Introduction
Breast cancer is a highly prevalent type of cancer,
which is associated with a high mortality rate and affects women
worldwide (1). Genetic, metabolic
and lifestyle-associated risk factors may be important in the onset
and progression of breast cancer. Obesity is a risk factor that
contributes to the initiation and progression of breast cancer, via
increased circulation of estrogen, insulin, insulin-like growth
factor and adipokines (1).
Mutations in oncogenes and growth regulatory genes are further
variable factors that may be associated with the development of the
disease. Aberrant activation of the Wnt/β-catenin signaling
pathway, resulting from accumulation of β-catenin, has been
observed to be critically associated with carcinogenesis, including
breast cancer (2–4). Potentially targeting the
Wnt/β-catenin signaling pathway may provide a novel molecular
approach to cancer therapy.
Adenosine monophosphate (AMP)-activated protein
kinase (AMPK) is a key regulator of the cellular energy system
(5). It regulates cellular
metabolism and protects living cells from the environmental
stressors they may be exposed to, including hypoxia and nutrient
deficiency, which lead to elevations in cellular AMP:adenosine
triphosphate ratio (6).
Structurally, mammalian AMPK is a heterotrimeric complex composed
of one catalytic α subunit (63 kDa) and two regulatory subunits
each, of β and γ (38 and 36 kDa, respectively); each of which has
multiple isoforms (α1 and α2; β1 and β2; γ1, γ2 and γ3). AMP binds
to the γ subunit of AMPK and induces its allosteric activation
(5,6). AMPK activation impairs the mammalian
target of rapamycin (mTOR) signaling pathway, and its confirmatory
p70S6 kinase and 4E-BP1 activity (7), inhibiting protein synthesis and cell
growth. Notably, low AMPK activity favors carcinogenesis (8,9).
Various AMPK activators have been investigated for their antitumor
effects in several types of human cancer, including A23187,
A769662, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) and
metformin (10–13). Metformin in particular has been
used in several clinical trials (13–15).
The underlying molecular mechanisms by which these agents exhibit
their antitumor effects remain to be elucidated. AMPK may prevent
tumorigenesis via various mechanisms (16), including activation of an upstream
kinase, liver kinase B1 (LKB1). LKB1 is a proven tumor suppressor
that coordinates cell polarity in part via AMPK signaling, which
leads to disorganized cell division due to cross-talk with the
pro-proliferative Wnt signaling pathway and growth-restrictive
AMPK-mTOR metabolic pathway, in various types of carcinoma,
including gastrointestinal, pancreatic, ovarian and breast cancer
(17).
The present study investigated the interaction of
AMPK with the Wnt/β-catenin signaling pathway and its associated
components in breast carcinogenesis. Dishevelled segment polarity
protein (DVL) 3 was observed to be significantly upregulated and
correlated with Wnt/β-catenin activity in breast cancer cell
growth. Furthermore, AMPK activators exhibited the potential to
inhibit breast cancer cell growth by diminishing the DVL3-mediated
upregulation of Wnt/β-catenin signaling. The activation of AMPK led
to DVL3-mediated Wnt/β-catenin reduction; however, this was
significantly abrogated by an AMPK inhibitor. The results of the
present study verified the importance of the role of DVL3 in breast
cancer tumorigenesis and emphasized the potential therapeutic value
of targeting DVL3 via AMPK activators in the treatment of breast
cancer.
Materials and methods
Cell culture and treatments
MCF-7, MDA-MB-231 and T-47D breast cancer cell
lines, and the MCF-10 healthy breast cell line were procured from
the American Type Culture Collection (Manassas, VA, USA). MCF-7 and
MDA-MB-231 cells were grown in Minimum Essential Media (MEM), T-47D
cells were grown in RPMI-1640 media and MCF-10 cells were grown in
Mammary Epithelial Cell Growth Medium (Lonza, Basel, Switzerland).
MEME and RPMI-1640 were purchased from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Media were supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and
cells were cultured in the presence of 1% penicillin-streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in an atmosphere
containing 5% CO2. AMPK activators AICAR (Sigma-Aldrich,
Merck Millipore, Darmstadt, Germany) and A23187 (Calbiochem, EMD
Millipore, Billerica, MA, USA) were used to treat MCF-7 cells.
AICAR was used at 0, 1 and 2 mM and A23187 at 0, 2 and 4 mM for 24
h followed by protein isolation and western blotting. Another AMPK
activator, metformin (Sigma-Aldrich, Merck Millipore) was used to
treat MCF-7 cells at 0, 20 and 40 mM concentrations for 24 h
followed by subsequent analyses. An AMPK inhibitor, Compound C
(Sigma-Aldrich, Merck Millipore) was used to pre-treat MCF-7 cells
at 5 mM for 2 h followed by subsequent treatments and analyses.
Plasmids and cell transfection
For expression of green fluorescent protein
(GFP)/DVL3 protein, a GFP-tagged-DVL3 expressing construct was
generated by amplifying DVL3 using primer 1
(5′-GTGCTGGAATTCCCGAGGCC-3′) and primer 2
(5′-GCTCACATTGGATCCACAAAG-3′), with pEGFP-C1 plasmid (Addgene,
Cambridge, MA, USA) serving as the negative control.
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for MCF-7 and MDA-MB-231 cell
transfection according to the manufacturer's protocol. Transfected
cells were selected with antibiotic G418 (Sigma-Aldrich, Merck
Millipore) for 2 weeks. Western blotting was performed to verify
the cells that stably expressed GFP-DVL3.
Total RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Reverse
transcription reagent kit (Invitrogen; Thermo Fisher Scientific,
Inc.) was used to synthesize cDNA according to the manufacturer's
protocol. The expression of DVL genes was assessed by RT-qPCR using
the ABI PRISM™ 7500 system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with Taqman Gene Expression Assays, as per
manufacturer's protocol. The TaqMan gene expression assays
contained prevalidated primers and TaqMan probes for the individual
genes specifically custom synthesized by Applied Biosystems. For
amplification of DVL genes, two sets of primers and Taqman probes
were used. The probes were labeled at the 5′ end with the reporter
molecule 6-carboxy-fluorescein and labeled at the 3′ end with the
quencher molecule 6-carboxy-tetramethylrhodamine. Primer sequences
were: DVL2: 5′-TGAGCAACGATGACGCTGTG-3′ (forward) and
5′-GCAGGGTCAATTGGCTGGA-3′ (reverse); DVL3:
5′-ACAATGCCAAGCTACCATGCTTC-3′ (forward) and
3′-AGCTCCGATGGGTTATCAGCAC-5′ (reverse); and DVL-1:
5′-CCTTCCATCCAAATGTTGC-3′ (forward) and
5′-GTGACTGACCATAGACTCTGTGC-3′ (reverse). The RT-PCR amplification
conditions were set at: 95°C for 5 min, 95°C for 1 min, 58°C for 30
sec, and 72°C for 1 min, for 40 cycles. Relative quantification of
gene expression was performed as described by Applied Biosystems
using 18S rRNA as an internal control. The comparative CT (cycle
threshold) method was used for relative quantification of DVL
genes' mRNA (18). PCR was
performed in accordance with the manufacturer's protocol, in
triplicate, on an ABI 7500 real-time PCR system (Applied
Biosystems).
