Introduction
Aniridia is characterized by partial or complete
absence of the iris and iris hypoplasia. It is a rare congenital
disorder with a frequency of 1 in 64,000–96,000 newborns. Aniridia
may occur isolated or as part of a syndrome, such as 'Wilms tumor,
Aniridia, Genitourinary abnormalities, Retardation' (WAGR), Rieger
syndrome and Peters syndrome. 85% of patients with aniridia have an
autosomal-dominant inheritance pattern (1).
Approximately 90% of patients with aniridia have
mutations in the human PAX6 gene, a member of the paired box gene
family. It is located on chromosome 11p13 and contains 14 exons.
The PAX6 gene encodes a transcriptional regulator that has crucial
roles in the development of the eye, central nervous system and
pancreatic islets (1,2).
Non-sense mutations or deletions leading to complete
absence of the PAX6 protein primarily cause aniridia, while
missense mutations of this gene are associated with other ocular
phenotypes such as Rieger anomaly, cataract or glaucoma (3).
The present study performed a clinical and genetic
analysis of two families containing several cases of aniridia. The
heterozygous deletion in exon 8, which was identified in one of the
families, is a novel PAX6 mutation.
Materials and methods
Patients
Two Turkish families were enrolled in the present
study, each including five affected and 12 unaffected members,
whose health status was confirmed by ophthalmological examination.
Clinical data were collected and detailed family surveys were
performed. Written informed consent was obtained from every adult
patient and from the parents of all patients under the age of 18.
The study was approved by the ethics committee of Dokuz Eylul
University (Izmir, Turkey).
DNA isolation and molecular analysis
Genomic DNA from the peripheral blood lymphocytes of
all individuals were extracted using the QIAamp DNA Blood Mini kit
(Qiagen, Hilden, Germany) using standard procedures. Concentration
and purity of isolated DNA were determined using a NanoDrop
spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
All coding exons and exon-intron boundaries of the PAX6 gene were
amplified using polymerase chain reaction (PCR) with the HelixAmp™
Ready-2X-Multiplex version 2.0 PCR mix (NanoHelix, Upper Heyford,
UK). Each reaction mixture contained 12.5 µl master mix, 2
µl distilled water, 0.5 µl forward primer, 0.5
µl reverse primer and DNA (20–30 ng). Primers were
synthesized by Metabion (Planegg, Germany) and are listed in
Table I. PAX6 gene regions are
listed in Table I. Amplifications
were performed using an ABI GeneAmp® PCR System 9700
thermal cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.)
with the following thermocycling conditions: Initial denaturation
at 94°C for 15 min, followed by 35 cycles of denaturation at 94°C
for 30 sec, annealing at 57°C for 45 sec (or 59°C for 45 sec for
exon 4) and extension at 72°C for 45 sec, followed by a final
extension at 72°C for 7 min. PCR products were verified by 2%
agarose gel electrophoresis and ethidium bromide staining.
Accomplished amplicons were continued for second PCR using the
BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems).
Second PCR conditions were as follows: Initial denaturation step at
96°C for 1 min, followed by 25 cycles including denaturation at
96°C for 10 sec, annealing at 50°C for 5 sec and extension at 60°C
for 4 min. After purification using the Zymo Research purification
kit (Zymo Research Corp, Irvin, CA, USA), samples were analyzed
using ABI 3100 and ABI 3130 capillary electrophoresis systems
(Applied Biosystems). The sequences were evaluated using the CLC
Genomics Workbench 3 sequencing software (Qiagen). The Ensembl
database (www.ensembl.org; GRCh38.p3,
GCA_000001405.18) with the transcript ID ENST00000419022 of the
PAX6 gene was used to compare the individual sequences with the
reference sequence taken from the Ensembl database. All variations
were analyzed using mutation and single nucleotide polymorphism
(SNP) databases [Human Genome Mutation Database (http://www.hgmd.cf.ac.uk), National Center for
Biotechnology Information/SNP (http://www.ncbi.nlm.nih.gov/snp), Ensembl and Mutation
Taster (http://www.mutationtaster.org/)]. Each variation was
confirmed by bidirectional sequencing. Variations were described
according to the nomenclature recommended by the Human Genomic
Variation Society (http://www.hgvs.org/mutnomen/).
 | Table IPrimers of the PAX6 gene exons. |
Table I
Primers of the PAX6 gene exons.
