Introduction
LEOPARD syndrome (LS, OMIM 151100), an autosomal
dominant inherited disorder, presents with phenotypes that strongly
overlap with Noonan syndrome (NS, OMIM 163950). These features
include ocular hypertelorism, pulmonary stenosis, growth
retardation, sensorineural deafness, genitourinary abnormalities
and in particular multiple lentigines (1). The majority of the clinical features
of LS appear to be age-dependent (2), similar to that observed in
neurofibromatosis type 1 (NF1, OMIM 162200). The cutaneous
appearance is also similar to Legius syndrome, formerly termed
neurofibromatosis type 1-like syndrome (NFLS, OMIM 611431),
characterized by multiple café-au-lait spots (CALS) and skin-fold
freckling. Thus, NF1, NFLS, NS and LS, which belong to a new class
of genetic disorders called the 'RASopathies', may be clinically
indistinguishable at early stages.
LS harbors certain genetic heterogeneity.
PTPN11 (proportion, ~85%), RAF1 and BRAF (~0%)
are the major pathogenic genes known to cause LS. Two recurrent
mutations, Tyr279Cys and Thr468Met in PTPN11, were found in
~65% of the cases examined in a previous large study (3). Moreover, recently, a novel
heterozygous MAP2K1 mutation (c.305A>G) was reported to
be associated with LS (4).
To date there have been few cases of LS described in
the Chinese population, except for a recent study that demonstrated
cardiovascular complications in a patient with sporadic LS caused
by a Tyr279Cys mutation in the PTPN11 gene (5) and four previous Taiwanese cases with
Thr468Met and Gly464Ala mutations (6). The current study presented another
case of LS with atypical symptoms diagnosed via identification of a
common Thr468Met mutation in the PTPN11 gene and reviewed
the literature for cases of LS associated with this mutation.
Materials and methods
Case history
A 5 year-old male patient was admitted to the
Department of Dermatology, Xinhua Hospital (Shanghai, China) in
December 2013. His parents were concerned about several CALS and
freckles presenting over the trunk and face of the child from
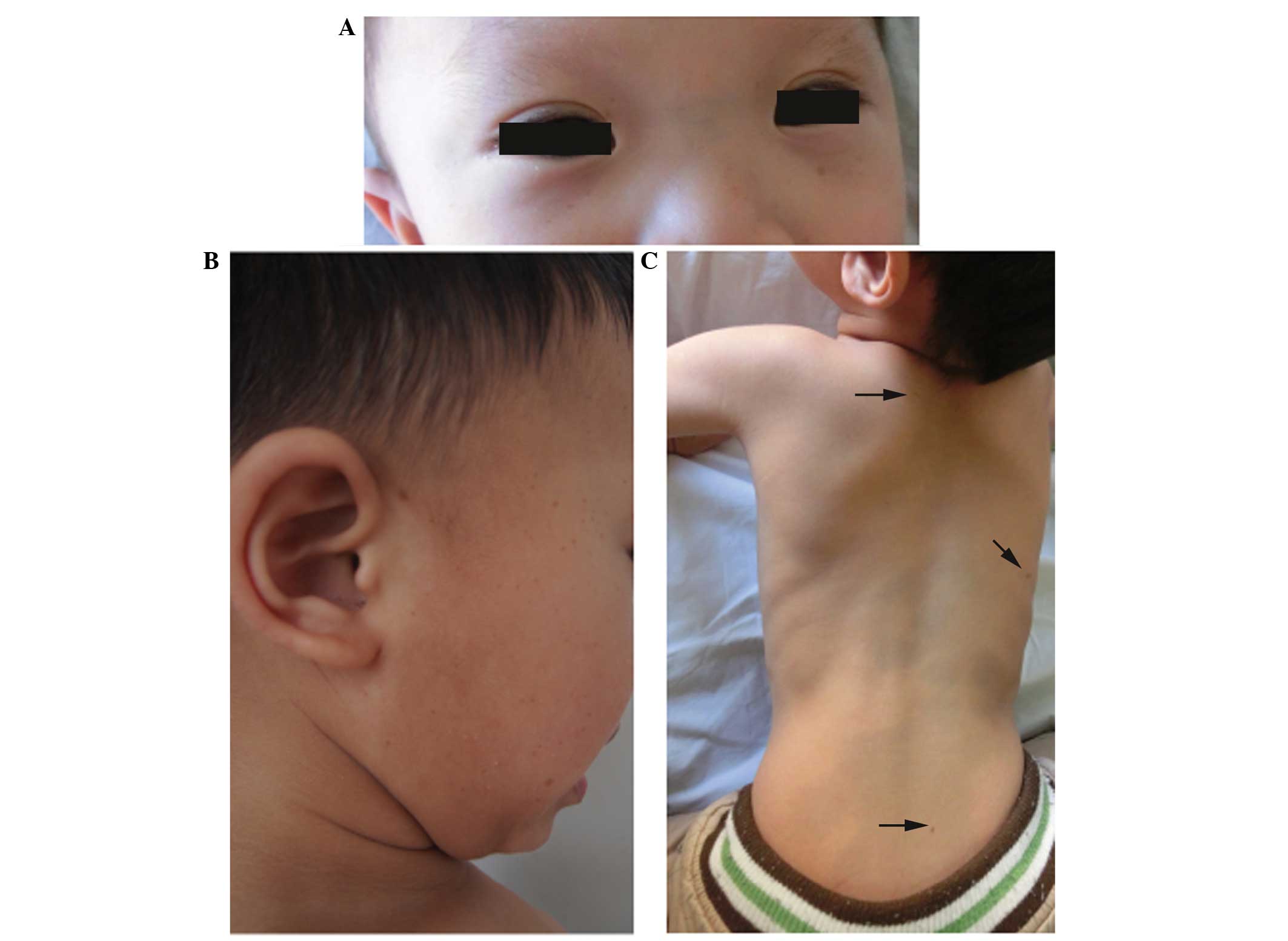
birth. Physical examination revealed 4 CALS with a diameter >0.5
cm and several freckle-like lesions scattered over the body. The
