Introduction
Multiple myeloma (MM) is the second most common
hematological malignancy characterized by the proliferation of
clonal plasma cells in the bone marrow (1). The first-line treatment strategies
for MM are combination regimens, including proteasome inhibitors
and immunomodulatory agents. Bortezomib (BTZ) is the first
proteasome inhibitor to be approved by the Food and Drug
Administration (USA) for clinical use in patients with MM. The
degradation of damaged or misfolded proteins is crucial in myeloma
plasma cells, and BTZ treatment leads to a wide number of
intracellular pleiotropic effects, including cell cycle arrest,
induction of apoptosis and deregulation of nuclear factor-κB
(NF-κB) activity by preventing the normal breakdown of proteins in
MM (2,3). In addition, the incorporation of BTZ
and novel immunomodulatory therapeutic agents, particularly
thalidomide and lenalidomide, has markedly improved the rate of
complete remission, progression-free survival, and overall survival
in patients with MM (4,5). However, despite available therapeutic
strategies, MM remains incurable (6).
MM has a complex pathogenesis, which involves
chromosomal instability (7),
microRNA expression (8) and DNA
methylation (9). DNA methylation
is an epigenetic event that occurs at the cytosine residues in CpG
dinucleotides and is catalyzed by DNA methyltransferase enzymes
(DNMTs). DNA methylation is associated with gene silencing, which
is important for the occurrence and development of MM (10). Therefore, it is possible that DNA
methyltransferase inhibitors may be of benefit in the current
clinical therapy of MM. Decitabine (5-aza-2′-deoxycytidin; DAC) is
an epigenetic therapeutic agent that inhibits DNA methylation,
which has been approved for the treatment of myelodysplastic
syndrome and older patients with acute myeloid leukemia (AML)
(11,12). In addition to its use in
hematological malignancy, DAC also exhibits antineoplastic effects
against solid tumors (13–16). The molecular mechanism underlying
its antitumor activity is that DAC induces DNMT inhibition, which
contributes to DNA hypomethylation, gene activation, cell
differentiation and apoptosis (17,18).
Therapeutic agents used in combination often have
synergistic anti-tumor effects and reduced side effects as lower
concentrations of each individual agent are used. The present study
investigated whether DAC may synergize with the frequently used
anti-myeloma agent, BTZ. The results of the current study indicated
that the anti-MM activity of the two agents was mutually
reinforced, which resulted in an enhanced effect of proliferation
inhibition, increased apoptosis and G0-G1
cell cycle arrest in the RPMI 8226 myeloma cell line. These events
were associated with poly(ADP-ribose) polymerase 1 (PARP-1)
cleavage, caspase-3 and-9 activation and DNA
(cytosine-5-)-methyltransferase 1 (DNMT1) downregulation.
Materials and methods
Cell culture, proliferation inhibition
assay and cell morphology analysis
RPMI 8226 cells (American Type Culture Collection,
Manassas, VA, USA) were routinely cultured in RPMI-1640 media
(Sigma-Aldrich, St. Louis, MO, USA), supplemented with 10% (v/v)
heat-inactivated fetal bovine serum (GE Healthcare Life Sciences,
Logan, UT, USA) in a humidified atmosphere with 5% CO2
at 37°C. Inhibition of cell proliferation was estimated using a
Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). Half inhibitory concentration (IC50)
of DAC in RPMI-8226 cells for 48 h was further calculated by a
modified Kou-type method: lgIC50 = Xm-I
[∑P-(3-Pm-Pn)/4], in which Xm, lg maximum dose; I, lg (maximum
dose/adjacent dose); ∑P, sum of positive response rate; Pm, the
largest positive response rate; Pn, the smallest positive response
rate. Cells were observed and images were captured directly in
flasks using a phase contrast microscope.
Chemicals and antibodies
BTZ was purchased from Millennium Pharmaceuticals,
Inc. (Cambridge, MA, USA) and DAC was obtained from Sigma-Aldrich.
These chemicals were dissolved in dimethyl sulfoxide (DMSO) and
stored at −20°C. The primary antibodies used were as follows:
Rabbit polyclonal anti-DNMT1 (cat. no. M0231L; New England Biolabs,
Inc., Ipswich, MA, USA; 1:1,000); mouse monoclonal anti-PARP-1
(cat. no. sc-365315; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA; 1:1,000); rabbit polyclonal anti-caspase-9 (cat. no. 9502;
1:1,000), rabbit polyclonal anti-caspase-3 (cat. no. 9661; 1:1,000)
and mouse monoclonal anti-β-actin (cat. no. 3700; 1:1,000) (Cell
Signaling Technology, Danvers, MA, USA). The secondary antibodies
used were as follows: Horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG (cat. no. 7074; 1:1,000) and HRP-conjugated horse
anti-mouse IgG (cat. no. 7076; 1:1,000) (Cell Signaling
Technology).
Cell apoptosis assays
Cell apoptosis was determined using a fluorescein
isothiocyanate (FITC) Annexin V Apoptosis Detection kit (BD
Biosciences, Franklin Lakes, NJ, USA) following the manufacturer's
protocol. Following treatment with DAC and BTZ, RPMI 8226 MM cells
were harvested (2×105), washed twice with ice-cold
phosphate-buffered saline (PBS) and resuspended with binding
buffer. Next, 5 μl Annexin V-FITC was added to the suspended
cells, mixed gently and incubated for 5 min in the dark. Following
incubation, 5 μl propidium iodide (PI; Sigma-Aldrich) was
added, gently mixed and incubated for a further 3 min. Cells were
analyzed using flow cytometry with CellQuest software version 5.1
(BD Biosciences).