Protein isolation and western
blotting
Cells were prepared as per experimental requirement,
and harvested and pelleted for protein isolation. Cell pellets were
lysed with 1X lysis buffer composed of 20 mM Tris pH 7.5, 1 mM
EDTA, 150 mM NaCl, 2.5 mM sodium pyrophosphate, 1% Triton X-100, 1%
sodium vanadate, 1 mM PMSF together with protease inhibitor
cocktail (1:100; including phenylmethane sulfonyl fluoride). The
cell lysates were centrifuged at 12, 000 × g for 20 min at 4°C,
supernatant was collected and total protein content was estimated
by using BCA protein assay kit (Sigma-Aldrich, Merck Millipore).
For western blot analysis, 20 µg protein for each sample was
separated by 12% SDS-PAGE and transferred onto polyvinylidene
fluoride membranes. These membranes were then subsequently blocked
with 5% skimmed milk (fat-free) for 30 min and incubated with
specific primary antibodies overnight at 4°C. The primary
antibodies used were procured from Cell Signaling Technology, Inc.
(Danvers, MA, USA), Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA) and EMD Millipore. Anti-phosphorylated (p) AMPKα (catalog no.
cs 2531; 1:1,000), anti-AMPKα (catalog no. cs 2532; 1:1,000),
anti-c-Myc (catalog no. cs 5605; 1:1,000), anti-cyclin D1 (catalog
no. cs 2978; 1:1,000), anti-DVL1 (catalog no. sc-8025; 1:1,000),
anti-DVL2 (catalog no. sc-8026; 1:1,000), anti-DVL3 (catalog no.
sc-8027; 1:1,000), anti-β-catenin (catalog no. sc-7963; 1:1,000),
and anti-GAPDH antibody (catalog no. EMD AB2302; 1:2,000) as a
loading control. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary anti-mouse (catalog no.
cs 7076) or anti-rabbit antibodies (catalog no. cs 7074), obtained
from Cell Signaling Technology, Inc. in accordance with the
manufacturer's protocol. An enhanced chemiluminescence solution
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for signal
detection using photographic film followed by membrane
scanning.
Immunohistochemistry (IHC)
IHC was performed in healthy and cancerous breast
tissue (n=5/each). Cancerous breast tissue samples and their
corresponding healthy tissue samples were procured at the time of
surgical resection, from patients presenting at the Department of
Breast Surgery, The Third Hospital of Nanchang, Jiangxi, China. The
study was approved by the ethics committee of The First Affiliated
Hospital of Nanchang University, (Nanchang, China; Reference no.
2123432524AK) and written informed consent was obtained from
patients or their family. Patients included in the study were
Chinese adult women, mean age, 40.2±5.67. The patients had never
received chemotherapy prior to surgical resection. For IHC
analysis, surgically resected tissue parts were fixed in normal
buffered formalin followed by serial dehydration in graded alcohol
then embedded in paraffin. Tissues embedded in paraffin blocks were
cut into 4 µM thick sections. Tissue sections were de-paraffinized
and treated with primary antibodies for anti-DVL3 (1:200 dilution)
and anti-β-catenin (1:200 dilution) for 24 h at 4°C, while
Tris-buffered saline served as a negative control. Antibodies were
procured from Novus Biologicals, LLC (Littleton, CO, USA) and BD
Biosciences (Franklin Lakes, NJ, USA), respectively. Sections were
visualized under a light microscope (Leica Microsystems GmbH,
Wetzlar, Germany) at 40x magnification and manual scoring of the
expression levels of proteins was calculated from immunopositive
staining area (0–100%). The intensity of staining was recorded on
the scale of: +1, weak; +2, moderate; +3, intense; and +4, very
intense. The fold-change in the expression level of each protein
was calculated by normalization to the expression level of the same
protein in the healthy breast section. The IHC analysis from each
tissue section was examined and scored independently by two
investigators. The breast tumor array (BC08022) analysis for DVL3
and β-catenin was performed by US Biomax, Inc. (Rockville, MD,
USA). The expression levels of DVL3 and β-catenin were represented
as fold-change of each gene as compared to normal tissue. The
cutoff point (6-folds) of both genes was determined by its
expression levels and statistical significance.
Cell viability assay
The XTT cell proliferation kit (Invitrogen; Thermo
Fisher Scientific, Inc.) was used to measure cell viability. The
assay was performed according to the manufacturer's protocol. Each
cell line was assayed in triplicate and three independent
experiments were performed.
Clonogenic assay
Cells were grown and seeded for 24 h. Media was
removed after 24 h, followed by incubation with methanol for 30
min. Cells were then stained with crystal violet for 1 h at room
temperature, and subsequently washed with PBS followed by cell
counting under a light microscope (Leica Microsystems GmbH) at 40x
magnification using Image J software version 1.4 (U.S. National
Institutes of Health, Bethesda, USA). The experiment was performed
in triplicate and three independent experiments were performed.
Statistical analysis
Data were analyzed using one-way analysis of
variance followed by Tukey's post hoc test, and the Chi-square
test. Data are presented as the mean ± standard deviation. SPSS v16
was used for statistical analyses. P<0.05 was considered to
indicate a statistically significant difference.
Results
DVL3 is recurrently upregulated in
breast cancer cells
It has previously been demonstrated that DVLs are
key in the upregulation of the expression of β-catenin and
promotion of cell growth in various cancers, such as colorectal
cancer (19), malignant pleural
mesothelioma (20) and
non-small-cell lung cancer (21).
The present study first examined the expression patterns of DVL1, 2
and 3, in MCF-7, MDA-MB-231 and T-47D breast cancer cell lines, and
the MCF-10 healthy breast cell line by western blot analysis. The
results demonstrated that DVL3 was notably upregulated in breast
cancer cell lines, compared with the healthy breast cell line
(Fig. 1A). These data suggested
that DVL3 is upregulated in breast cancer cells. The MCF-7 cells
revealed greater levels of DVL3 compared with the other breast
cancer cell lines. The mRNA expression levels of DVL genes in
breast cancer cell lines and the healthy breast cell line were then
determined by RT-qPCR. The gene expression analysis demonstrated
similar results to those presented in Fig. 1A. The mRNA expression levels of
DVL3 were considerably greater in breast cancer cell lines compared
with healthy breast cells (Fig.