patient also presented with short stature (height, 102 cm) and
ocular hypertelorism (Fig. 1).
Results of neuropsychological examination and hearing tests were
normal. No Lisch nodules were found through slit-lamp examination.
Following examination seven months later, it was observed that the
freckle-like lesions had increased. Moreover, the patient had a
medical history of pulmonary stenosis at 5 months and a follow-up
surgical history of percutaneous balloon pulmonary valvuloplasty.
Therefore, RASopathies, such as NS and LS, were investigated as
potential diagnoses using genetic testing. This study was approved
by the Institutional Review Board of Xinhua Hospital, Shanghai
JiaoTong University School of Medicine and written informed consent
was obtained from the parents of the patient. Peripheral blood was
collected for DNA extraction using a TIANamp Blood DNA kit (Beijing
Tiangen Biochemical Co., Ltd., Tiangen, China).
DNA sequencing
Primers flanking all coding exons and intron-exon
boundaries of NF1, SPRED1 and PTPN11 were
designed by software Primer Premier 5.0 (Premier Biosoft
International, Palo Alto, CA, USA) (Table I). The extracted genomic DNA (gDNA)
samples were amplified by polymerase chain reaction (PCR). Thermal
cycling conditions were as follows: Denaturation at 94°C for 5 min;
31 cycles of denaturation at 94°C for 30 sec, annealing for 30 sec
at a temperature determined by the primers of each fragments and
extension at 72°C for 1 min; followed by extension at 72°C for 1
min and an extension of 4°C for 5 min. The experiment was repeated
10–20 times After PCR, products were purified with AxyPrep DNA Gel
Extraction Kit (Axygen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) according to the manufacturer's protocol. Sanger
sequencing was conducted using an ABI PRISM 3730 automated
sequencer (Applied Biosystems; Thermo Fisher Scientific Inc.) to
identify the mutation in the proband and verify it in his
unaffected family members. When no pathogenic mutations were found
in NF1 by Sanger sequencing, gDNA samples of the patient
were further analyzed using multiplex ligation-dependent probe
amplification (MLPA) kits P122, P081 P082 and P295 (MRC-Holland,
Amsterdam, The Netherlands) as previously described (7,8).
