Introduction
Oculocutaneous albinism (OCA) is a group of
heterogeneous and autosomal recessive disorders characterized by a
reduction or complete loss of melanin biosynthesis in melanocytes
(1). All types of OCA demonstrate
a lack of melanin pigment in the skin, hair and eyes, and are often
accompanied by ocular abnormalities, including varying degrees of
nystagmus, hypopigmentation of the iris, foveal hypoplasia, poor
vision and refractive errors, photophobia and occasionally color
vision impairment (2). The
prevalence of OCA subtypes varies considerably between ethnicities.
OCA has a prevalence of ~1:20,000 individuals worldwide, or
~1:18,000 in the Chinese Han population (3). At least 17 genes, including four
non-syndromic and 13 syndromic, have been identified to be
associated with OCA. The tyrosinase (TYR), oculocutaneous
albinism II, tyrosinase-related protein 1 and solute carrier family
45, member 2 genes are the four non-syndromic OCA genes and are
responsible for OCA type 1 (OCA1; MIM 203100), type 2 (OCA2; MIM
203200), type 3 (OCA3; MIM 203290), and type 4 (OCA4; MIM 606574),
respectively. The 13 syndromic OCA genes include the
Hermansky-Pudlak syndrome 1 (HPS1), adaptor-related protein
complex 3, β 1 subunit, HPS3, HPS4, HPS5, HPS6,
dystrobrevin binding protein 1, biogenesis of lysosomal organelles
complex-1 subunit 3 (BLOC1S3), BLOC1S6, lysosomal
trafficking regulator, myosin VA, ras-related protein and
melanophilin genes (4,5). Due to the phenotypic variation and
overlap in clinical presentation among different OCA subtypes,
genetic analysis is essential to identify the gene defect and the
subtype of OCA (6).
OCA1 is the most severe and common form of OCA, and
is caused by mutations of the TYR gene (7). It is often divided into two
categories: OCA1A and OCA1B. Patients with OCA1A are characterized
by a lifelong and complete absence of pigment in the skin, hair and
eyes. Patients with OCA1B may have a reduced activity of tyrosinase
and may produce some melanin over time, resulting in a darkening of
hair (3,8). OCA1 affects ~1 in 40,000 individuals
in the majority of populations, and has been reported to be the
most common subtype of OCA in Japanese, non-Hispanic Caucasian and
Danish populations (9). It
accounts for ~64.3% of patients with OCA in China. However, OCA1 is
rare among African-Americans, reflecting a population-specific
distribution of different OCA subtypes (6).
The present study aimed to identify the genetic
mutation responsible for OCA1 in a four-generation family of
Chinese origin. Patients in the family presented with typical OCA1
characteristics, including white hair and skin, nystagmus, impaired
visual acuity, photophobia, color vision impairment, refractive
errors, foveal hypoplasia and iris hypopigmentation and
translucency. The p.R299H variant of the TYR gene, predicted
to disrupt the overall integrity of tyrosinase, was revealed to
co-segregate with patients in this family and was absent in healthy
controls, indicating that it is a pathogenic mutation.