| Exon | Forward primer | Reverse primer | Amplicon length
(bp) |
|---|
| 4 |
5′-TTGGGAGTTCAGGCCTACCT-3′ |
5′-CCAGTATCGAGAAGAGCCAAG-3′ | 324 |
| 5 |
5′-TCTTCTTCCTCTTCACTCTGC-3′ |
5′-TGAAAGAGATAGGGAAGGATG-3′ | 392 |
| 6–7 |
5′-ATTTATCTACTTCGTTTTGATGC-3′ |
5′-AGAGGGTGGGAGGAGGTAAAG-3′ | 633 |
| 8 |
5′-AGCTGAGATGGGTGACTGTGT-3′ |
5′-AAGGGATGCACATATGGAGAG-3′ | 281 |
| 9 |
5′-GGGAATGTTTTGGTGAGGCT-3′ |
5′-ACCTCCAACCAATTCCCTTTA-3′ | 698 |
| 10 |
5′-GGAACCAGTTTGATGCACAGT-3′ |
5′-GCAGCAGAGCATTTAGCAGAC-3′ | 302 |
| 11–12 |
5′-GTCTGCTAAATGCTCTGCTGC-3′ |
5′-AGCTCTCAAGGGTGCAGACAC-3′ | 579 |
| 13 |
5′-TGGCTGTGTGATGTGTTCCTC-3′ |
5′-AGAAAACTTGCAGTCTCAGGC-3′ | 462 |
| 14 |
5′-CCATGTCTGTTTCTCAAAGGGA-3′ |
5′-CCCCAGTGGTACAATACAGGA-3′ | 295 |
Results
First family comprised a female patient (age, three
months) with bilateral congenital cataracts and partial aniridia as
well as her affected mother, who were referred to the Department of
Pediatrics (Izmir, Turkey) for genetic evaluation of aniridia. The
mother's first child (male aged 2.5 years old), was healthy and her
second pregnancy had been terminated due to cleft palate and
hydrocephaly of the fetus. The patient's brother, parents, all
uncles and grandparents were examined. The mother, two uncles and
grandmother (on mother's side) presented with bilateral congenital
cataracts and partial aniridia (Fig.
1A).
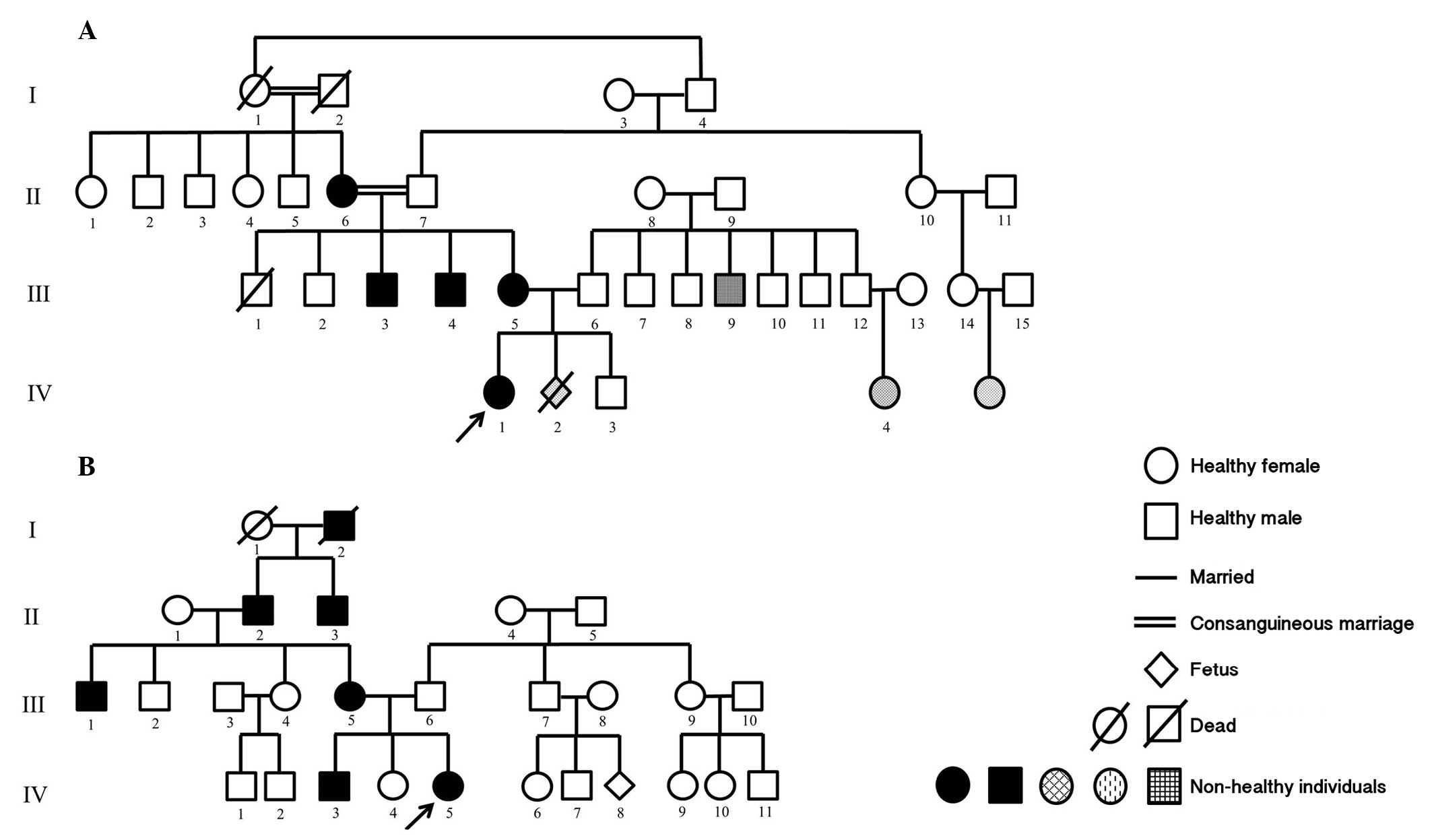 | Figure 1Pedigrees of the two families examined
in the present study. (A) Family 1. II-6, III-3, III-4, III-5 and
IV-1: Aniridia, congenital cataract; III-9: Stutter; IV-2:
Hydrocephalus, cleft lip-palate; IV-4: Congenital heart disease.
(B) Family 2. III-1, III-5, IV-3 and IV-5: Aniridia, congenital
cataract. The arrows indicate the main probands. Circle, female;
Square, male. |
The second family included a female patient (nine