Cell cycle analysis
Cells (2×105) were harvested, washed
twice with ice-cold PBS and fixed with 75% cold ethanol at 4°C
overnight. Then cells were incubated with RNase (100 mg/ml) for 30
min at 37°C and stained with PI (250 mg/ml) for another 30 min on
ice and in the dark. The resulting cell suspension was subjected to
flow cytometry. The fluorescent intensity of stained DNA was
quantified using CellQuest software 5.1 (BD Biosciences).
Western blotting
Cells were harvested, washed with PBS and lysed with
lysis buffer (62.5 mM Tris-HCl, pH 6.8; 100 mM DTT, 2% SDS and 10%
glycerol). Following incubation at 95°C for 10 min, lysates were
cooled on ice and centrifuged at 20,000 × g for 10 min at 4°C. The
Bradford method was used to quantify protein. Equal quantities (10
μg) of protein extract were resolved by 6–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
nitrocellulose membranes. Following blocking with 5% non-fat milk
in PBS, the membranes were probed with specific primary antibodies
overnight at 4°C. The membranes were washing in Tris Buffered
Saline with Tween-20, and subsequently incubated with the
HRP-conjugated secondary antibodies for 1 h at room temperature.
The signals were detected by the chemiluminescence Phototope-HRP
kit (EMD Millipore, Billerica, MA, USA), and imaged using a
commercial imaging system (Fuji LAS400; Fuji Medical Systems,
Stamford, CT, USA). The western blots were stripped using Restore
Western Blot Stripping Buffer (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and re-probed with anti-β-actin antibody as a
loading control. All experiments were repeated three times and
similar results were obtained.
RNA extraction and reverse
transcription-quantitative poly- merase chain reaction
(RT-qPCR)
Total RNA was isolated using the TRIzol kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. The RNA was treated with DNase I (Promega
Corporation, Madison, WI, USA). Complementary DNA was synthesized
according to the manufacturer's protocol of the High-Capacity cDNA
Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The analysis of DNMT1 and β-actin mRNA
expression levels was performed by qPCR using SYBR Green PCR Master
mix reagents (Applied Biosystems; Thermo Fisher Scientific, Inc.)
on an Applied Biosystems 7300 Real Time PCR system (Thermo Fisher
Scientific, Inc.). The specific primers used for detecting DNMT1
were as follows: Forward, 5′-GATTTGTCCTTGGAGAACGGTG-3′ and reverse,
5′-TGAGATGTGATGGTGGTTTGCC-3′. The primers were synthesized by
Sangon Biotech Co., Ltd. (Shanghai, China). Thermal cycler
conditions were as follows: An initial incubation step of 95°C for
5 min, followed by 50 cycles of 95°C for 15 sec, 60°C for 15 sec
and 72°C for 45 sec, with a final 5 min extension step at 94°C to
generate a melting curve. All experiments were performed in
triplicate. Data were normalized to the housekeeping gene β-actin,
and the relative abundance of transcripts was calculated using the
comparative 2−∆∆Cq method (19).
