Introduction
Ocular albinism type 1 (OA1) is an X-linked disorder
(1), which is the most common form
of ocular albinism, with an estimated prevalence of 1 in 60,000 at
birth (2). OA1 is characterized by
a severe reduction in visual acuity, refractive errors, nystagmus,
iris translucency, fundus hypopigmentation, foveal hypoplasia and
loss of stereoscopic vision due to misrouting of the optic fibers
at the optic chiasm (3–6).
In the sclera, the performance of the iris and
fundus is crucial. The majority of patients have evident clinical
features of pronounced pigment loss in the iris and retina, which
may be distinguished from patients with congenital motor nystagmus
(CMN). However, the performance of the iris and retinal
depigmentation is not typical and varies greatly among patients.
Additionally, congenital nystagmus is frequently clinically
misdiagnosed as OA1 or CMN in China, although the genetic and
clinical manifestations of these diseases differ significantly.
OA1 occurs due to mutations in the ocular albinism I
G protein-coupled receptor 143 (GPR143) gene (Online Mendelian
Inheritance in Man no. 300500). This G protein-coupled receptor
protein is expressed in ocular and epidermal melanocytes and may be
a melanosomal transmembrane protein (7,8).
GPR143 is a pigment cell-specific intracellular glycoprotein
consisting of 404 amino acid residues that is mutated in patients
with OA1 (9).
The aim of the present study was to identify
mutations in the exons and exon/intron junctions of the OA1 gene
using cycle sequencing, clinically evaluate the characteristics of
the OA1 patients, including female carriers and provide additional
evidence for future clinical and differential diagnoses, and
genetic counseling of this disease. A total of five mutations in
the GPR143 gene were identified in the present study, four of these
were previously unknown mutations.
Materials and methods
Patients and clinical data
A total of 8 patients with OA1 and 96 normal
controls, mainly from the south of China, participated in the
present study. These individuals, recruited between August 2008 and
July 2011, were identified by the Pediatric and Genetic Clinic of
Zhongshan Ophthalmic Center (Sun Yat-sen University, Guangzhou,
China). Informed consent conforming to the Declaration of Helsinki
was obtained from each participant prior to the study. The medical
and ophthalmic histories of the patients were obtained. A detailed
ophthalmological examination, including slit lamp photography of
the anterior segment, fundus photography, optical coherence
tomography (OCT) and examination of visual acuity, were used to
identify the clinical features of OA1.
Genetic mutation screening
Genomic DNA was prepared from leukocytes, collected
from 5 ml peripheral venous blood obtained from the patients, using
the phenol-chloroform extraction method (10). The primers (Takara Bio, Inc., Otsu,
Japan) for GPR143 (Table I) were
used with a polymerase chain reaction (PCR) Amplification kit
(Takara Bio, Inc.) to amplify the coding exons (exon 1 to exon 9)
of GPR143 and adjacent intronic sequences of the gene (human genome
build 36.3, NC_000023.10 for gDNA, NM_000273.2 for cDNA,
NP_000264.2 for protein). The PCR reaction was performed in a
thermocycler (Biometra GmbH, Göttingen, Germany) under the
following conditions: Initial denaturation at 94°C for 5 min
followed by 35–37 cycles of 94°C for 30 sec, primer-specific
annealing temperature (Table I)
for 30 sec, and 72°C for 30 sec. The final extension cycle was
performed at 72°C for 5 min. The PCR products of the exons and
adjacent intronic sequences for each patient were sequenced with
the ABI BigDye Terminator cycle sequencing kit v3.1 (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol, using an ABI Prism 3100
sequencer and the results were manually confirmed. The sequencing
results from each patient and the consensus sequences of GPR143
from the National Centre for Biotechnology Information (NCBI) human
genome database (NM_000273.2) were imported into the SeqMan II
software (DNAStar, Inc., Madison, WI, USA), using the Lasergene 8.0
package (DNAStar, Inc.) and were aligned to identify genetic
variations. Each mutation was confirmed by bidirectional
sequencing, and novel mutations were named according to the
nomenclature recommended by the Human Genome Variation Society
(http://www.hgvs.org/).
 | Table I.Oligonucleotides used for GPR143
amplification. |
Table I.
