNE2 axis may reduce tumorigenicity and tumor progression
in a nude mouse model.
Discussion
Understanding the underlying molecular mechanisms of
prostate tumorigenesis and cancer progression is crucial to improve
clinical treatment and the management of patients with CRPC. In
previous studies, it was reported that the loss of miR-146a is a
critical mechanism for the overexpression of epidermal growth
factor (EGF) receptor in CRPC (10), and miR-361-5p can act as a tumor
suppressor by targeting signal transducer and activator of
transcription 6 in CRPC (7). Based
on the preliminary microarray analysis, miR-30a was focused upon
for subsequent investigation, which indicated it was down-regulated
in CRPC tissues. miR-30a expression was markedly downregulated in
CRPC tissues compared with the ADPC and BPH tissues, however, there
was no significant difference between the ADPC and BPH tissues,
which is consistent with previous reports (8,11).
These results suggest that miR-30a may serve a role in the
progression of ADPC to CRPC. Previous studies have reported miR-30a
to be a tumor suppressor in renal cell carcinoma (12), non-small cell lung cancer (13), breast cancer (14) and colorectal carcinoma (15), however, a tumor inducer in glioma
(16). These contradictory reports
indicate that there may be disease-specific modulation of miR-30a.
For prostate cancer, there has been a single previous study, which
indicated that miR-30 suppresses epithelial-to-mesenchymal
transition (EMT) phenotypes and inhibits migration and invasion in
prostate cancer cells by connecting EGF/Src signal to Ets-related
gene and EMT (17). In the present
study, functional analyses showed that the restoration of miR-30a
expression in CRPC cells significantly suppressed proliferation,
migration and invasion ability, induced cell cycle arrest and
apoptosis in vitro, and in addition, reduced tumorigenicity
and tumor progression in vivo. Furthermore, the present
study reported another oncogene, CCNE2, as a target gene of miR-30a
in CRPC.
Cyclin E is composed of cyclin E1 and E2, which are
encoded by separate genes located at chromosomes 19q12 (CCNE1) and
8q22.1 (CCNE2) in humans, however, they share high sequence
identity and functional redundancy (18). Cyclin E activates Cdk2 in late
G1 phase, driving the transition from G1 to S
phase, with Cdk2 phosphorylating the Rb protein and other targets
necessary for the initiation of DNA replication (19). Unlike CCNE1, which is expressed in
most proliferating normal and tumor cells, CCNE2 levels are low to
undetectable in nontransformed cells, with levels increasing
significantly in tumor-derived cells (20), suggesting that mechanisms distinct
from CCNE1 induce tumorigenesis and progression (21). Previous studies indicate that CCNE2
overexpression is associated with pathogenesis (22), endocrine resistance (23), metastasis and reduced survival
(24) in breast cancer. A previous
study examined PCa array data (25) and showed higher expression levels
of CCNE2 probe sets in metastatic samples compared with benign and
localized samples. This is supported by results of an independent
analysis using Oncomine (26).
Furthermore, CCNE2 is phosphatase and tensin homolog-regulated and
is associated with cell cycle arrest in G1 phase and
metastasis in PCa (27). In the
current study, it was observed that CCNE2 was overexpressed in
patients with CRCP, and had an inverse correlation with the level
of miR-30a. Furthermore, CCNE2 was identified as a direct target of
miR-30a. Overexpression of miR-30a reduced the malignant
progression of PCa cells and reduced the expression of CCNE2 in
vitro and in vivo.
In conclusion, these data provide evidence that
miR-30a, which is frequently downregulated in CRPC, supports the
multi-step process of CRPC development via modulating CCNE2
expression, which in turn alters cancer development and
progression. Therefore, miR-30a and CCNE2 may be regarded as
potential targets for CRPC therapy.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81370849, 81300472,
81070592 and 81202034), the Natural Science Foundation of Jiangsu
Province (grant nos. BK2013032 and BK2012336), Nanjing City (grant
no. 201201053) and Southeast University (grant no. 3290002402), and
the Science Foundation of the Ministry of Education of China (grant
no. 20120092120071).
References
|
1
|
Siegel R, Ma J, Zhou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feldman BJ and Feldman D: The development
of androgen independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar
|
|
3
|
Lai EC: MicroRNAs are complementary to
3′UTR sequence motifs that mediate negative post-transcriptional
regulation. Nat Genet. 30:363–364. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang L, Xul B, Chen S, Lu K, Liu C, Wang
Y, Zhao Y, Zhang X, Liu D and Chen M: The complex roles of
microRNAS in the metastasis of renal cell carcinoma. J Nanosci
Nanotechnol. 13:3195–3203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coppola V, De Maria R and Bonci D:
MicroRNAs and prostate cancer. Endocr Relat Cancer. 17:F1–F17.