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experiments. All statistical analyzes were
performed using SPSS version 20 software (IBM SPSS, Armonk, NY,
USA). One-way analysis of variance and least significant difference
tests were used to analyze the differences between groups.
P<0.05 was considered to indicate a statistically significant
difference. Data are presented as the mean ± standard
deviation.
Results
Combination treatment with BTZ and DAC
inhibits cell proliferation
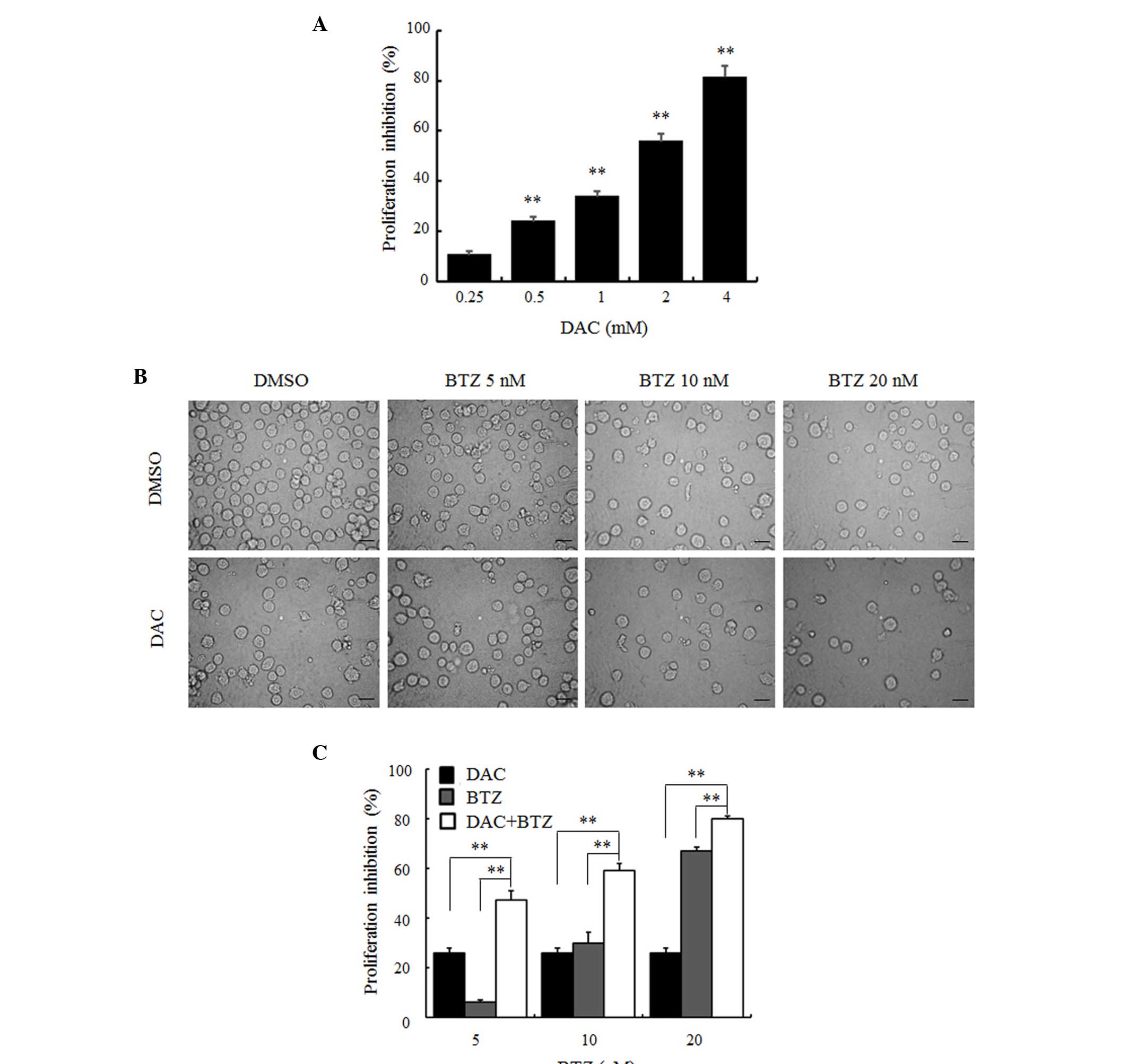
A CCK-8 assay was performed to evaluate the effects
of DAC on the proliferation of RPMI-8226 cells. As presented in
Fig. 1A, exposure to increasing
concentrations of DAC for 48 h inhibited cell proliferation in a
dose-dependent manner. The inhibition rate was significantly
increased to 10.69±0.41%, 26.04±0.91%, 33.80±2.09%, 55.79±1.04% and
81.37±2.36% with increasing doses of 0.25, 0.5, 1, 2 and 4 mM,
respectively (P<0.01; Fig. 1A).
The half maximal inhibitory concentration (IC50) of DAC
for 48 h was 1.35 mM. To assess whether DAC enhanced the anti-MM
effect of BTZ, the low dose (0.5 mM) of DAC and different
concentrations (5, 10, and 20 nM) of BTZ were selected to treat
myeloma cells. Inverted phase contrast microscopic examination
demonstrated that, compared with the DMSO control group, cell
density of the BTZ or DAC single agent groups was decreased and
cellular debris was evident. This was also observed in the combined
group (Fig. 1B). The CCK-8 assay
was used to confirm the qualitative observations (Fig. 1C). Following treatment with 5, 10,
and 20 nM BTZ for 48 h, the inhibitory rate of cell growth was
6.21±0.75%, 29.80±4.48% and 67.15±1.51%, respectively. When 0.5 mM
DAC was used in combination with the aforementioned concentrations
of BTZ, the inhibitory rate was increased to 47.24±3.79%,
59.06±3.01% and 79.92±1.21%. These results demonstrated that
combined treatment with DAC and BTZ led to a significantly higher
inhibition of cell growth compared with each agent alone
(P<0.01; Fig. 1C).
DAC promotes BTZ-induced apoptosis of
RPMI 8226 MM cells
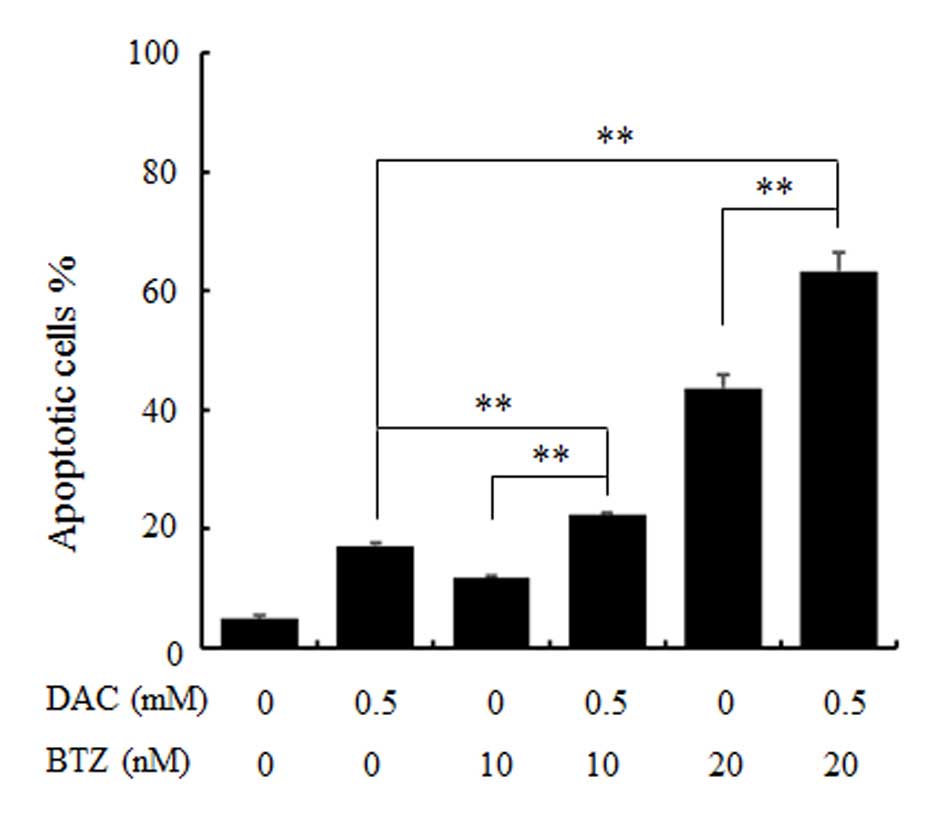
To analyze the action between DAC and BTZ, the
apoptotic rate of RPMI 8226 cells treated with these two
therapeutic agents was examined by flow cytometry analysis using
Annexin V and PI staining. As demonstrated in Fig. 2, the rates of Annexin V-positive
cells in the BTZ groups (10 and 20 nM) were increased gradually
with increasing dose, 11.82±0.31% and 43.69±4.21%, respectively,
compared with the control group, 4.86±0.71% (P<0.01). In
addition, in the combination treatment group (0.5 mM of DAC with
each concentration of BTZ), a significant increase in the
percentage of Annexin V positive cells was observed compared with
the single treatment group (P<0.01; 22.38±0.38% and 63.33±3.08%,
respectively). The results of apoptosis assay indicated that DAC
may enhance the apoptotic rate of RPMI 8226 MM cells induced by
BTZ.