Materials and methods
Subjects
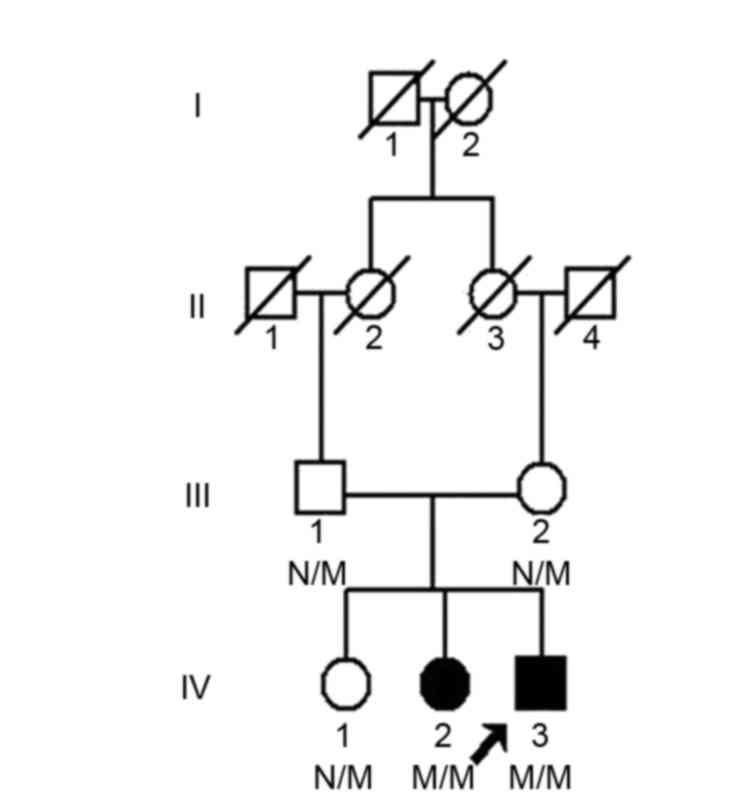
A four-generation, 11-member consanguineous Chinese
Han family with OCA from Hunan, China was recruited from The Third
Xiangya Hospital, Central South University (Hunan, China; Fig. 1). Blood samples from the five
living members of the family, including two affected patients (IV:2
and IV:3) and three unaffected members (III:1, III:2 and IV:1) were
collected. Additionally, blood samples were collected from 100
unrelated healthy controls (male, n=50; female, n=50; age, 45.8±3.4
years) with normal pigmentation and with no family history of
ocular abnormalities, from the same region of China. All
participants provided written informed consent. The present study
was approved by the Institutional Review Board of The Third Xiangya
Hospital, Central South University.
Exome capture
Genomic DNA was extracted from peripheral blood
samples according to the standard phenol-chloroform extraction
method (10). Exome capture was
performed on genomic DNA from the proband (Fig. 1) by the Novogene Bioinformatics
Institute (Beijing, China). The SureSelect Human All Exon V5 kit
(Agilent Technologies, Inc., Santa Clara, CA, USA) was used for
exome capture and the HiSeq 2000 platform (Illumina, Inc., San
Diego, CA, USA) was used for sequencing, following the
manufacturer's protocol. A total of 1.5 µg genomic DNA was used to
construct the exome library and the genomic DNA was sheared into
180–280 bp for enrichment. Enrichment libraries for target regions
were sequenced by the HiSeq 2000 platform, which generated 100 bp
pair-end reads.
Read mapping and variant analysis
All the clean reads were aligned to the human
reference genome (University of California, Santa Cruz; Build 37.1,
hg19) using Burrows-Wheeler Aligner (bio-bwa.sourceforge.net). High quality alignment was
required to guarantee variant calling accuracy (>0) to detect
single nucleotide polymorphisms (SNPs) and insertions-deletions
(indels). Picard software (sourceforge.net/projects/picard/), the Genome Analysis
Toolkit software version 2.1 (software.broadinstitute.org/gatk/) and SAMtools
(samtools.sourceforge.net) were used for
base quality recalibration. Following this, the binary
alignment/map results were obtained, ready for analysis, and
ANNOVAR software (Annotate Variation; annovar.openbioinformatics.org/enllatest/user-guider/download)
was used to annotate SNPs and indels. Variant lists were filtered
against the dbSNP build 137 (dbSNP137), database of SNPs
(www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi),
the 1000 Genomes Project (1000genomes release_20100804; www.1000genomes.org/) and the National Heart, Lung and
Blood Institute Exome Sequencing Project (NHLBI ESP) 6500.
Polymorphism Phenotyping version 2 (PolyPhen-2; genetics.bwh.harvard.edu/pph2/) and
Sorting Intolerant from Tolerant (SIFT; sift.jcvi.org/) software were used to predict protein
functions.