Oligonucleotides used for GPR143
amplification.
| Exon | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) | Product length
(bp) | Annealing temperature
(°C) |
|---|
| GPR143-exon1-a |
CTCCTCCGCCCGCCCAAGCATCAC |
CCCAGGCAGCCGAGAAGGTC | 464 | 67.2 |
| GPR143-exon1-b |
CCGCGCCTAGGGACCTTCTGCT |
AACCCGCGGGCCTCTCGTCCTCAC | 399 | 67.2 |
| GPR143-exon2 |
CTTTCTTCCTTTTCCCTCCTTGTC |
GTTTGCTGCTGCTGCGATTTG | 360 | 60.8 |
| GPR143-exon3 |
CACGTGCGGCTTCCTGAC |
TTGGCCTCTTATAAAAATGA | 385 | 63.8 |
| GPR143-exon4 |
GGGCTTTCCTCTGTGTACATTTTC |
CCCTGAGACAACGGCCTAACC | 334 | 60.8 |
| GPR143-exon5 |
GCATTTCCCTTTTTGTTCTCATCC |
AGGCCTGCACATTTTCATTTATTG | 406 | 60.8 |
| GPR143-exon6 |
TTGCTTCCTGCCCCTCTGG |
ACTTGCTCCCCTGTCCTCTGT | 400 | 62.3 |
| GPR143-exon7 |
TGCACCTGGCCCTCTTAGTTTC |
TCAGGAGGCCAAGACAGAGGAT | 441 | 66.6 |
| GPR143-exon8a |
AAACCAACCCACCAACCAGTCAAC |
GCATGCTCAGGGCTTCGTCA | 395 | 67.2 |
| GPR143-exon8b |
CCAGCCCAGGGATTTCTCTT |
ACCCCGCCATGCACAGGAC | 329 | 67.2 |
| GPR143-exon9 |
AGCTGATGACAAACCTGCTAG |
CCCTTTCTCCTATCCTAAAG | 330 | 60.8 |
| GPR143-mutation
(c.275_276int CGCTGC) |
CAGCTCGTGCTGAGCTTCC |
CTCACTCCATCACGGAACA | 335 | 57.2 |
| GPR143-mutation
(c.658+2T>G) |
TCAGCAGCAGATTTATGCATTTCCC |
CTGGAGAATTCCAGGACAACATGTG | 330 | 62.9 |
| GPR143-mutation (c.17
T>C) |
CCGGCGGGGTCCTGGCACAC |
CAGGCAGAGCGCGTGGAAGG | 148 | 61.8 |
| GPR143- mutation
(c.333G>A) |
ACAGCGTCTCGGATATGAACCAC |
CTGCTGCTGCGATTTGAGGA | 125 | 60.4 |
Determination of changes in genetic
information
The sequences containing genetic variations and the
standard sequences obtained from the NCBI human genome database
were imported into MapDraw of the Lasergene package to identify the
impact on amino acid coding. To estimate the conservation of the
mutation sites using ClustalW, protein sequences from different
species were identified from the NCBI website and entered into the
Lasergene package MegAlign program (DNAStar, Inc.). Differences in
the amino acids and the pathogenicity of each mutation were
evaluated using the Blosum 62 matrix (http://www.uky.edu/Classes/BIO/520/BIO520WWW/blosum62.htm)
and the PolyPhen (http://genetics.bwh.harvard.edu/pph/) analytical tool,
respectively.
Heteroduplex-single strand
conformational polymorphism (HA-SSCP) analysis
The genetic variations that were identified in the
GPR143 gene were evaluated in the 96 normal controls and in the
patients that screened positive for genetic variations using
HA-SSCP as previously described (11–13).
DNA fragments of the mutated sites were PCR-amplified
aforementioned. The PCR products were mixed with an equal volume of
gel loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol
blue and 0.05% xylene cyanol), denatured at 95°C for 5 min and
immediately placed on ice for 5 min. Samples (3 µl/well) were
loaded directly onto 8% polyacrylamide gels and gel electrophoresis
was conducted at 40 W for 8 h, at room temperature, in a solution
of 0.5X Tris/borate/EDTA buffer.
Results
Mutation analysis
In the present study, five patients with OA1 were
determined to have genetic mutations in the GPR143 gene, with four
out of the five mutations being novel. The screening rate was 62.5%
for the following mutations: C.333G>A (p.W111X), c.353G>A
(p.G118E) (known mutation), C.658+2T>G (splice mutation),
c.215_216 insCGCTGC (p.71-72insAA) and c.17T>C (p. L6P)
(Fig. 1).