1B). Furthermore, the mRNA expression levels of DVL1 and DVL2
were reduced in each cell line, compared with DVL3. These findings
primarily indicated that DVL3 was the major isoform among DVLs that
was recurrently upregulated in breast cancer cells. The results
also suggested that MCF-7 and MDA-MB-231 cell lines were more
susceptible to alterations in DVL3 levels, compared with T-47D
cells. MCF-7 and MDA-MB-231 cell lines were therefore selected for
further studies.
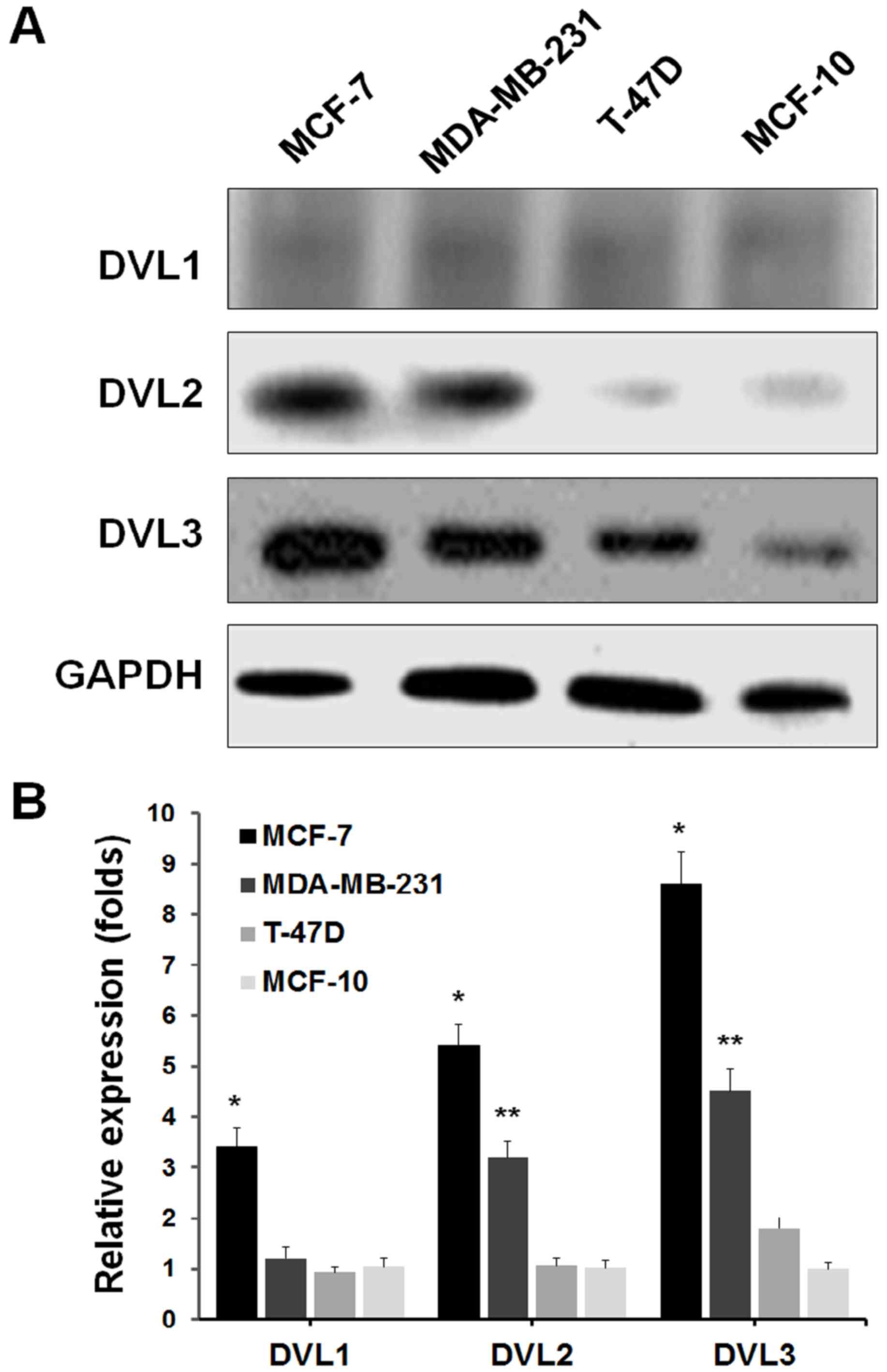 | Figure 1.DVL3 upregulation in breast cancer
cell lines. (A) Western blot analysis of the expression of DVL1, 2
and 3 in MCF-7, MDA-MB-231 and T-47D breast cancer cells, and the
MCF-10 healthy breast cell line. GAPDH served as a loading control.
(B) Reverse transcription-quantitative polymerase chain reaction
analysis of human breast cancer and healthy breast cell lines, to
quantify mRNA expression levels of DVL1, 2 and 3. DVL-1, *P=0.003
vs. MCF-10; DVL-2, *P=0.002 vs. MCF-10, **P=0.004 vs. MCF-10;
DVL-3, *P=0.0006 vs. MCF-10, **P=0.002 vs. MCF-10. DVL, dishevelled
segment polarity protein. |
DVL3 and β-catenin are overexpressed
in breast cancer
The significance of DVL3 and β-catenin expression
level alterations in breast cancer was investigated by examining
the expression levels of these proteins using IHC and tissue array
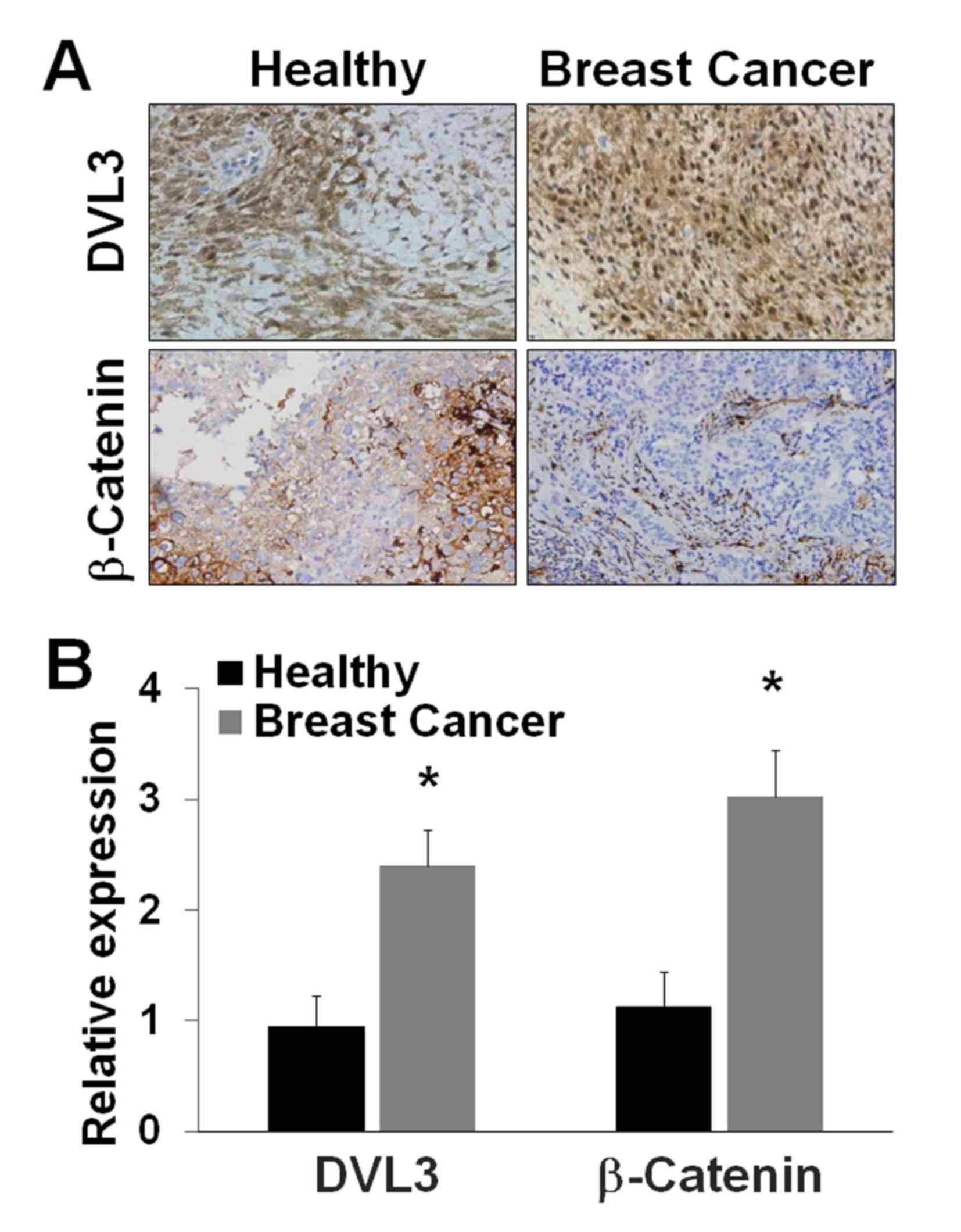
analysis in breast cancer and healthy breast tissues. The IHC
analysis of breast cancer and healthy breast tissues revealed a
greater level of staining of DVL3 and β-catenin in breast cancer
tissues compared with healthy breast tissue (Fig. 2A). The relative protein expression
levels of DVL3 and β-catenin were estimated from tissue sections.
DVL3 expression exhibited a >2-fold (P=0.03) increase in breast
cancer tissues compared with in healthy breast tissue (Fig. 2B). β-Catenin expression exhibited a
>3-fold (P=0.022) increase in breast cancer tissues compared
with in healthy breast tissue (Fig.
2B). DVL3 and β-catenin were revealed to be overexpressed in