Mutation validation
Sanger sequencing was used to confirm the presence
and identity of potential disease-causing variants, using the 3500
Genetic Analyzer sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Polymerase chain reaction
amplification and Sanger sequencing were conducted as described
previously (11), using the
following primers: Forward, 5′-GTCTGTAGCCGATTGGAGGA-3′ and reverse,
5′-GCAGCTTTATCCATGGAACC-3′.
The National Centre for Biotechnology Information
Basic Local Alignment Search Tool (blast.ncbi.nlm.nih.gov/Blast.cgi) was used to perform
multiple sequence alignments. The pathogenic potential of amino
acid changes as possible disease-causing mutations was further
examined using the MutationTaster online tool (http://www.mutationtaster.org/).
Results
Clinical findings
Complete physical examinations, including detailed
ophthalmic examination (best-corrected visual acuity testing,
slit-lamp examination, dilated fundus examination and optical
coherence tomography) were performed. The two patients had typical
OCA skin, hair and eye symptoms; they presented with white hair
that had not altered in color over time. More detailed descriptions
of clinical features of the two OCA patients are presented in
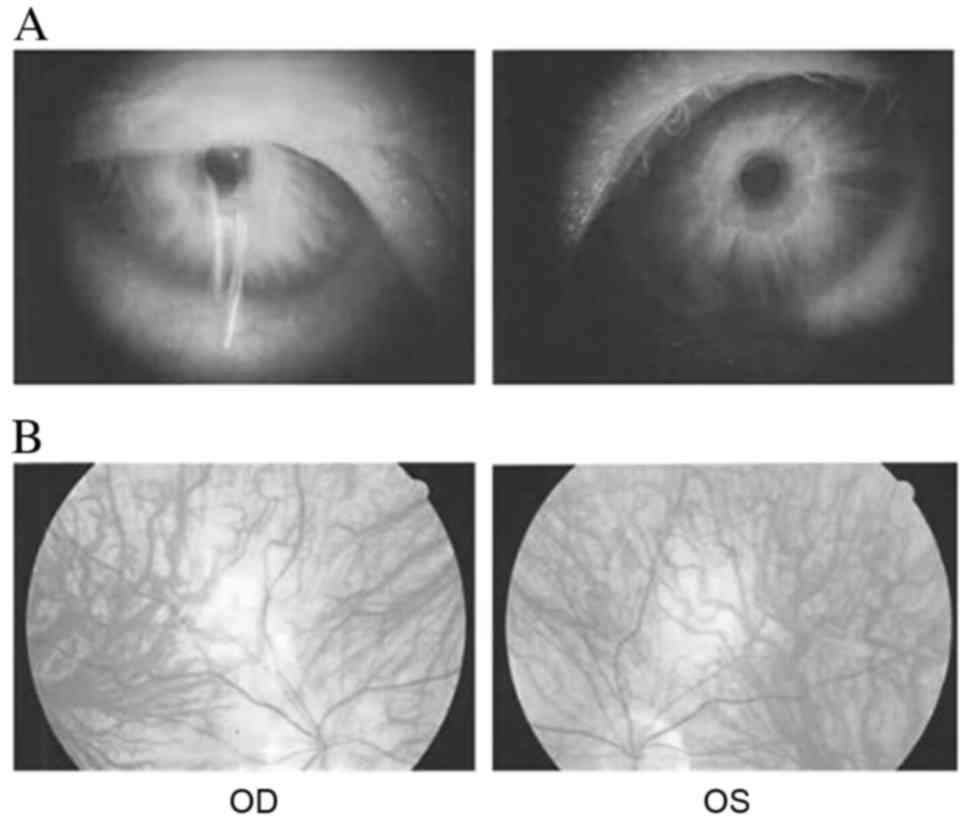
Table I. The proband (IV:3)
presented with evident horizontal nystagmus, photophobia, impaired
visual acuity, absence of the foveal pit, visible choroidal macula
vessels, and iris hypopigmentation and translucency (Fig. 2). Additionally, he had refractive
errors (−3.00D) and color vision impairment.
 | Table I.Clinical and genetic data of the two
patients with the tyrosinase gene c.896G>A (p.R299H)
mutation. |
Table I.