2010. View Article : Google Scholar
|
|
7
|
Liu D, Tao T, Xu B, Chen S, Liu C, Zhang
L, Lu K, Huang Y, Jiang L, Zhang X, et al: MiR-361-5p acts as a
tumor suppressor in prostate cancer by targeting signal transducer
and activator of transcription-6(STAT6). Biochem Biophys Res
Commun. 445:151–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Porkka KP, Pfeiffer MJ, Waltering KK,
Vessella RL, Tammela TL and Visakorpi T: MicroRNA expression
profiling in prostate cancer. Cancer Res. 67:6130–6135. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
National Research Council: Guide for the
care and use of laboratory animals. 7th edition. National Academy
Press; Washington DC: 1996
|
|
10
|
Xu B, Wang N, Wang X, Tong N, Shao N, Tao
J, Li P, Niu X, Feng N, Zhang L, et al: MiR-146a suppresses tumor
growth and progression by targeting EGFR pathway and in a
p-ERK-dependent manner in castration-resistant prostate cancer.
Prostate. 72:1171–1178. 2012. View Article : Google Scholar
|
|
11
|
Ozen M, Creighton CJ, Ozdemir M and
Ittmann M: Widespread deregulation of microRNA expression in human
prostate cancer. Oncogene. 27:1788–1793. 2008. View Article : Google Scholar
|
|
12
|
Huang QB, Ma X, Zhang X, Liu SW, Ai Q, Shi
TP, Zhang Y, Gao Y, Fan Y, Ni D, et al: Down-Regulated miR-30a in
clear cell renal cell carcinoma correlated with tumor hematogenous
metastasis by targeting angiogenesis-specific DLL4. PLoS One.
8:e672942013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kumarswamy R, Mudduluru G, Ceppi P,
Muppala S, Kozlowski M, Niklinski J, Papotti M and Allgayer H:
MicroRNA-30a inhibits epithelial-to-mesenchymal transition by
targeting Snai1 and is downregulated in non-small cell lung cancer.
Int J Cancer. 130:2044–2053. 2012. View Article : Google Scholar
|
|
14
|
Fu J, Xu X, Kang L, Zhou L, Wang S, Lu J,
Cheng L, Fan Z, Yuan B, Tian P, et al: MiR-30a suppresses breast
cancer cell proliferation and migration by targeting Eya2. Biochem
Biophys Res Commun. 445:314–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baraniskin A, Birkenkamp-Demtroder K,
Maghnouj A, Zöllner H, Munding J, Klein-Scory S, Reinacher-Schick
A, Schwarte-Waldhoff I, Schmiegel W and Hahn SA: MiR-30a-5p
suppresses tumor growth in colon carcinoma by targeting DTL.
Carcinogenesis. 33:732–739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jia Z, Wang K, Wang G, Zhang A and Pu P:
MiR-30a-5p antisense oligonucleotide suppresses glioma cell growth
by targeting SEPT7. PLoS One. 8:e550082013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kao CJ, Martiniez A, Shi XB, Yang J, Evans
CP, Dobi A, de Vere White RW and Kung HJ: MiR-30 as a tumor
suppressor connects EGF/Src signal to ERG and EMT. Oncogene.
33:2495–2503. 2014. View Article : Google Scholar
|
|
18
|
Caldon CE and Musgrove EA: Distinct and
redundant functions of cyclin E1 and cyclin E2 in development and
cancer. Cell Div. 5:22010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gudas JM, Payton M, Thukral S, Chen E,
Bass M, Robinson MO and Coats S: Cyclin E2, a novel G1 cyclin that
binds Cdk2 and is aberrantly expressed in human cancers. Mol Cell
Biol. 19:612–622. 1999. View Article : Google Scholar
|
|
21
|
Caldon CE, Sergio CM, Burgess A, Deans AJ,
Sutherland RL and Musgrove EA: Cyclin E2 induces genomic
instability by mechanisms distinct from cyclin E1. Cell Cycle.
12:606–617. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Payton M, Scully S, Chung G and Coats S:
Deregulation of cyclin E2 expression and associated kinase activity
in primary breast tumors. Oncogene. 21:8529–8534. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Caldon CE, Sergio CM, Kang J,
Muthukaruppan A, Boersma MN, Stone A, Barraclough J, Lee CS, Black
MA, Miller LD, et al: Cyclin E2 overexpression is associated with
endocrine resistance but not insensitivity to CDK2 inhibition in
human breast cancer cells. Mol Cancer Ther. 11:1488–1499. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sieuwerts AM, Look MP, Meijer-van Gelder
ME, Timmermans M, Trapman AM, Garcia RR, Arnold M, Goedheer AJ, de
Weerd V, Portengen H, et al: Which cyclin E prevails as prognostic
marker for breast cancer? Results from a retrospective study
involving 635 lymph node-negative breast cancer patients. Clin
Cancer Res. 12:3319–3328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Varambally S, Yu J, Laxman B, Rhodes DR,
Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, et
al: Integrative genomic and proteomic analysis of prostate cancer
reveals signatures of metastatic progression. Cancer Cell.
8:393–406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu YP, Landsittel D, Jing L, Nelson J, Ren
B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S, et al: Gene
expression alterations in prostate cancer predicting tumor
aggression and preceding development of malignancy. J Clin Oncol.
22:2790–2799. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu Z, Cho H, Hampton GM and Theodorescu D:
Cdc6 and cyclin E2 are PTEN-regulated genes associated with human
prostate cancer metastasis. Neoplasia. 11:66–76. 2009. View Article : Google Scholar
|