breast cancer tissue compared with healthy breast tissue. The
tissue array analysis revealed an increase in the expression of
DVL3 and β-catenin in breast tumor samples (Table I). The data obtained were
categorized ≤ or >6-fold for a comparative analysis with healthy
tissue. A greater level of β-catenin (>6-fold) was noted as
statistically significant in breast cancer tissue samples.
Statistical analysis revealed that expression levels of DVL3
(P=0.044) and β-catenin (P=0.038) were significantly increased
(>6-fold) in breast cancer tissue at an advanced stage of the
disease. In addition, the levels of DVL3 and β-catenin were
significantly increased (>6-fold) in breast tissue with
metastatic cancer (Table I).
 | Table I.Clinical and pathological
correlations of DVL3 and β-catenin expression in breast cancer
tissue. |
Table I.
Clinical and pathological
correlations of DVL3 and β-catenin expression in breast cancer
tissue.
|
|
| DVL3
expression | β-catenin
expression |
|---|
|
|
|
|
|
|---|
|
Characteristics | Total number | ≤6-fold | >6-fold |
P-valuea | ≤6-fold | >6-fold |
P-valuea |
|---|
| All cases | 48 | 22 (45.8%) | 26 (54.2%) |
| 18 (37.5%) | 30 (62.5%) |
|
| Cancer stage |
|
Early | 29 | 12 (41.4%) | 17 (58.6%) | 0.048 | 8 (27.6%) | 21 (72.4%) | 0.041 |
|
Late | 19 | 8 (42.1%) | 11 (57.9%) | 0.044 | 7 (36.8%) | 12 (63.2%) | 0.038 |
| Metastasis |
| No | 35 | 22 (62.9%) | 13 (37.1%) | 0.032 | 19 (54.3%) | 16 (45.7%) | 0.030 |
|
Yes | 13 | 4 (30.8%) | 9 (69.2%) | 0.022 | 3 (23.1%) | 10 (76.9%) | 0.012 |
DVL3 enhances breast cancer cell
proliferation
DVL3 has previously been suggested to act as an
important signal transduction molecule, which mediates the
Wnt/β-catenin signaling pathway and controls cell growth (3,4). The
association of DVL3 and β-catenin signaling in breast
carcinogenesis was investigated, in order to examine the role of
DVL3 in cell proliferation via Wnt/β-catenin signal pathway
activation in breast cancer. MCF-7 and MDA-MB-231 breast cancer