These kits were able to detect deletions/duplications involving
single or multiple exons or the entire NF1 and SPRED1
genes. Sequencing results were analyzed using Geneious (version
5.6.7; Biomatters Ltd., Auckland, New Zealand). Full details on the
use of Geneious are available on the website (https://support.geneious.com/home). Identified
mutations were determined by comparing with the reported cDNA
reference sequences (NM_000267.3 for NF1, NM_152594.2 for
SPRED1 and NM_002834.2 for PTPN11).
 | Table IPrimers of the PTPN11 gene. |
Table I
Primers of the PTPN11 gene.
| Primer name | Forward primer
sequence | Reverse primer
sequence | Product size
(bp) | Annealing (°C) |
|---|
| PTPN11-E01 |
GCCAGCCCGATGTGACCGAG |
CTGGAGGGCGAGGGGACGAG | 245 | 64.0 |
| PTPN11-E02 |
ACTCTGCTCATAATGCGTCT |
ACTTCTATGACCTGCTCCAA | 452 | 55.0 |
| PTPN11-E03 |
TCCTTGGGTTTCTTTCAACA |
AGTCATACACAGACCGTCAT | 392 | 53.0 |
| PTPN11-E04 |
CCCTTGGAGGAATGTGTCTA |
GTGTTTGTCCTCTTCCAGCA | 552 | 57.0 |
| PTPN11-E05 |
TCCCAGGCTGAAGCACAGTTG |
GAAGCTGCAATGGGTACATGGAG | 677 | 62.4 |
| PTPN11-E06 |
CCTCTGTCCGTGCCTTTATG |
ACTCACTGCCAACTCCCTTC | 441 | 59.0 |
| PTPN11-E07 |
TTCTGTGACTCTTTGACACGT |
GATTATTTTGGAAACTGCTTG | 291 | 53.0 |
| PTPN11-E08 + 9 |
TGAATGAACAAAACTTGGAC |
CACCAAGGAATAACATAATCA | 625 | 51.0 |
| PTPN11-E10 |
AACCTAACAGATGCGAAACAG |
GATGAGGGCAGGAACACTAC | 478 | 57.0 |
| PTPN11-E11 |
GCCCAAAAGGAGACGAGTTC |
TGGGTAGGTAAAAGCAAGCC | 397 | 57.0 |
| PTPN11-E12 |
AATGGCTTGGTTTTGAGTCT |
TGTAAACAAGGTCAGGTGGC | 414 | 55.0 |
| PTPN11-E13 |
GAATCCTGACTTCTGCCACT |
CAAGAGAATGAGAATCCGCA | 405 | 57.0 |
| PTPN11-E14 |
TTGGTTCGGTACAGTAAGTT |
AGTCACAGATACACTAACAG | 526 | 53.0 |
| PTPN11-E15 |
GCGTTATTTCACTTCTGCCT |
TTAACCAATAGAGCACTTGCA | 337 | 55.0 |
Moreover, the literature was reviewed for the
reported LS cases with p.Thr468Met mutation to determine the
phenotypes of different LS patients with the same mutation
(6,9–15).
The Pubmed database was searched using the term ̔PTPN11 mutation',
and all studies investigating LS cases were downloaded prior to the
selection of LS cases with a p.Thr468Met mutation.
Results
Sanger sequencing
Sanger sequencing and MLPA analyses for NF1
and SPRED1 did not identify any pathogenic mutations.