DAC enhances BTZ-induced cell cycle
arrest of RPMI 8226 MM cells
The effect of DAC and BTZ on cell cycle progression
was further examined. As presented in Table I, the percentages of cells arrested
at G0-G1 phase were 40.93±1.49, 40.96±0.88
and 49.64±0.92%, following treatment with 0.5 mM DAC alone, and 10
and 20 nM BTZ alone, respectively. These results were significantly
increased compared with the control (P<0.05; 29.32±2.32%). This
indicates that DAC and BTZ single exposure induced high numbers of
G0-G1 phase cells, with fewer S and
G2/M phase cells. The combination treatment of DAC and
10 and 20 nM BTZ resulted in a significantly increased number of
cells in G0-G1 phase compared with the single
treatment groups (P<0.01; 50.61±1.18% and P<0.01;
59.07±2.25%, respectively). Overall, these findings suggested that
DAC promoted the BTZ-induced cell cycle arrest of MM cells.
 | Table ICell cycle distribution of the
different treatment groups was determined by flow cytometry
analysis of DNA content. |
Table I
Cell cycle distribution of the
different treatment groups was determined by flow cytometry
analysis of DNA content.
| Group |
G0/G1 (%) | S (%) | G2/M
(%) |
|---|
| DMSO | 29.32±2.32 | 63.24±1.40 | 7.45±0.94 |
| DAC | 40.93±1.49a | 53.60±0.81a | 5.47±1.31 |
| BTZ 10 nM | 40.96±0.88a | 54.20±1.57a | 4.82±0.94 |
| DAC + BTZ 10
nM | 50.61±1.18b | 45.03±1.24b | 4.36±0.10 |
| BTZ 20 nM | 49.64±0.92a | 45.31±1.41a | 5.05±1.19 |
| DAC + BTZ 20
nM | 59.07±2.25b | 38.01±2.51b | 2.92±1.43b |
Combined treatment with BTZ and DAC leads
to increased PARP-1 cleavage, caspase-3 and -9 activation
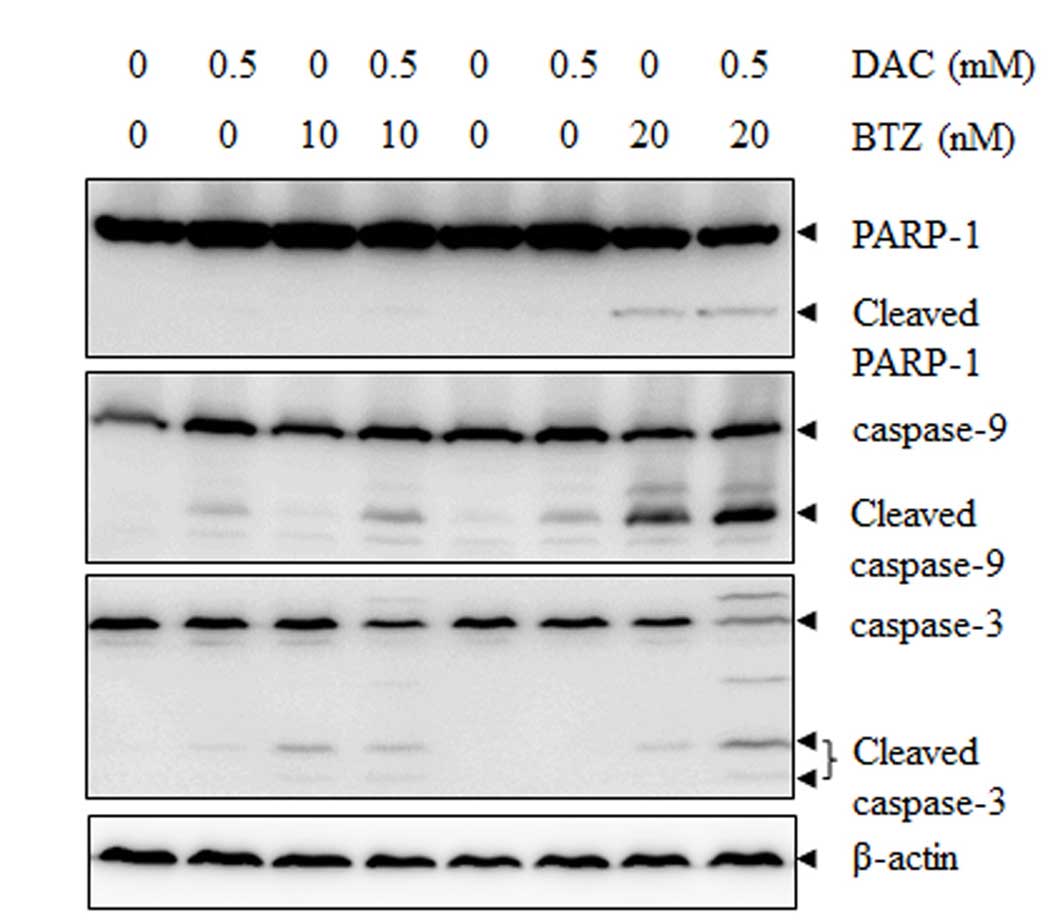
In order to determine the underlying mechanism of
the increasing apoptotic rate induced by BTZ and DAC combination
treatment, western blot analysis was performed. It was demonstrated
that treatment with BTZ or DAC alone resulted in minimal effects,
whereas combined treatment for up to 48 h resulted in an increase
in caspase-3 and caspase-9 activation and enhanced PARP-1 cleavage
(Fig. 3).