cell lines were transfected with a human GFP-tagged DVL3 expression
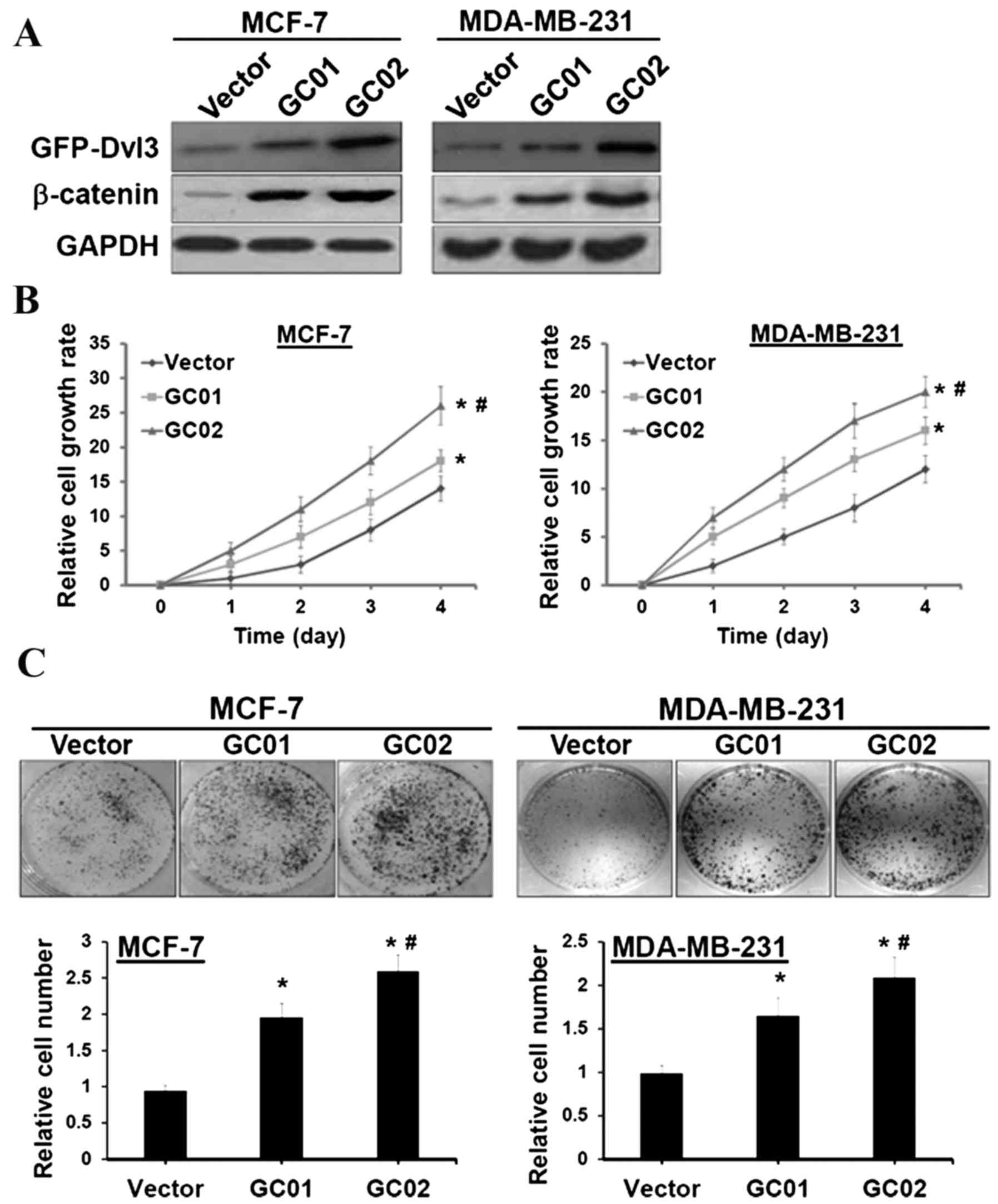
plasmid for stable expression of DVL3. Western blotting revealed
that β-catenin was significantly elevated in cells transfected with
GFP-DVL3 stable clones (termed GC01 and GC02) (Fig. 3A). GC01 demonstrated a reduced
expression of GFP-DVL3, compared with GC02; however, the expression
analysis was statistically significant in the two cell lines
(Fig. 3B). The XTT cell
proliferation assay revealed that MCF-7 and MDA-MB-231 cells with
ectopic expression of DVL3 demonstrated a more significant
time-dependent rate of proliferation compared with the vector
control (Fig. 3B). Furthermore, a
clonogenic assay revealed a 1.6–2.5-fold increase in the number of
colonies in MCF-7 and MDA-MB-231 cells stably expressing GFP-DVL3
compared with their respective vector controls (Fig. 3C). These results suggested that
breast cancer cell proliferation is promoted by DVL3-mediated
increases in Wnt/β-catenin signaling activity.
AMPK activators suppress DVL3 in
breast cancer cells
It has previously been demonstrated that AMPK
activators induce growth inhibitory effects against human cancers,
including cervical and breast cancer (13,22,23).
The AMPK activators, AICAR and A23187, were used in the present
study to analyze the effects of AMPK activation on the expression
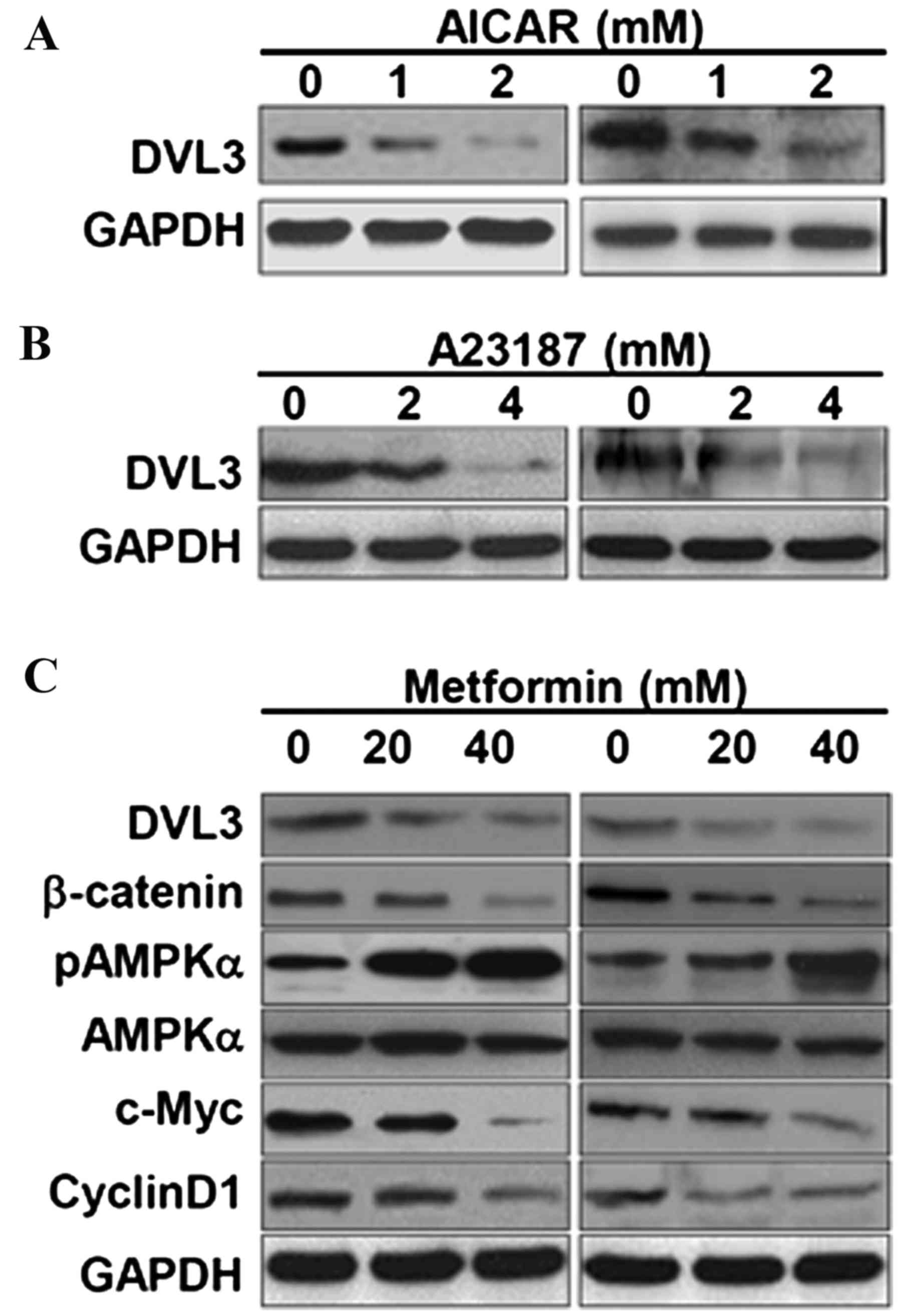
levels of DVL3 and β-catenin in breast cancer cells. MCF-7 and
MDA-MB231 cell lines treated with AICAR demonstrated a marked
reduction in DVL3 expression levels (Fig. 4A). A23187, a calcium ionophore,
activates AMPK via its upstream calmodulin dependent protein kinase
domain by increasing cytosolic calcium levels (24). In the present study, A23187 reduced
DVL3 expression in MCF-7 and MDA-MB-231 cells in a dose-dependent
manner (Fig. 4B). Treatment of
breast cancer cell lines with these two AMPK activators resulted in
an inhibition of DVL3 expression levels, which may act to further
modulate the signaling events in cancer cell growth. The present