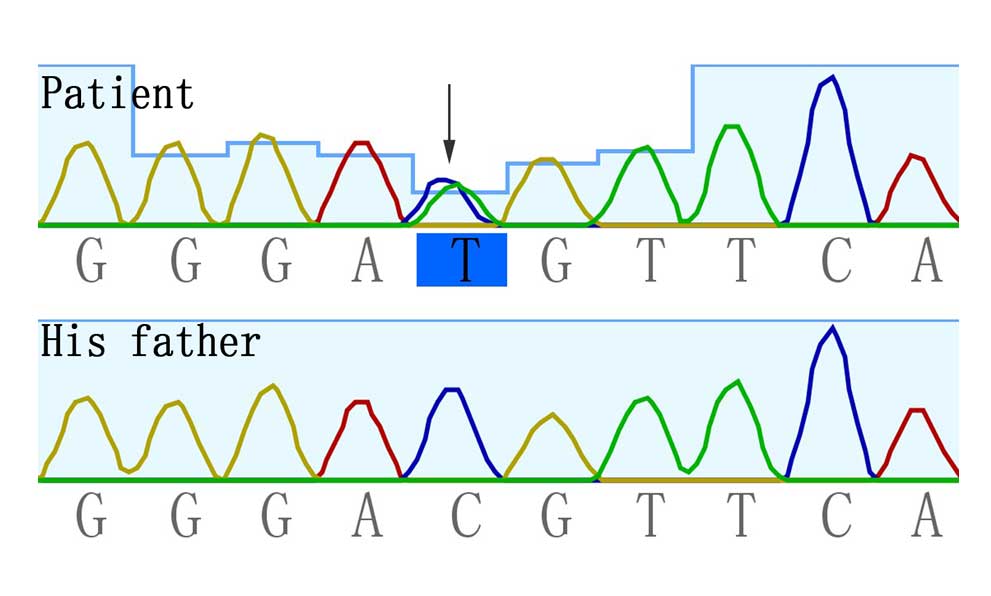
However, a common heterozygous missense mutation c.1403C>T
(p.Thr468Met) in PTPN11 was identified in the proband and
was absent in his unaffected parents (Fig. 2).
Discussion
PTPN11 mutations can cause LS and NS.
Currently, there are twelve missense mutations in PTPN11
that are known to be responsible for LS: Tyr279Cys/Ser,
Ala461Thr/Ser, Gly464Ala, Thr468Met/Pro, Arg498Leu/Trp, Gln506Pro
and Gln510Glu/Pro (3,5,16,17).
Mutation loci are important in the pathogenic mechanisms of NS and
LS. The genetic discrimination between these two syndromes is
primarily dependent on LS-related mutants all located at a PTP
domain (amino acid residues 221-524) (18), exerting a dominant negative effect
on PTPN11 (19). Moreover,
LS-associated loss-of-function mutations in PTPN11 result in
sustained extracellular signal-regulated kinases 1/2 activation by
enhanced specific substrate dephosphorylation and cause
hypertrophic cardio-myopathy by dysregulating mechanistic target of
rapamycin signaling (20–24). Conversely, NS was associated with
excessive PTPN11 activity by gain-of-function changes predominantly
located at the N-SH2 domain (amino acid residues 3-104) (25).
Typical lentigines were flat, black-brown
hyperpigmented macules that appeared all over the body (primarily
on the face, neck and upper part of the trunk) during late
childhood (Table II), and
increased in number and darkened in color with age (2,3).
However, the present case presented with light-brown freckle-like
lesions (probably atypical lentigines) predominantly on the face
since birth, which developed gradually to the current state at 4
years old. In consideration of the presence of several CALS, the
most common and relevant disorders NF1 and NF1-like syndrome (i.e.,
Legius syndrome) were initially suspected. Following exclusion by
molecular genetic testing, although there was no diffuse pattern of
lentigines, the DNA was sequenced for mutations in the second most
common pathogenic gene of RASopathies, PTPN11. Finally, a
mutation in PTPN11 that accounts for NS and LS was
identified. While patients with NF1 usually have >6 CALS
(>0.5 cm in childhood, >1.5 cm in puberty), this study
indicated that children with light brown scattered freckling on the
face, several CALS and congenital cardiac defects such as pulmonary
stenosis or hypertrophic cardiomyopathy should be considered for a
diagnosis of LS or NS. Moreover, PTPN11 should analyzed in
these patients.
| Table IIReview of patients with LEOPARD
syndrome with Thr468Met mutation in literature. |
Table II
Review of patients with LEOPARD
syndrome with Thr468Met mutation in literature.