DNMT1 mRNA and protein expression levels
are downregulated by combined treatment with DAC and BTZ
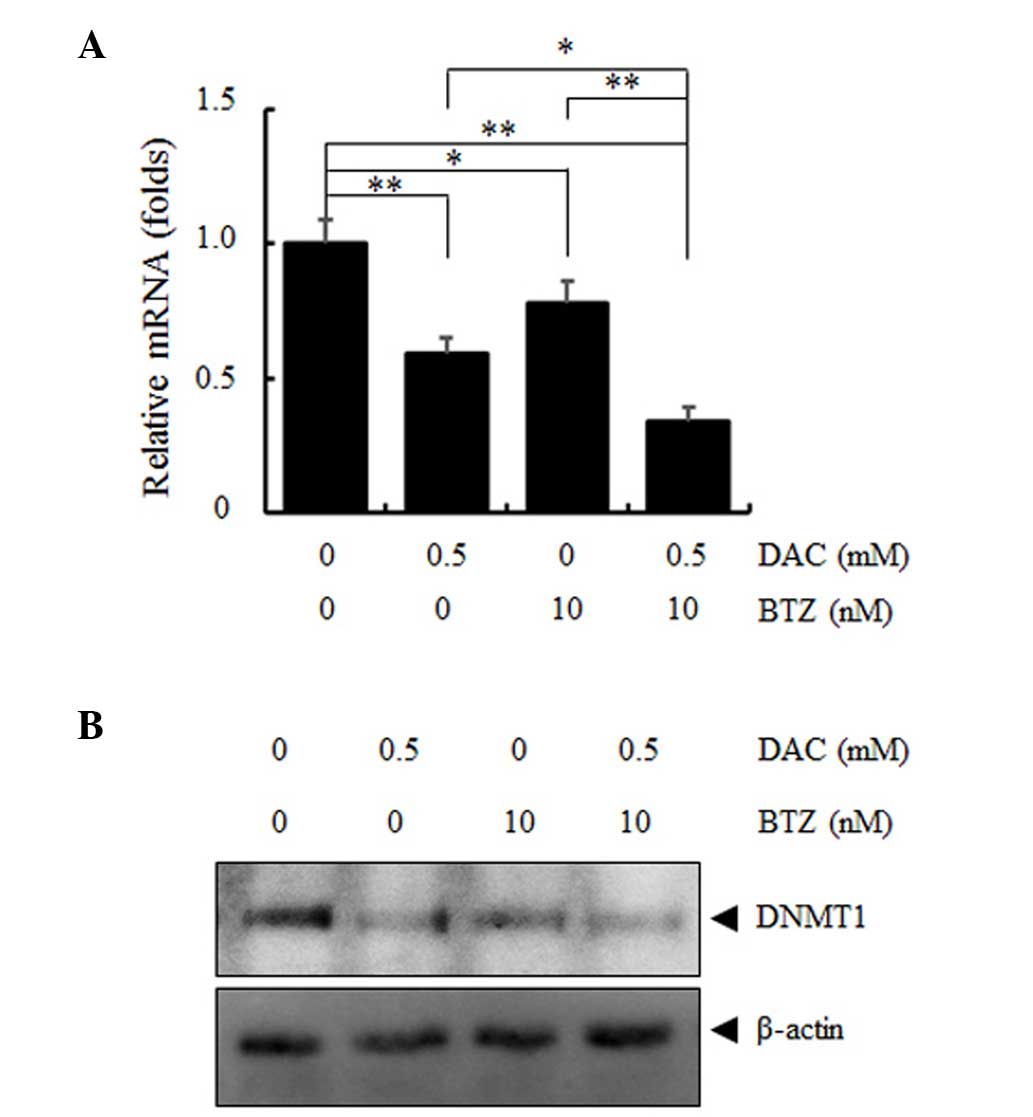
The clinical efficacy of DAC is due to its
demethylating epigenetic action; therefore, combination treatment
with DAC and BTZ may synergistically downregulate the mRNA and
protein expression levels of DNMT1. RPMI 8226 MM cells were
incubated with DAC and BTZ for 48 h and DNMT1 mRNA levels were
determined using RT-qPCR (Fig.
4A). The single treatment with either BTZ or DAC alone was able
to reduce DNMT1 mRNA expression to a certain extent; however, the
combined treatment with the two agents resulted in a significant
decrease in DNMT1 mRNA expression (P<0.01; Fig. 4A). This effect was also confirmed
by the downregulation of DNMT1 protein expression levels analyzed
by western blotting (Fig. 4B), the
results were consistent with the findings in previous studies in
AML and MCL cells, which reported BTZ as a potent inhibitor of DNA
methylation (20,21).