study then aimed to decipher the underlying molecular mechanism of
breast cancer cell growth inhibition, and its association with DVL3
reduction and AMPK activators by using metformin (another AMPK
activator). Metformin is a commonly used AMPK activator and potent
anticancer drug (13). Treatment
of MCF-7 and MDA-MB-231 breast cancer cells with metformin resulted
in a depletion of DVL3 in the cell lines in a dose-dependent manner
(Fig. 4C). Treatment with
metformin reduced the levels of DVL3 and β-catenin in a
dose-dependent manner in the two cell lines. Metformin activated
the levels of pAMPKα in a dose-dependent manner, but did not result
in alteration to total AMPK levels (Fig. 4C). In addition, metformin
dose-dependently inhibited expression of the two downstream
transcriptional products of β-catenin, c-Myc and cyclin D1
(Fig. 4C). These results suggested
that AMPK activators enhanced the phosphorylation of AMPK,
resulting in an inhibition of the DVL3 and β-catenin crosslink, and
suppressed cell growth in breast cancer cells as indicated by
reduced colony number.
AMPK activator suppresses breast
cancer cell growth by reducing DVL3 and β-catenin
The AMPK activators primarily activate AMPK
activity; however, it has previously been demonstrated that AMPK
activators may be involved in cellular metabolic processes, such as
energy metabolism (25,26). The present study subsequently
examined whether AMPK activator-mediated DVL3 reduction is
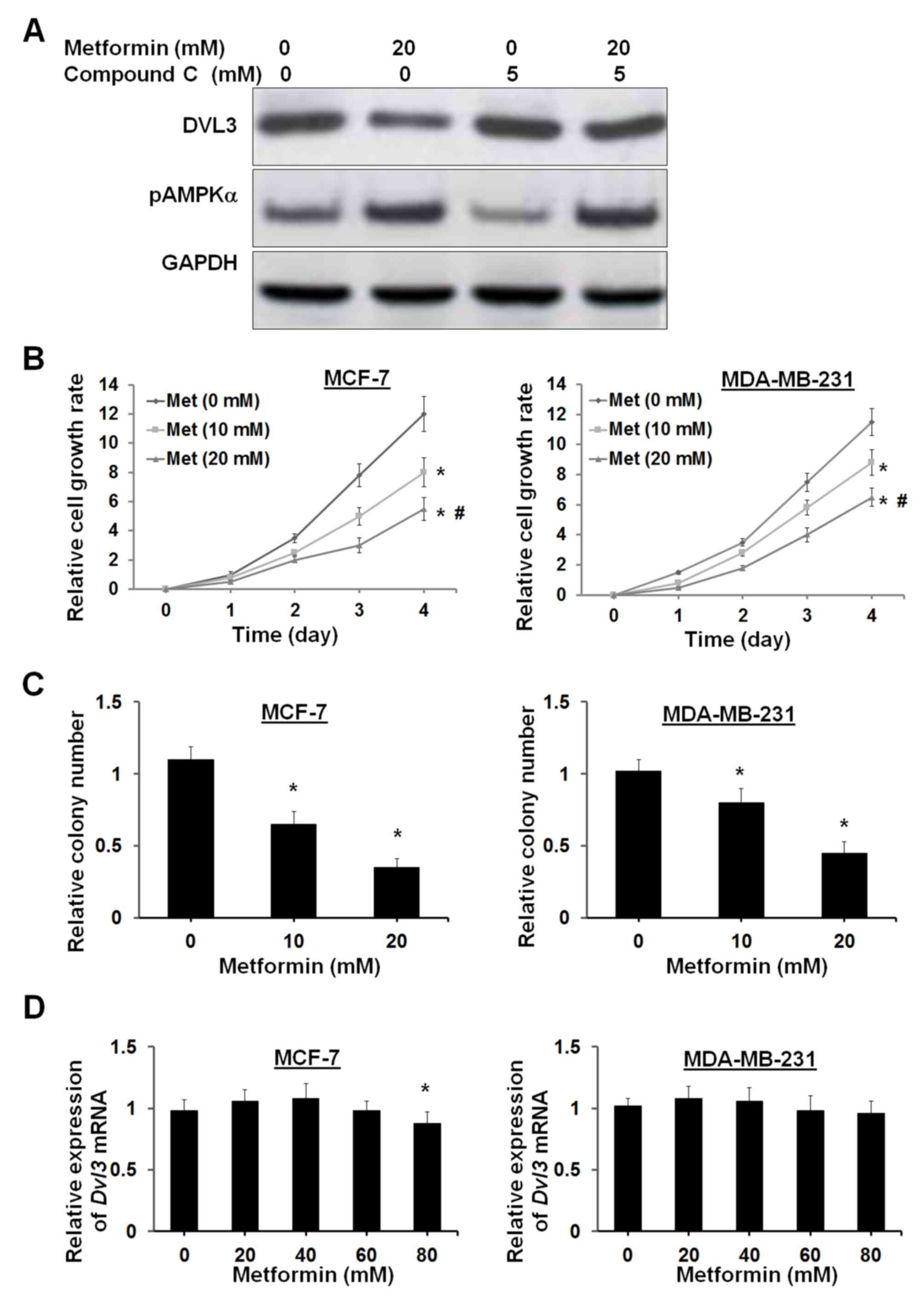
dependent on AMPK signaling. Compound C, which is a potent AMPK
inhibitor, was used to counteract the effects of metformin on AMPK
activation. Treatment of MFC-7 cells with 5 µM compound C resulted
in a small increase in DVL3 levels, with a small inhibition of
pAMPKα levels (Fig. 5A). Treatment
with 20 mM metformin in MCF-7 cells induced a reduction in DVL3
expression and an increase in pAMPKα levels (Fig. 5A). The metformin-induced decrease
in DVL3 and increase in pAMPKα levels were reversed following the
addition of compound C (Fig. 5A).