Clinical and genetic data of the two
patients with the tyrosinase gene c.896G>A (p.R299H)
mutation.
| Parameter | IV:2 | IV:3 |
|---|
| Gender | Female | Male |
| Age (years) | 42 | 39 |
| Genotype | Homozygous | Homozygous |
| Hair color (at
birth/at present) | White/white | White/white |
| Skin color | White | White |
| Nystagmus | + | + |
| Iris hypopigmentation
and translucency | + | + |
| Foveal
hypoplasia | + | + |
| Refractive
errors | + | + |
| Color vision
impairment | + | + |
| Photophobia | + | + |
| Impaired visual
acuity | + | + |
Mutation screening
Exome sequencing of the proband (IV:3; Fig. 1) was performed in the Chinese
family with OCA. A total of 46.37 million reads with an average
read length of 100 bp were generated from the patient; 46.33
million bases (99.92%) were aligned to the human reference
sequence. A total of 38,626 genetic variants in the coding regions
and splice sites, and 36,091 SNPs, including 17,241 in the exon
regions and 1,595 in the splice sites, were identified.
Furthermore, 2,535 indels, including 410 in the exon regions and
190 in the splice sites, were detected. A prioritization scheme was
used to detect the potential pathogenic mutation in the patient, as
previously described (12). Known
variants identified in the dbSNP137, 1,000 Genomes Project and
NHLBI ESP 6500 with a minor allele frequency >0.50% were
excluded. SIFT and PolyPhen-2 were used to predict the function of
non-synonymous variants. By applying the above filtration criteria,
the number of candidate genes was reduced by >99.03%; only 238
novel variants were identified to be potentially disease-causing,
therefore were selected for further analysis.
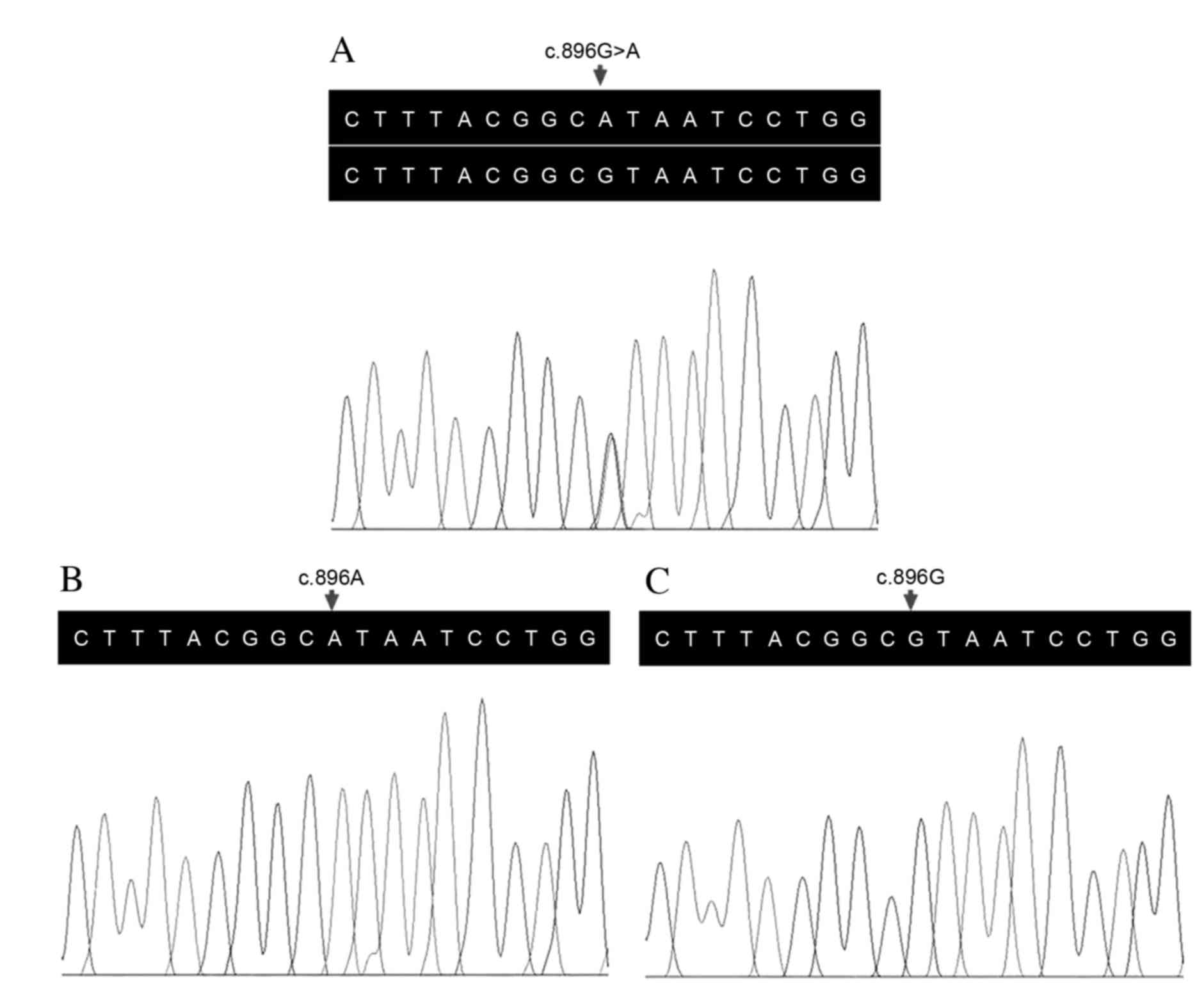
The parents of the proband were identified to carry
a heterozygous variant in the TYR gene (Fig. 3A). A c.896G>A (p.R299H) variant
in the two alleles of the TYR gene was identified in the
proband (Fig. 3B) following
validation by Sanger sequencing. One female patient (IV:2) in the
family was subsequently identified to carry the same homozygous
mutation. This variant co-segregated with disease phenotype. The