Discussion
The present study investigated the combined effect
of DAC and BTZ on the RPMI 8226 MM cell line and the underlying
mechanism of action. It was determined that DAC and BTZ induce
proliferation inhibition, apoptosis and G0-G1
arrest in RPMI 8226 cells. Notably, the combination treatment
markedly enhanced these effects. DAC and BTZ used in combination
also enhanced PARP-1 cleavage, caspase-3 and -9 activation and
downregulated the mRNA and protein expression levels of DNMT1.
MM remains an almost incurable disease, it is
characterized by a high rate of relapse and drug resistance,
despite the recent introduction of therapies, including proteasome
inhibitors and immunomodulatory agents. Therefore, novel agents
with improved efficacy and lower toxicities are required to develop
the therapeutic strategies used in MM treatment. Epigenetic DNA
methylation is involved in the pathogenesis and progression of MM
(9), which has stimulated
considerable interest in research on combinations of epigenetic
agents with traditional chemotherapy. DAC and BTZ have long been
approved for use in clinical treatment of specific diseases for,
thus, their safety, efficacy, therapeutic window and side effects
are well described. A previous study performed a phase 1 trial of
BTZ and DAC in patients with poor-risk AML and has revealed their
feasibility and preliminary clinical activity for use in AML
treatment (22). However, little
is known regarding their combined effects on MM. The present study
determined, that DAC markedly enhanced the anti-MM effect of the
frequently used proteasome inhibitor, BTZ. Therefore, a novel
combined therapeutic strategy using BTZ and DAC may potentially
enhance the effectiveness of BTZ in MM. However, this approach
requires further investigation.
BTZ is a highly selective inhibitor of 26S
proteasome by reversibly binding to its active site. Proteasomes
are protein complexes present in all eukaryotic cells located in
the nucleus and the cytoplasm. The predominant function of a
proteasome is to regulate the concentration of particular proteins
and degrade unnecessary or damaged ones. This is important for the
modulation of differentiation, apoptosis and cell cycle progression
(23–26). Disorders in the proteasomal
degradation system are involved in the pathogenesis of malignancy;
therefore, protein degradation pathways are investigated as targets
for cancer therapy, as confirmed by the successful clinical use of
BTZ (27,28). However, BTZ is not a perfect
therapeutic agent. Previous studies indicated that BTZ has
relatively narrow therapeutic window and may result in side
effects, including cutaneous adverse reactions (29), neurotoxicity (30), and thrombocytopenia (31) during the treatment of MM.
Therefore, the combination of BTZ with other novel therapeutic
agents may alleviate its adverse effects by reducing the dosage
used.
DNA methylation is an epigenetic regulation
mechanism, which occurs at CpG islands. CpG islands frequently
contain gene promoters or exons in normal mammalian cells. They are
usually unmethylated, which allows transcriptional activity to
control cell differentiation and phenotype (32). Methylation of CpG islands occurs at
the carbon 5 position of the cytosine ring within CpG dinucleotides
by a transfer of the methyl group from
S-adenosyl-l-methionine (33). Various types of cancer often
exhibit aberrant CpG methylation as this confers a growth advantage
to neoplastic cells, which is associated with condensed chromatin,
gene silencing and microRNA regulation. In MM, DNA methylation is
important for the pathogenesis, progression and prognosis of the
disease. Previous studies have suggested that numerous genes are
hypermethylated in MM, including glutathione peroxidase 3, retinol
binding protein 1, secreted protein acidic and cysteine rich,
transforming growth factor β induced, cyclin-dependent kinase
inhibitor 2A, suppressor of cytokine signaling 1, fragile histidine
triad 1, death associated protein kinase 1 and transforming growth
factor β receptor 2 (34–38). Therefore, targeting DNA methylation
may permit optimized therapy for MM, which warrants further
investigation.
DNA methylation is catalyzed by DNMT1, 3a and 3b.
DNMT3a and 3b are important for de novo methylation, and
DNMT1 is an important housekeeping gene for the maintenance of the
established methylation pattern (39). Previous studies have determined
that inhibition of DNMT1 by DAC induced DNA hypomethylation and
reactivated silenced tumor-suppressor genes in leukemia cells and
epithelial tumor cells (40,41).
The current study demonstrated that DAC and BTZ induced a
significant decrease in DNMT1 expression levels, which may
contribute to their antitumor activity. Information on the DNMT1
downregulation effect of BTZ in MM is scarce. The present study
defined DNMT1 as a novel target, which mediated the BTZ antitumor
effect. A significant reduction in the expression levels of DNMT1
was observed in MM cells following exposure to BTZ, this is
consistent with the results reported in mantle cell lymphoma (MCL)
and AML cells (20,21). In MCL models, BTZ has been reported
as a potent inhibitor of DNA methylation, which decreased DNMT1
expression and induced global DNA hypomethylation (21). In AML models, BTZ was able to
decrease Sp1 transcription factor (Sp1) protein levels and
interfere with Sp1/NF-κB complex, which leads to DNMT1
downregulation and transcription of methylation-silenced genes
(20). Overall, the present study
supported BTZ as a novel and effective non-azanucleoside inhibitor
of DNA methylation. In addition, as BTZ induced DNA hypomethylation
with mechanisms different from those currently established
hypomethylating azanucleosides, the combination of BTZ with
azanucleosides, including DAC, may achieve synergistic anti-tumor
activity with lower toxicity. However, the exact underlying
mechanisms by which BTZ downregulates DNMT1 expression levels in MM
require further investigation.