| Reference | Gender/age
(years) | Population | Major clinical
features
| Ref no. |
|---|
| Cutaneous
features | Dysmorphic face | Skeletal
anomalies | Hearing loss | Tumors | Pulmonary
stenosis | HCM | Other cardio vascular
anomalies |
|---|
| Present study | M/5 | Chinese | CLS AL | + | − | − | − | + | − | − | |
| Lin et al
2009 | M/8 | Chinese | ML | + | + | − | − | − | + | − | 6 |
| M/8 | Chinese | ML | + | + | − | − | − | + | − | |
| F/7 | Chinese | ML | + | + | − | − | + | − | − | |
| Digilio et
al 2002 | F/12.8 y | Italian | CLS ML | + | − | − | − | − | − | − | 18 |
| M/15 | Italian | CLS ML | + | + | − | − | − | − | + | |
| F/15.1 | Italian | CLS ML | + | + | − | − | − | − | + | |
| F/39 | Italian | CLS ML | + | + | − | − | − | − | − | |
| F/8.9 | Italian | CLS ML | + | − | − | − | − | − | + | |
| F/3 | Italian | CLS | + | + | − | − | + | + | − | |
| M/4.9 | Italian | CLS | + | + | − | − | − | + | + | |
| Sarkozy et
al 2004 | ?/4 | Italian | – | + | ? | − | − | − | − | − | 14 |
| ?/34 | Italian | ML | + | ? | − | − | − | + | − | |
| Limongelli et
al 2008 | ?/13 | Italian | CLS ML | + | ? | ? | − | + | − | + | 10 |
| ?/1 | Italian | ML | + | ? | ? | − | − | − | + | |
| ?/9 | Italian | CLS ML | + | ? | ? | − | − | − | + | |
| ?/4 | Italian | ML | + | ? | ? | − | − | − | + | |
| Santoro et
al 2014 | M/8 | Italian | CLS AL | + | + | − | − | − | − | + | 9 |
| M/12 | | CLS ML | | | | | | | | |
| M/50 | Italian | CLS ML | + | + | + | − | − | + | + | |
| Carcavilla et
al 2011 | M/4 | Spanish | CLS AL | + | − | − | − | − | + | + | 11 |
| M/9 | Spanish | CLS AL | + | − | − | − | − | + | − | |
| M/16 | Spanish | CLS AL | + | − | − | − | − | + | + | |
| Carcavilla et
al 2013 | M/49 | Spanish | ML | + | − | − | − | − | + | − | 13 |
| F/3 | Spanish | CLS ML | + | + | − | − | + | − | − | |
| M/11 | Spanish | CLS ML | + | − | − | − | − | + | − | |
| M/4 | Spanish | ML | + | + | − | − | − | + | − | |
| M/1.11 | Spanish | – | + | − | − | − | + | − | − | |
| F/0.8 | Spanish | CLS | + | − | − | − | + | − | − | |
| F/2 | Spanish | – | + | − | − | − | − | − | − | |
| M/14 | Spanish | – | + | + | − | − | − | + | − | |
| Rankin et al
2013 | M/23 | British | CLS ML | + | + | − | + | − | − | − | 15 |
| Keren et al
2004 | Mean: 19 | French | ML (7/7)
CLS (3/7) | (4/7) | (5/7) | (0/6) | ? | (2/7) | (4/7) | (4/7) | 12 |
| Total (38) | | | CLS (23/38) | (35/38) | (19/32) | (1/34) | (1/31) | (9/38) | (17/38) | (16/38) | |
A previous genotype-phenotype study suggested that
hypertrophic cardiomyopathy is characteristic of PTPN11
mutation-positive LS patients (13), and was particularly associated with
mutations in exon 7 and exon 12 (26), and cardiovascular anomalies were
also prevalent in previous cases (Table II). Therefore, a long-term cardiac
evaluation regarding cardiomyopathy should be concerned in our
case. Moreover, overactive RAS signals can result in cell growth
and division, ultimately leading to tumors. Therefore, we still
should pay attention to the tumor risk of 'RASopathies' (27), and the potential tumor spectrum of
LS, such as hematologic malignancies (28), medulloblastoma (15), which have been demonstrated in
numerous LS cases.
In conclusion, the present study presents a young
Chinese patient with LS with CALS and atypical lentigines who was
successfully diagnosed by a series of molecular genetic testing.