variant was absent in the control cohort, consisting of 100
ethnicity-matched unrelated controls (Fig. 3C). Arginine at position 299 was
phylogenetically conserved among various species (Fig. 4). MutationTaster predicted that the
substitution was disease-causing. These data indicated that the
TYR p.R299H variant may be the disease-associated variant in
the family investigated in the present study.
Discussion
Albinism was one of the earliest genetic disorders
to be studied. In 1903, Farabee (13) first suggested that human albinism
may be recessively inherited. The mouse and human TYR genes
were isolated in 1987. In 1989, the first mutation (nonsense
mutation) in the TYR gene responsible for human OCA was
reported by Tomita et al (14). There are 349 TYR sequence
variants recorded in the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php). A wide
spectrum of mutations, including gross and small deletions, small
insertions, small indels, and splicing, missense and nonsense
mutations, have been described, with missense mutations being the
most common.
The TYR gene (MIM 606933) is located on
chromosome 11q14.3, and contains 5 exons spanning ~65 kb of genomic
DNA. It encodes a 529-amino acid protein, tyrosinase. Tyrosinase
consists of an 18-amino acid signal peptide, two copper binding
sites, a transmembrane region at the C-terminal end and an
epidermal growth factor-like motif (15). It is a glycoprotein and a
copper-containing oxidase, and is expressed in melanocytes.
Tyrosinase serves an important role in melanin biosynthesis by
catalyzing the rate-limiting conversions of tyrosine to
dihydroxy-phenylalanine (DOPA) and from DOPA to DOPA-quinone. In
addition, it may catalyze the conversion of 5,6-dihydroxyindole to
indole-5,6 quinone (16).
Mutations in the TYR gene lead to decreased or even absent
tyrosinase enzyme activity, and subsequently, a decreased or
complete loss of melanin synthesis. Numerous missense mutations are
located in or adjacent to the copper binding sites, and interrupt
the normal function of tyrosinase, by affecting copper binding or
by disrupting the substrate binding site (17).