years old) with bilateral congenital cataracts and aniridia. Her
parents, all siblings and grandparents were assessed. Her mother,
brother (7 years old), grandfather and uncle presented with
bilateral congenital cataracts and aniridia and her father suffered
from loss of vision, as a result of glaucoma (Fig. 1B).
Karyotype analysis of the patients and their parents
from peripheral blood lymphocytes yielded normal results. Mutation
screening in all coding exons of PAX6 were performed using the
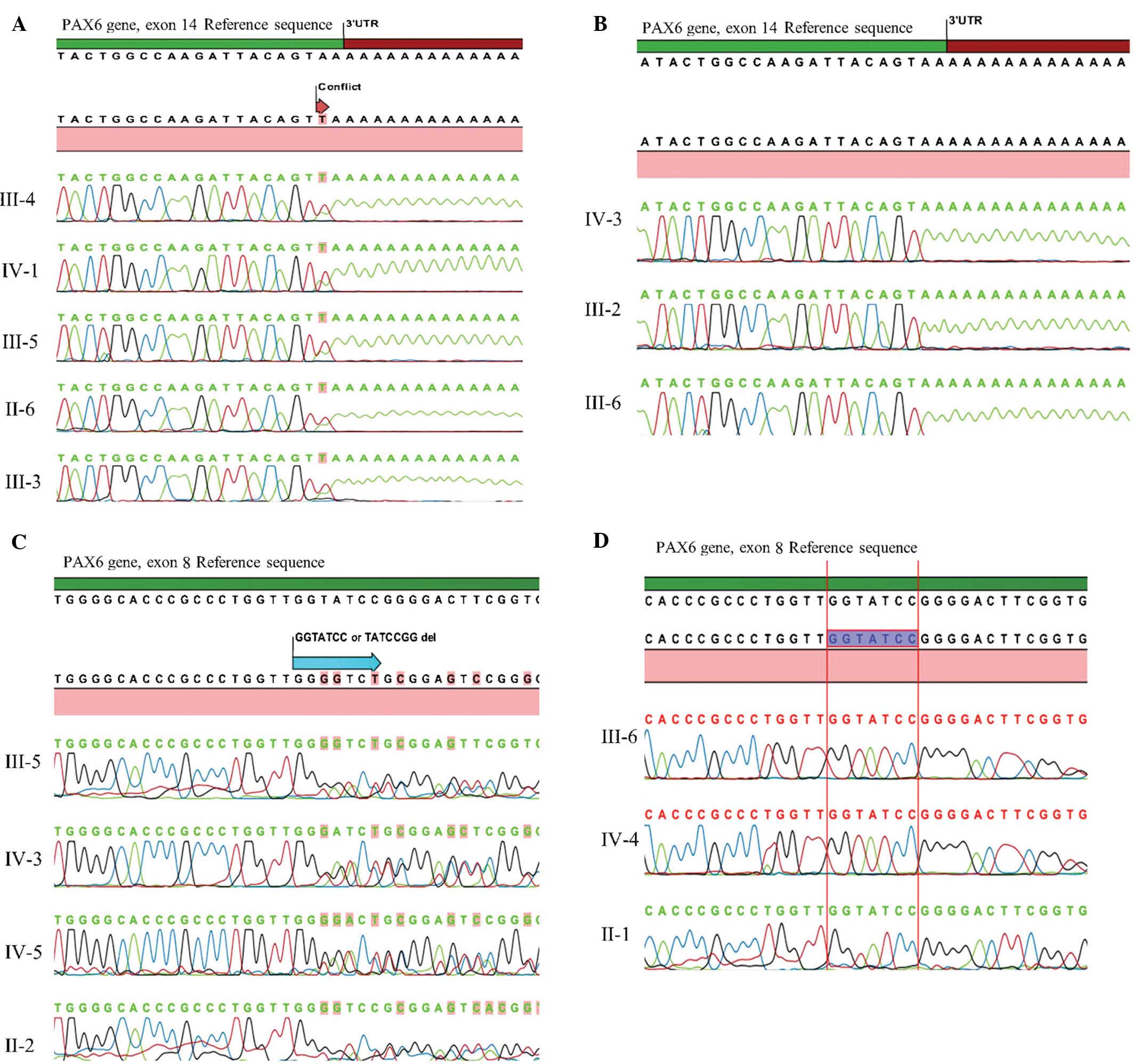
Sanger sequencing technique. In the first family, a heterozygous
non-stop mutation in exon 14 (c.1290 A>T, X437L) was identified
in all affected members and was inherited in an autosomal dominant
inheritance pattern.(Fig. 2A).
Conversely, this mutation was not identified in the unaffected
members.
A novel heterozygous deletion in exon 8 (c.529_535
TATCCGGdel or c.527_533 GGTATCCdel) that causes a frameshift was
identified in all affected members of the second family (Fig. 2B), except for the male patient with
loss of vision due to glaucoma. Unaffected members of the family
did not have this mutation. To the best of our knowledge, the
present study was the first to report on this deletion mutation in
the PAX6 gene patients with aniridia.
Discussion
The present study presented two pedigrees with two
different heterozygous PAX6 mutations that cause aniridia and
congenital cataracts. The affected members of the first family
carried a heterozygous non-stop mutation in exon 14 that causes
on-going translation of mRNA into 3′-untranslated region. The
mutation identified in the second family was a heterozygous
deletion in exon 8 causing a frameshift, which has not been
reported previously.
Aniridia is a rare congenital panocular disorder
with a frequency of 1 in 64,000–96,000 newborns and is