Considering that two Taiwanese LS cases harbored an identical
mutation, Thr468Met is likely to be a mutation hotspot in the
Chinese population. Generally, children with NS or NF1 are prone to
developing malignancies, while the prognosis of LS is relatively
favorable only if no severe cardiac defects and adverse cardiac
events, which underline the necessity of definite diagnosis by
genetic methods along with subsequent early prevention of
congenital cardiac defects. A more recent follow up of the patient
showed nearly normal pulmonary artery pressure and no obvious
electrocardiogram abnormalities; however, regular monitoring
concerning potential complications are required.
Acknowledgments
This study was supported by a grant from the Ph.D.
Programs Foundation of Ministry of Education of China (grant no.
20130073120014) and a grant from the Natural Science Foundation of
Shanghai Jiaotong University School of Medicine (grant no.
13XJ10023).
References
|
1
|
Voron DA, Hatfield HH and Kalkhoff RK:
Multiple lentigines syndrome: Case report and review of the
literature. Am J Med. 60:447–456. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Digilio MC, Sarkozy A, de Zorzi A, Pacileo
G, Limongelli G, Mingarelli R, Calabrò R, Marino B and Dallapiccola
B: LEOPARD syndrome: Clinical diagnosis in the first year of life.
Am J Med Genet A. 140:740–746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martínez-Quintana E and Rodríguez-González
F: LEOPARD syndrome: Clinical features and gene mutations. Mol
Syndromol. 3:145–157. 2012.PubMed/NCBI
|
|
4
|
Nishi E, Mizuno S, Nanjo Y, Niihori T,
Fukushima Y, Matsubara Y, Aoki Y and Kosho T: A novel heterozygous
MAP2K1 mutation in a patient with Noonan syndrome with multiple
lentigines. Am J Med Genet A. 167A:407–411. 2015. View Article : Google Scholar
|
|
5
|
Wang Y, Chen C and Wang DW: Leopard
syndrome caused by heterozygous missense mutation of Tyr 279 Cys in
the PTPN11 gene in a sporadic case of Chinese Han. Int J Cardiol.
174:e101–e104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin IS, Wang JN, Chao SC, Wu JM and Lin
SJ: PTPN11 mutations in LEOPARD syndrome: Report of four cases in
Taiwan. J Formos Med Assoc. 108:803–807. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valero MC, Martín Y, Hernández-Imaz E,
Marina Hernández A, Meleán G, Valero AM, Javier Rodríguez-Álvarez
F, Tellería D and Hernández-Chico C: A highly sensitive genetic
protocol to detect NF1 mutations. J Mol Diagn. 13:113–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Tong H, Fu X, Zhang Y, Liu J,
Cheng R, Liang J, Peng J, Sun Z, Liu H, et al: Molecular
Characterization of NF1 and Neurofibromatosis type 1
genotype-phenotype correlations in a Chinese population. Sci Rep.