In conclusion, the results of the present study
demonstrated that the epigenetic agent DAC promoted BTZ treatment
in RPMI 8226 multiple myeloma cells. The combination of DAC and BTZ
resulted in enhanced inhibition of proliferation, increased
apoptosis and G0-G1 cell cycle arrest. Overall, these data
supported BTZ as a novel and effective non-azanucleoside inhibitor
of DNA methylation. In addition, as BTZ induced DNA hypomethylation
with mechanisms different from those of currently established
hypomethylating azanucleosides, the combination of BTZ with
azanucleosides, including DAC, may achieve synergistic antitumor
activity with lower toxicity. However, the exact underlying
mechanisms by which BTZ downregulates DNMT1 expression levels in MM
require further investigation.
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
BTZ
|
bortezomib
|
|
DAC
|
decitabine
|
|
DNMTs
|
DNA methyltransferase enzymes
|
|
HRP
|
horseradish peroxidase
|
|
MDS
|
myelodysplastic syndrome
|
|
MM
|
multiple myeloma
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PBS
|
phosphate-buffered saline
|
Acknowledgments
The current study was supported in part by grants
from the Funding of Science and Technology Bureau of Changzhou
(grant no. CJ20130035) and Health and Research Funding of Changzhou
(grant no. KY201442).
References
|
1
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chauhan D, Hideshima T, Mitsiades C,
Richardson P and Anderson KC: Proteasome inhibitor therapy in
multiple myeloma. Mol Cancer Ther. 4:686–692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hideshima T, Mitsiades C, Akiyama M,
Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi
NC, Mitsiades N and Anderson KC: Molecular mechanisms mediating
anti-myeloma activity of proteasome inhibitor PS-341. Blood.
101:1530–1534. 2003. View Article : Google Scholar
|
|
4
|
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy
MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust
JA, et al: Improved survival in multiple myeloma and the impact of
novel therapies. Blood. 111:2516–2520. 2008. View Article : Google Scholar
|
|
5
|
Moreau P, Attal M and Facon T: Frontline
therapy of multiple myeloma. Blood. 125:3076–3084. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ludwig H, Bolejack V, Crowley J, Bladé J,
Miguel JS, Kyle RA, Rajkumar SV, Shimizu K, Turesson I, Westin J,
et al: Survival and years of life lost in different age cohorts of
patients with multiple myeloma. J Clin Oncol. 28:1599–1605. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Decaux O, Lodé L, Magrangeas F, Charbonnel
C, Gouraud W, Jézéquel P, Attal M, Harousseau JL, Moreau P,
Bataille R, et al: Prediction of survival in multiple myeloma based
on gene expression profiles reveals cell cycle and chromosomal
instability signatures in high-risk patients and hyperdiploid
signatures in low-risk patients: A study of the Intergroupe
Francophone du Myélome. J Clin Oncol. 26:4798–4805. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gutiérrez NC, Sarasquete ME,
Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV,
Fermiñán E, Martín-Jiménez P, Chillón C, Risueño A, et al:
Deregulation of microRNA expression in the different genetic
subtypes of multiple myeloma and correlation with gene expression
profiling. Leukemia. 24:629–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walker BA, Wardell CP, Chiecchio L, Smith
EM, Boyd KD, Neri A, Davies FE, Ross FM and Morgan GJ: Aberrant
global methylation patterns affect the molecular pathogenesis and
prognosis of multiple myeloma. Blood. 117:553–562. 2011. View Article : Google Scholar
|
|
10
|
San-Miguel J, García-Sanz R and
López-Pérez R: Analysis of methylation pattern in multiple myeloma.
Acta Haematol. 114(Suppl 1): 23–26. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Curran MP: Decitabine: A review of its use
in older patients with acute myeloid leukaemia. Drugs Aging.
30:447–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jabbour E, Issa JP, Garcia-Manero G and
Kantarjian H: Evolution of decitabine development: Accomplishments,
ongoing investigations, and future strategies. Cancer.
112:2341–2351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nie J, Liu L, Li X and Han W: Decitabine,
a new star in epigenetic therapy: The clinical application and
biological mechanism in solid tumors. Cancer Lett. 354:12–20. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Primeau M, Gagnon J and Momparler RL:
Synergistic antineoplastic action of DNA methylation inhibitor
5-AZA-2′-deoxycytidine and histone deacetylase inhibitor
depsipeptide on human breast carcinoma cells. Int J Cancer.
103:177–184. 2003. View Article : Google Scholar
|
|
15
|
Numoto K, Yoshida A, Sugihara S, Kunisada
T, Morimoto Y, Yoneda Y, Fujita Y, Nishida K, Ouchida M and Ozaki
T: Frequent methylation of RASSF1A in synovial sarcoma and the
anti-tumor effects of 5-aza-2′-deoxycytidine against synovial
sarcoma cell lines. J Cancer Res Clin Oncol. 136:17–25. 2010.
View Article : Google Scholar
|
|
16
|
Wang L, Amoozgar Z, Huang J, Saleh MH,
Xing D, Orsulic S and Goldberg MS: Decitabine enhances lymphocyte
migration and function and synergizes with CTLA-4 blockade in a
murine ovarian cancer model. Cancer Immunol Res. 3:1030–1041. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Turcan S, Fabius AW, Borodovsky A, Pedraza
A, Brennan C, Huse J, Viale A, Riggins GJ and Chan TA: Efficient
induction of differentiation and growth inhibition in IDH1 mutant
glioma cells by the DNMT inhibitor Decitabine. Oncotarget.