characterized by partial or complete absence of the iris and
verying degrees of iris hypoplasia. It may be associated with other
ocular abnormalities, including foveal hypoplasia, cataracts,
nystagmus, glaucoma, optic nerve hypoplasia, ectopia lentis and
keratopathy. Cataracts have been reported in 50–85 % of cases with
aniridia (1). Congenital cataracts
were identified in affected members of each of the two
families.
Aniridia may occur in a familial or sporadic form.
Approximately two-thirds of cases with aniridia have an affected
parent and the inheritance pattern of familial aniridia is known to
be almost exclusively autosomal dominant.
Aniridia can be an isolated disorder or part of a
syndrome, such as WAGR, Rieger syndrome or Peters syndrome
(1,2). WAGR syndrome is a contiguous gene
deletion syndrome that is caused by constitutional deletions in
11p13 leading to loss of one copy of PAX6 and WT (1). Approximately 90% of cases with
aniridia carry mutations in the human PAX6 gene. Two-thirds of all
aniridia cases have intragenic mutations, while chromosomal
re-arrangements have been identified in one-third of cases
(2).
Mutations in the PAX6 gene, a member of the paired
box gene family, have been reported as a cause of aniridia in the
early 1990s (3,4). The human PAX6 gene is located at
chromosome 11p13 and consists of 14 exons encoding a
transcriptional regulator protein. PAX6 has crucial roles in the
morphogenesis of the eye, the central nervous system and pancreatic
islets (1,2). Lack of the homolog PAX6 gene causes
an eyeless phenotype in Drosophila and in rats. Complete
loss of the function of PAX6 can cause anophthalmia, nasal
hypoplasia and prenatal death as a result of central nervous system
defects in vertebrates, while PAX6 haploinsufficiency is associated
with the pathologies including aniridia, foveal hypoplasia and
Peters anomaly (4).