5:112912015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santoro C, Pacileo G, Limongelli G,
Scianguetta S, Giugliano T, Piluso G, Ragione FD, Cirillo M, Mirone
G and Perrotta S: LEOPARD syndrome: Clinical dilemmas in
differential diagnosis of RASopathies. BMC Med Genet. 15:442014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Limongelli G, Sarkozy A, Pacileo G,
Calabrò P, Digilio MC, Maddaloni V, Gagliardi G, Di Salvo G,
Iacomino M, Marino B, et al: Genotype-phenotype analysis and
natural history of left ventricular hypertrophy in LEOPARD
syndrome. Am J Med Genet A. 146A:620–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carcavilla A, Pinto I, Muñoz-Pacheco R,
Barrio R, Martin-Frías M and Ezquieta B: LEOPARD syndrome (PTPN11,
T468M) in three boys fulfilling neurofibromatosis type 1 clinical
criteria. Eur J Pediatr. 170:1069–1074. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Keren B, Hadchouel A, Saba S, Sznajer Y,
Bonneau D, Leheup B, Boute O, Gaillard D, Lacombe D, Layet V, et
al: PTPN11 mutations in patients with LEOPARD syndrome: A French
multicentric experience. J Med Genet. 41:e1172004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carcavilla A, Santomé JL, Pinto I,
Sánchez-Pozo J, Guillén-Navarro E, Martín-Frías M, Lapunzina P and
Ezquieta B: LEOPARD syndrome: A variant of Noonan syndrome strongly
associated with hypertrophic cardiomyopathy. Rev Esp Cardiol (Engl
Ed). 66:350–356. 2013. View Article : Google Scholar
|
|
14
|
Sarkozy A, Conti E, Digilio MC, Marino B,
Morini E, Pacileo G, Wilson M, Calabrò R, Pizzuti A and
Dallapiccola B: Clinical and molecular analysis of 30 patients with
multiple lentigines LEOPARD syndrome. J Med Genet. 41:e682004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rankin J, Short J, Turnpenny P, Castle B
and Hanemann CO: Medulloblastoma in a patient with the PTPN11
p.Thr468Met mutation. Am J Med Genet A. 161A:2027–2029. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Conti E, Dottorini T, Sarkozy A, Tiller
GE, Esposito G, Pizzuti A and Dallapiccola B: A novel PTPN11
mutation in LEOPARD syndrome. Hum Mutat. 21:6542003. View Article : Google Scholar
|
|
17
|
Osawa R, Akiyama M, Yamanaka Y, Ujiie H,
Nemoto-Hasebe I, Takeda A, Yanagi T and Shimizu H: A novel PTPN11
missense mutation in a patient with LEOPARD syndrome. Br J
Dermatol. 161:1202–1204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Digilio MC, Conti E, Sarkozy A, Mingarelli
R, Dottorini T, Marino B, Pizzuti A and Dallapiccola B: Grouping of
multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11
gene. Am J Hum Genet. 71:389–394. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kontaridis MI, Swanson KD, David FS,
Barford D and Neel BG: PTPN11 (Shp2) mutations in LEOPARD syndrome
have dominant negative, not activating, effects. J Biol Chem.
281:6785–6792. 2006. View Article : Google Scholar
|
|
20
|
Schramm C, Fine DM, Edwards MA, Reeb AN
and Krenz M: The PTPN11 loss-of-function mutation Q510E-Shp2 causes
hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am J
Physiol Heart Circ Physiol. 302:H231–H243. 2012. View Article : Google Scholar
|
|
21
|
Marin TM, Keith K, Davies B, Conner DA,
Guha P, Kalaitzidis D, Wu X, Lauriol J, Wang B, Bauer M, et al:
Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of
LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest.
121:1026–1043. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Motegi S, Yokoyama Y, Ogino S, Yamada K,
Uchiyama A, Perera B, Takeuchi Y, Ohnishi H and Ishikawa O:
Pathogenesis of multiple lentigines in LEOPARD syndrome with PTPN11
gene mutation. Acta Derm Venereol. 95:978–984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu ZH, Zhang RY, Walls CD, Chen L, Zhang
S, Wu L, Liu S and Zhang ZY: Molecular basis of gain-of-function
LEOPARD syndrome-associated SHP2 mutations. Biochemistry.
53:4136–4151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu ZH, Xu J, Walls CD, Chen L, Zhang S,
Zhang R, Wu L, Wang L, Liu S and Zhang ZY: Structural and
mechanistic insights into LEOPARD syndrome-associated SHP2
mutations. J Biol Chem. 288:10472–10482. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tartaglia M, Mehler EL, Goldberg R,
Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A,
Jeffery S, et al: Mutations in PTPN11, encoding the protein
tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet.
29:465–468. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sarkozy A, Digilio MC and Dallapiccola B:
Leopard syndrome. Orphanet J Rare Dis. 3:132008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kratz CP, Rapisuwon S, Reed H, Hasle H and
Rosenberg PS: Cancer in Noonan, Costello, cardiofaciocutaneous and
LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 157C:83–89.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tartaglia M, Niemeyer CM, Fragale A, Song
X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD and Gelb BD:
Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia,
myelodysplastic syndromes and acute myeloid leukemia. Nat Genet.
34:148–150. 2003. View
Article : Google Scholar
|