The present study examined a Chinese Han family with
OCA1, characterized by white hair that had not altered in color
with age, white skin, nystagmus, impaired visual acuity,
photophobia, color vision impairment, refractive errors, foveal
hypoplasia and iris hypopigmentation and translucency. The
homozygous c.896G>A variant (p.R299H, rs61754375) in the
TYR gene was identified in the two patients in the family
and was absent in the 100 unrelated ethnicity-matched controls. The
mutation was predicted to be ‘probably damaging’ by PolyPhen-2,
‘damaging’ by SIFT and ‘disease-causing’ by MutationTaster. These
results suggested that the c.896G>A variant (p.R299H) may be
pathogenic. Multiple sequence alignment demonstrated that the
arginine residue is highly conserved among vertebrates, suggesting
that it may be important for function. The parents of the patients,
with a heterozygous p.R299H mutation in the TYR gene,
presented a typical phenotype with no symptoms of OCA. Notably,
homozygous and heterozygous p.R299H mutations in the TYR
gene have been frequently identified in Chinese, Caucasian, Korean
and Arabian patients with OCA1, indicating that this location is a
mutation hotspot in OCA1 (7,17–20).
Albinism is understood to occur in the majority of
fish, birds, reptiles, amphibians and mammals (21). Molecular abnormalities in the
Tyr gene that are associated with OCA have been described in
whales, chickens, mice, rats, cats, rabbits, cattle, buffalo,
American mink, ferrets and rhesus monkeys (22). In 2005, Blaszczyk et al
(23) identified the mutation
p.R299H in the albino Wistar rat, emphasizing the functional
significance of this mutation leading to OCA.
In conclusion, the results of the present study
confirmed the clinical diagnosis of OCA1 in a Chinese family by
identifying a p.R299H mutation in the TYR gene. These
findings suggested that exome sequencing may be a cost-effective
tool for accurate diagnosis of the disease, therefore providing
genetic counseling options for individuals with OCA. Further
studies using appropriate TYR-deficient animal models may
facilitate the development of novel therapeutic strategies for the
treatment of this disease.
Acknowledgements
The authors thank the participating subjects for
their cooperation; and investigators for collecting clinical,
radiological and genetic information and DNA specimens. The present
study was supported by the Talent Projects of New Xiangya (to
H.D.).
References
|
1
|
Kamaraj B and Purohit R: Mutational
analysis of oculocutaneous albinism: A compact review. Biomed Res
Int. 2014:9054722014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khordadpoor-Deilamani F, Akbari MT,
Karimipoor M and Javadi G: Sequence analysis of tyrosinase gene in
ocular and oculocutaneous albinism patients: Introducing three
novel mutations. Mol Vis. 21:730–735. 2015.PubMed/NCBI
|
|
3
|
Wang Y, Wang Z, Chen M, Fan N, Yang J, Liu
L, Wang Y and Liu X: Mutational analysis of the TYR and OCA2 genes
in four Chinese families with oculocutaneous albinism. PLoS One.
10:e01256512015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ghodsinejad Kalahroudi V, Kamalidehghan B,
Arasteh Kani A, Aryani O, Tondar M, Ahmadipour F, Chung LY and
Houshmand M: Two novel tyrosinase (TYR) gene mutations with
pathogenic impact on oculocutaneous albinism type 1 (OCA1). PLoS
One. 9:e1066562014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gargiulo A, Testa F, Rossi S, Di Iorio V,
Fecarotta S, de Berardinis T, Iovine A, Magli A, Signorini S, Fazzi
E, et al: Molecular and clinical characterization of albinism in a
large cohort of Italian patients. Invest Ophthalmol Vis Sci.
52:1281–1289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grønskov K, Ek J and Brondum-Nielsen K:
Oculocutaneous albinism. Orphanet J Rare Dis. 2:432007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Guo X, Li W and Lian S: Four novel
mutations of TYR gene in Chinese OCA1 patients. J Dermatol Sci.