The PAX6 gene contains a serine, proline and
threonine-rich trans-activation domain and two DNA-binding domains;
a paired box (PD) and a homeobox (HD) that bind to each other via a
glycine-rich linker region (5). PD
consists of two different DNA-binding sub-domains: The N-terminal
sub-domain (NTS) and C-terminal sub-domain (CTS). The NTS region
has a primary role in DNA-binding. As NTS and CTS have different
effects on the transactivation of HD, missense mutations in these
regions may lead to distinct pathologies (1).
To date, 404 unique PAX6 gene variants have been
reported (http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6) and
>90% of them are known/likely pathogenic mutations (2). The types of disease-associated
mutations found in the patients are non-sense mutations, splicing
mutations, frame-shifting insertions or deletions, in-frame
insertions or deletions, missense mutations and run-on mutations.
Mutations that introduce a premature termination codon into the
open reading frame are predominantly associated with aniridia;
while non-aniridia phenotypes are typically associated with
missense mutations (6). The
classical pathology of aniridia is found in individuals carrying a
mutation that introduces a premature termination codon, while
missense mutations frequently cause non-aniridia pathologies,
including cataracts, glaucoma, optic nerve hypoplasia, foveal
hypoplasia, Peters anomaly or microphthalmia (2,6).
C-terminal extensions (CTE) or run-on mutations are
caused by a change in the termination codon to an amino acid codon,
therefore leading to uninterrupted translation into the 3′
untranslated region (2,4,6).
Hingorani et al (7)
reported that patients carrying CTE mutations have more severe
pathologies than others. In the present study, a heterozygous
non-stop mutation in exon 14 (c.1290 A>T, X437L) was identified
in the first family. This mutation was first reported by Baum et
al (8) in a female patient
(age, three months) with poor vision, absence of the inner margin
of the iris and nystagmus.
In the present study, a female patient with aniridia
carrying the heterozygous non-stop mutation in exon 14 (c.1290
A>T, X437L) had a pregnancy termination due to cleft palate and
hydrocephaly of the fetus; however, genetic analysis of the fetus
could not be performed. Takagi et al (9) reported on a young female patient with
a heterozygous 310-kb deletion of the downstream flanking region of
PAX6, isolated GH deficiency, cleft palate and bilateral optic disc
cupping. It was indicated that PAX6 is involved in palate
development through Sonic hedgehog.
Of note, the present study identified a novel
heterozygous deletion in exon 8 (c.529_535 TATCCGGdel or c.527_533
GGTATCCdel) causing a frameshift in all affected members of the
second family, except for the male patient with loss of vision due
to glaucoma. To the best of our knowledge, the present study was
the first to report on this frameshift deletion. Further studies
are required to define the phenotype associated this deletion.
In conclusion, the present study identified a
heterozygous deletion and a run-on mutation in PAX6 in two families
with autosomal dominant aniridia. In one of the families, a novel
frameshift heterozygous deletion in exon 8 was identified. It is
recommended that cases with aniridia refer to genetics departments
for clinical assessment and genetic counselling.
References
|
1
|
Kokotas H and Peterson MB: Clinical and
molecular aspects of aniridia. Clin Genet. 77:409–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hingorani M, Hanson I and van Heyningen V:
Aniridia. Eur J Hum Genet. 20:1011–1017. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Axton R, Hanson I, Danes S, Sellar G, van
Heyningen V and Prosser J: The incidence of PAX6 mutation in
patients with simple aniridia: An evaluation of mutation detection
in 12 cases. J Med Genet. 34:279–286. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee HJ and Colby KA: A review of the
clinical and genetics aspects of aniridia. Semin Ophthalmol.
28:306–312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh S, Chao LY, Mishra R, Davies J and
Saunders GF: Missense mutation at the C-terminus of PAX6 negatively
modulates homeodomain function. Hum Mol Genet. 10:911–918. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tzoulaki I, White IM and Hanson IM: PAX6
mutations: Genotype-phenotype correlations. BMC Genet. 6:272005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hingorani M, Williamson KA, Moore AT and
van Heyningen V: Detailed ophthalmologic evaluation of 43
individuals with PAX6 mutations. Invest Ophthalmol Vis Sci.
50:2581–2590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baum L, Pang CP, Fan DS, Poon PM, Leung
YF, Chua JK and Lam DS: Run-on mutation and three novel nonsense
mutations identified in the PAX6 gene in patients with aniridia.
Hum Mutat. 14:272–273. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takagi M, Nagasaki K, Fujiwara I, Ishii T,
Amano N, Asakura Y, Muroya K, Hasegawa Y, Adachi M and Hasegawa T:
Heterozygous defects in PAX6 gene and congenital hypopituitarism.
Eur J Endocrinol. 172:37–45. 2015. View Article : Google Scholar
|