These results suggested that metformin-induced DVL3 reduction may
be dependent on AMPK signaling. It was previously demonstrated that
DVL3 ectopic expression may increase breast cancer cell
proliferation. The present study elucidated the molecular basis of
DVL3 reduction and Wnt/β-catenin signaling by AMPK activators and
suppression of cell proliferation. MCF-7 and MDA-MB-231 cells were
treated with metformin, and an XTT assay was performed to analyze
cell proliferation. Cell proliferation analysis demonstrated that
metformin significantly suppressed cell proliferation in MCF-7 and
MDA-MB-231 cells in a dose- and time-dependent manner (Fig. 5B). The clonogenic assay supported
the results of the cell proliferation assay by demonstrating a
reduction in the number of colonies by 41 and 68% in MCF-7 cells,
and 22 and 56% in MDA-MB-231 cells at 10 and 20 mM concentrations
of metformin, respectively (Fig.
5C). Notably, metformin did not modulate the levels of DVL3
mRNA in the two breast cancer cell lines (Fig. 5D). A very high concentration of
metformin (80 mM) resulted in a small decrease in DVL3 mRNA in
MCF-7 cells (P=0.042). This observation suggested that metformin
may have no interaction with the DVL3 gene and it may act at the
post-transcriptional level. These results indicated that breast
cancer cell growth inhibition via AMPK is associated with a
reduction in DVL3 protein expression levels.
Discussion
The present study demonstrated that DVL3 aberrant
overexpression was significantly correlated with increased
β-catenin in breast cancer. DVL3 may activate the Wnt/β-catenin
signaling pathway and promote growth of breast cancer cells. The
potential use of AMPK activators in the reduction of DVL3 was
investigated to suppress the growth of breast cancer cells. The
molecular mechanism underlying the effects of AMPK activators may
be an intervention in the interaction between AMPK and the
Wnt/β-catenin signaling pathway to impair breast cancer cell
growth. The present study primarily demonstrated that AMPK
activators exhibit therapeutic potential against breast
carcinoma.
The Wnt/β-catenin signaling pathway has been
aberrantly activated in various types of human cancer, including
breast cancer. It has previously been demonstrated that DVLs, which
are positive regulators of the Wnt/β-catenin signaling pathway, are
frequently upregulated in several human cancer types, such as lung,
prostate, breast, liver and colon cancer (20,27–30).
The overexpression of DVLs is associated with an increase in
Wnt/β-catenin activity in human cancer (20,21,31).
However, the functional role of the DVL-β-catenin-AMPK interaction
in breast cancer remains to be elucidated. The present study
demonstrated that DVL3 was significantly upregulated and associated
with increased activity of the Wnt/β-catenin signaling pathway in
breast cancer cells. However, a similar association was not
observed for DVL1 and 2. It has previously been suggested that the
upstream kinases of AMPK (including LKB1) are frequently mutated
and deleted in various human cancers, including breast cancer
(23,32,33).
LKB1 reduces AMPK activities, which promote cancer cell growth
(8,9). AMPK is therefore a critical target in
cancer therapy. It has also been previously suggested that several
AMPK activators may suppress cancer cell growth, including breast
cancer (13,23,34).
However, the molecular mechanisms underlying the effects of DVL3
suppression and AMPK activation on the prevention of breast cancer
cell growth remain to be elucidated.
The present study investigated AMPK activation as a
potential cancer therapy via the application of the antidiabetic
drug metformin (N,N-dimethylbiguanide). Metformin has previously
been suggested to inhibit tumor growth via a reduction of serum
glucose levels and insulin/insulin-like growth factors, and via
intratumoral AMPK activation (34,35).
The results of the present study verified the previous findings
that metformin administration may reduce cancer formation via AMPK
activation, and suggest a possible strategy for the treatment of
breast cancer. Metformin was previously observed to reduce
β-catenin protein levels by regulating its phosphorylation in human
osteoblasts (36). These
observations indicated that upstream regulators of the
Wnt/β-catenin signaling cascade may be regulated by metformin and
thus may control the growth of cancer cells. The present study
suggested that AMPK activators may markedly reduce DVL3 expression
and subsequently result in breast cancer cell growth inhibition via
Wnt/β-catenin signaling. Previous reports using metformin have
indicated that its antitumor effects are mediated by the
downregulation of cyclin D1 in prostate and breast cancer (37,38).
In the present study, the reduction in DVL3 levels was associated
with the reduced levels of β-catenin and its downstream targets,
cyclin D1 and c-Myc. c-Myc regulates cell proliferation whereas
cyclin D1 maintains cell cycle progression; therefore, these
targets may be essential transcriptional products of β-catenin in
cell growth regulation. Notably, metformin altered DVL3 expression
at the protein level, but not at the mRNA level. This result
indicated that the regulation of DVL3 mediated by AMPK activators
may be dependent on post-transcriptional modification of proteins
rather than gene expression regulation. Numerous AMPK activators
have been revealed to reduce DVL3, indicating that AMPK activity is
essential in regulation of cell proliferation (11,13,22,23,37,38).
The effects of AMPK activators on DVL3 expression were assessed by
co-treating breast cancer cells with an AMPK inhibitor (compound C)
and metformin, to counteract the effects of AMPK activation.
Compound C abrogated the effects of metformin on the suppression of
DVL3 expression. These results suggested that AMPK activators may
suppress cell growth by reducing DVL3 levels, and similar results
were observed from in vitro cell proliferation assays.
In conclusion, the results of the present study
demonstrated that AMPK activators, including metformin,
downregulated DVL3 expression and thus reduced the expression
levels of β-catenin, c-Myc and cyclin D1, resulting in suppression
of cell proliferation. DVL3 is a key oncoprotein, which activates
the Wnt/β-catenin signaling pathway and mediates growth of breast
cancer cells. A reduction of DVL3 via AMPK activation is important
in suppressing tumor development in breast cancer. These results
therefore emphasize the therapeutic value of AMPK activators in
targeting DVL3 and Wnt/β-catenin signaling in breast cancer
treatment. The results of the present study demonstrated that
administration of AMPK activators reduced growth of breast cancer
cells via AMPK activation, thus suggesting a novel and potential
therapeutic strategy for the treatment of breast cancer.
References
|
1
|
Evans DG and Howell A: Breast cancer
risk-assessment models. Breast Cancer Res. 9:2132007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ilyas M: Wnt signalling and the
mechanistic basis of tumour development. J Pathol. 205:130–144.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
King TD, Suto MJ and Li Y: The
Wnt/b-catenin signaling pathway: A potential therapeutic target in
the treatment of triple negative breast cancer. J Cell Biochem.