53:80–81. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei AH, Yang XM, Lian S and Li W: Genetic
analyses of Chinese patients with digenic oculocutaneous albinism.
Chin Med J (Engl). 126:226–230. 2013.PubMed/NCBI
|
|
9
|
Liu N, Kong XD, Shi HR, Wu QH and Jiang M:
Tyrosinase gene mutations in the Chinese Han population with OCA1.
Genet Res (Camb). 96:e142014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo Y, Song Z, Xu H, Yi J, Zheng W, Xiang
H, Deng X, Lv H, Gao K, Qi Y and Deng H: Heterogeneous phenotype in
a family with the FERM domain-containing 7 gene R335X mutation. Can
J Ophthalmol. 49:50–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yuan L, Wu S, Xu H, Xiao J, Yang Z, Xia H,
Liu A, Hu P, Lu A, Chen Y, et al: Identification of a novel PHEX
mutation in a Chinese family with X-linked hypophosphatemic rickets
using exome sequencing. Biol Chem. 396:27–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng W, Chen H, Deng X, Yuan L, Yang Y,
Song Z, Yang Z, Wu Y and Deng H: Identification of a novel mutation
in the Titin gene in a Chinese family with limb-girdle muscular
dystrophy 2J. Mol Neurobiol. 53:5097–5102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Farabee WC: Notes on Negro albinism.
Science. 17:751903. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomita Y, Takeda A, Okinaga S, Tagami H
and Shibahara S: Human oculocutaneous albinism caused by a single
base insertion in the tyrosinase gene. Biochem Biophys Res Commun.
164:990–996. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oetting WS and King RA: Molecular basis of
albinism: Mutations and polymorphisms of pigmentation genes
associated with albinism. Hum Mutat. 13:99–115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shah SA, Din SU, Raheem N, Daud S, Mubeen
J, Nadeem A, Tayyab M, Baloch DM, Babar ME and Ahmad J:
Identification of a novel mutation (p.Ile198Thr) in gene TYR in a
Pakistani family with nonsyndromic oculocutaneous albinism. Clin
Exp Dermatol. 39:646–648. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu J, Choy KW, Chan LW, Leung TY, Tam PO,
Chiang SW, Lam DS, Pang CP and Lai TY: Tyrosinase gene (TYR)
mutations in Chinese patients with oculocutaneous albinism type 1.
Clin Exp Ophthalmol. 38:37–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin YY, Wei AH, He X, Zhou ZY, Lian S and
Zhu W: A comprehensive study of oculocutaneous albinism type 1
reveals three previously unidentified alleles on the TYR gene. Eur
J Dermatol. 24:168–173. 2014.PubMed/NCBI
|
|
19
|
Wei AH, Zang DJ, Zhang Z, Yang XM and Li
W: Prenatal genotyping of four common oculocutaneous albinism genes
in 51 Chinese families. J Genet Genomics. 42:279–286. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei A, Wang Y, Long Y, Wang Y, Guo X, Zhou
Z, Zhu W, Liu J, Bian X, Lian S and Li W: A comprehensive analysis
reveals mutational spectra and common alleles in Chinese patients
with oculocutaneous albinism. J Invest Dermatol. 130:716–724. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anistoroaei R, Fredholm M, Christensen K
and Leeb T: Albinism in the American mink (Neovison vison) is
associated with a tyrosinase nonsense mutation. Anim Genet.
39:645–648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding B, Ryder OA, Wang X, Bai SC, Zhou SQ
and Zhang Y: Molecular basis of albinism in the rhesus monkey.
Mutat Res. 449:1–6. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Blaszczyk WM, Arning L, Hoffmann KP and
Epplen JT: A Tyrosinase missense mutation causes albinism in the
Wistar rat. Pigment Cell Res. 18:144–145. 2005. View Article : Google Scholar : PubMed/NCBI
|