4:1729–1736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding L, Qiu L, Zhang J and Guo B:
Camptothecin-induced cell proliferation inhibition and apoptosis
enhanced by DNA methyltransferase inhibitor,
5-aza-2′-deoxycytidine. Biol Pharm Bull. 32:1105–1108. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann
E, Huynh L, Vukosavljevic T, Takeki M, Klisovic RB, et al:
Bortezomib induces DNA hypomethylation and silenced gene
transcription by interfering with Sp1/NF-kappaB-dependent DNA
meth-yltransferase activity in acute myeloid leukemia. Blood.
111:2364–2373. 2008. View Article : Google Scholar
|
|
21
|
Leshchenko VV, Kuo PY, Jiang Z, Weniger
MA, Overbey J, Dunleavy K, Wilson WH, Wiestner A and Parekh S:
Harnessing Noxa demethylation to overcome Bortezomib resistance in
mantle cell lymphoma. Oncotarget. 6:27332–27342. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blum W, Schwind S, Tarighat SS, Geyer S,
Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R,
et al: Clinical and pharmacodynamic activity of bortezomib and
decitabine in acute myeloid leukemia. Blood. 119:6025–6031. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adams J: The proteasome: A suitable
antineoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cecarini V, Bonfili L, Cuccioloni M,
Mozzicafreddo M, Rossi G, Buizza L, Uberti D, Angeletti M and
Eleuteri AM: Crosstalk between the ubiquitin-proteasome system and
autophagy in a human cellular model of Alzheimer's disease. Biochim
Biophys Acta. 1822:1741–1751. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bassermann F, Eichner R and Pagano M: The
ubiquitin proteasome system-implications for cell cycle control and
the targeted treatment of cancer. Biochim Biophys Acta.
1843:150–162. 2014. View Article : Google Scholar
|
|
26
|
Neutzner A, Li S, Xu S and Karbowski M:
The ubiquitin/proteasome system-dependent control of mitochondrial
steps in apoptosis. Semin Cell Dev Biol. 23:499–508. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yerlikaya A and Yöntem M: The significance
of ubiquitin proteasome pathway in cancer development. Recent Pat
Anticancer Drug Discov. 8:298–309. 2013. View Article : Google Scholar
|
|
28
|
Gatto S, Scappini B, Pham L, Onida F,
Milella M, Ball G, Ricci C, Divoky V, Verstovsek S, Kantarjian HM,
et al: The proteasome inhibitor PS-341 inhibits growth and induces
apoptosis in Bcr/Abl-positive cell lines sensitive and resistant to
imatinib mesylate. Haematologica. 88:853–863. 2003.PubMed/NCBI
|
|
29
|
Patrizi A, Venturi M, Dika E, Maibach H,
Tacchetti P and Brandi G: Cutaneous adverse reactions linked to
targeted anticancer therapies bortezomib and lenalidomide for
multiple myeloma: New drugs, old side effects. Cutan Ocul Toxicol.
33:1–6. 2014. View Article : Google Scholar
|
|
30
|
Cho Y, Hori M, Okoshi Y, Fujisawa F,
Shinagawa A, Kudo D, Komeno T, Yoshida C, Katsura Y, Ota I, et al:
Measurement of proteasome activity in peripheral blood mononuclear
cells as an indicator of susceptibility to bortezomib-induced
severe neurological adverse events in patients with multiple
myeloma. Acta Haematol. 134:25–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Richardson PG, Sonneveld P, Schuster MW,
Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D,
Lonial S, San Miguel JF, et al: Safety and efficacy of bortezomib
in high-risk and elderly patients with relapsed multiple myeloma.
Br J Haematol. 137:429–435. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li E and Zhang Y: DNA methylation in
mammals. Cold Spring Harb Perspect Biol. 6:a0191332014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
34
|
Kaiser MF, Johnson DC, Wu P, Walker BA,
Brioli A, Mirabella F, Wardell CP, Melchor L, Davies FE and Morgan
GJ: Global methylation analysis identifies prognostically important
epigenetically inactivated tumor suppressor genes in multiple
myeloma. Blood. 122:219–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Carvalho F, Colleoni GW, Almeida MS,
Carvalho AL and Vettore AL: TGFbetaR2 aberrant methylation is a
potential prognostic marker and therapeutic target in multiple
myeloma. Int J Cancer. 125:1985–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Galm O, Yoshikawa H, Esteller M, Osieka R
and Herman JG: SOCS-1, a negative regulator of cytokine signaling,
is frequently silenced by methylation in multiple myeloma. Blood.
101:2784–2788. 2003. View Article : Google Scholar
|
|
37
|
Mateos MV, Garcia-Sanz R, López-Pérez R,
Moro MJ, Ocio E, Hernández J, Megido M, Caballero MD,
Fernández-Calvo J, Bárez A, et al: Methylation is an inactivating
mechanism of the p16 gene in multiple myeloma associated with high
plasma cell proliferation and short survival. Br J Haematol.
118:1034–1040. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Takada S, Morita K, Hayashi K, Matsushima
T, Sawamura M, Murakami H and Nojima Y: Methylation status of
fragile histidine triad (FHIT) gene and its clinical impact on
prognosis of patients with multiple myeloma. Eur J Haematol.
75:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Robert MF, Morin S, Beaulieu N, Gauthier
F, Chute IC, Barsalou A and MacLeod AR: DNMT1 is required to
maintain CpG methylation and aberrant gene silencing in human
cancer cells. Nat Genet. 33:61–65. 2003. View Article : Google Scholar
|
|
41
|
Tsai HC, Li H, Van Neste L, Cai Y, Robert
C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al:
Transient low doses of DNA-demethylating agents exert durable
antitumor effects on hematological and epithelial tumor cells.
Cancer Cell. 21:430–446. 2012. View Article : Google Scholar : PubMed/NCBI
|