113:13–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hardie DG: Minireview: The AMP-activated
protein kinase cascade: The key sensor of cellular energy status.
Endocrinology. 144:5179–5183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Richter EA and Ruderman NB: AMPK and the
biochemistry of exercise: Implications for human health and
disease. Biochem J. 418:261–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thomas G and Hall MN: TOR signalling and
control of cell growth. Curr Opin Cell Biol. 9:782–787. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vazquez-Martin A, Oliveras-Ferraros C,
Lopez-Bonet E and Menendez JA: AMPK: Evidence for an energy-sensing
cytokinetic tumor suppressor. Cell Cycle. 8:3679–3683. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carretero J, Medina PP, Blanco R, Smit L,
Tang M, Roncador G, Maestre L, Conde E, Lopez-Rios F, Clevers HC
and Sanchez-Cespedes M: Dysfunctional AMPK activity, signalling
through mTOR and survival in response to energetic stress in
LKB1-deficient lung cancer. Oncogene. 26:1616–1625. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Corton JM, Gillespie JG, Hawley SA and
Hardie DG: 5-aminoimidazole-4-carboxamide ribonucleoside. A
specific method for activating AMP-activated protein kinase in
intact cells? Eur J Biochem. 229:558–565. 1995.PubMed/NCBI
|
|
12
|
Yung MM, Chan DW, Liu VW, Yao KM and Ngan
HY: Activation of AMPK inhibits cervical cancer cell growth through
AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer. 13:3272013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alimova IN, Liu B, Fan Z, Edgerton SM,
Dillon T, Lind SE and Thor AD: Metformin inhibits breast cancer
cell growth, colony formation and induces cell cycle arrest in
vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiralerspong S, Palla SL, Giordano SH,
Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi
GN and Gonzalez-Angulo AM: Metformin and pathologic complete
responses to neoadjuvant chemotherapy in diabetic patients with
breast cancer. J Clin Oncol. 27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo Z, Zang M and Guo W: AMPK as a
metabolic tumor suppressor: Control of metabolism and cell growth.
Future Oncol. 6:457–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin-Belmonte F and Perez-Moreno M:
Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer.
12:23–38. 2011.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–428. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Metcalfe C, Ibrahim AE, Graeb M, de la
Roche M, Schwarz-Romond T, Fiedler M, Winton DJ, Corfield A and
Bienz M: Dvl2 promotes intestinal length and neoplasia in the
ApcMin mouse model for colorectal cancer. Cancer Res. 70:6629–6638.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Uematsu K, He B, You L, Xu Z, McCormick F
and Jablons DM: Activation of the Wnt pathway in non small cell
lung cancer: Evidence of dishevelled overexpression. Oncogene.
22:7218–7221. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Uematsu K, Kanazawa S, You L, He B, Xu Z,
Li K, Peterlin BM, McCormick F and Jablons DM: Wnt pathway
activation in mesothelioma: Evidence of Dishevelled overexpression
and transcriptional activity of beta-catenin. Cancer Res.
63:4547–4551. 2003.PubMed/NCBI
|
|
22
|
Liu B, Fan Z, Edgerton SM, Deng XS,
Alimova IN, Lind SE and Thor AD: Metformin induces unique
biological and molecular responses in triple negative breast cancer
cells. Cell Cycle. 8:2031–2040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu SY, Chan DW, Liu VW and Ngan HY:
Inhibition of cervical cancer cell growth through activation of
upstream kinases of AMP-activated protein kinase. Tumour Biol.
30:80–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hawley SA, Pan DA, Mustard KJ, Ross L,
Bain J, Edelman AM, Frenguelli BG and Hardie DG:
Calmodulin-dependent protein kinase kinase-beta is an alternative
upstream kinase for AMP-activated protein kinase. Cell Metab.
2:9–19. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Canto C, Gerhart-Hines Z, Feige JN,
Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P and Auwerx
J: AMPK regulates energy expenditure by modulating NAD+ metabolism
and SIRT1 activity. Nature. 458:1056–1060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guigas B, Sakamoto K, Taleux N, Reyna SM,
Musi N, Viollet B and Hue L: Beyond AICA riboside: In search of new
specific AMP-activated protein kinase activators. IUBMB Life.
61:18–26. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizutani K, Miyamoto S, Nagahata T,
Konishi N, Emi M and Onda M: Upregulation and overexpression of
DVL1, the human counterpart of the Drosophila dishevelled gene, in
prostate cancer. Tumori. 91:546–551. 2005.PubMed/NCBI
|
|
28
|
Nagahata T, Shimada T, Harada A, Nagai H,
Onda M, Yokoyama S, Shiba T, Jin E, Kawanami O and Emi M:
Amplification, up-regulation and over-expression of DVL-1, the
human counterpart of the Drosophila disheveled gene, in primary
breast cancers. Cancer Sci. 94:515–518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miyoshi Y, Iwao K, Nagasawa Y, Aihara T,
Sasaki Y, Imaoka S, Murata M, Shimano T and Nakamura Y: Activation
of the beta-catenin gene in primary hepatocellular carcinomas by
somatic alterations involving exon 3. Cancer Res. 58:2524–2527.
1998.PubMed/NCBI
|
|
30
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Holloway KR, Calhoun TN, Saxena M, Metoyer
CF, Kandler EF, Rivera CA and Pruitt K: SIRT1 regulates Dishevelled
proteins and promotes transient and constitutive Wnt signaling.
Proc Natl Acad Sci USA. 107:9216–9221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen Z, Wen XF, Lan F, Shen ZZ and Shao
ZM: The tumor suppressor gene LKB1 is associated with prognosis in
human breast carcinoma. Clin Cancer Res. 8:2085–2090.
2002.PubMed/NCBI
|
|
33
|
Zhou J, Huang W, Tao R, Ibaragi S, Lan F,
Ido Y, Wu X, Alekseyev YO, Lenburg ME, Hu GF and Luo Z:
Inactivation of AMPK alters gene expression and promotes growth of
prostate cancer cells. Oncogene. 28:1993–2002. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gonzalez-Angulo AM and Meric-Bernstam F:
Metformin: A therapeutic opportunity in breast cancer. Clin Cancer
Res. 16:1695–1700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takatani T, Minagawa M, Takatani R,
Kinoshita K and Kohno Y: AMP-activated protein kinase attenuates
Wnt/β-catenin signaling in human osteoblastic Saos-2 cells. Mol
Cell Endocrinol. 339:114–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in Metformin treated breast cancer cells involves activation
of AMPK, downregulation of cyclin D1, and requires p27Kip1 or
p21Cip1. J Mol Signal. 3:182008. View Article : Google Scholar : PubMed/NCBI
|