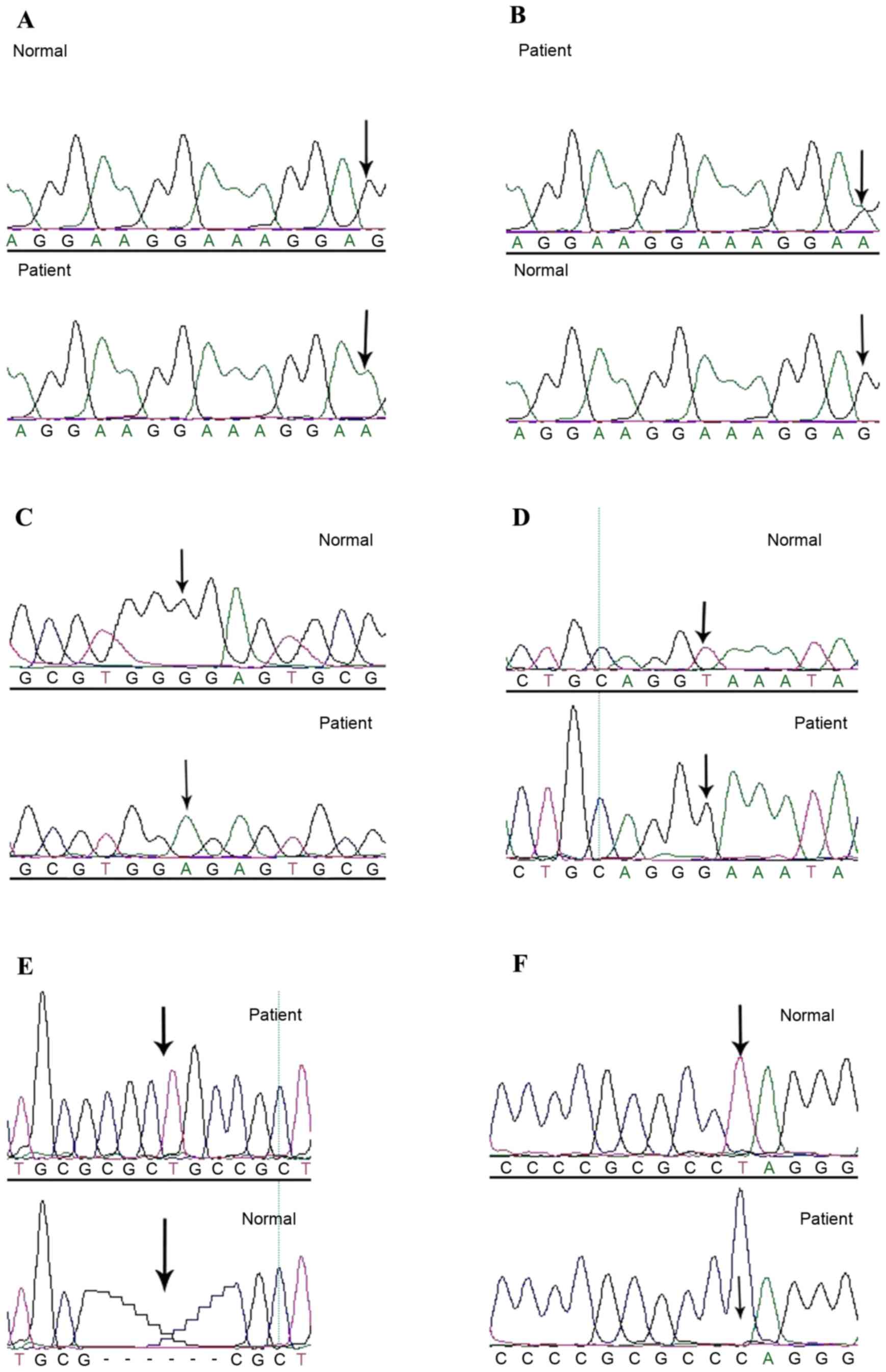 | Figure 1.GPR143 cycle sequencing results. (A)
Homozygous c.333G>A (p.W111X) mutation, indicated by the lower
arrow, was identified in the OA1 patient and confirmed by
bidirectional sequencing. The upper arrow represents the
corresponding normal allele from an unaffected control individual.
(B) Heterozygous c.333G>A (p.W111X) mutation, indicated by the
upper arrow, was identified in the OA1 patient and confirmed by
bidirectional sequencing. The lower arrow represents the
corresponding normal allele from an unaffected control individual.
(C) c.353G>A (p.G118E) mutation was identified in the OA1
patient, which is indicated by the lower arrow, and confirmed by
bidirectional sequencing. The upper arrow indicates the
corresponding normal sequence from an unaffected control
individual. (D) c.658+2T>G mutation, which is indicated by the
lower arrow, was identified in the OA1 patient and confirmed by
bidirectional sequencing. The upper arrow highlights the
corresponding normal sequence from an unaffected control
individual. (E) c.215_216 ins CGCTGC (p.71-72intAA) mutation, which
is indicated by upper arrow, was identified in the OA1 patient and
confirmed by bidirectional sequencing. The lower arrow represents
the corresponding normal sequence from an unaffected control
individual. (F) c.17T>C (p. L6P) mutation, which is indicated by
the lower arrow, was identified in the OA1 patient and confirmed by
bidirectional sequencing. The upper arrow indicates the
corresponding normal sequence from an unaffected control
individual. OA1, ocular albinism type 1. |
The protein in this position was highly conserved
based on the comparative analysis with 7 orthologs from different
mammalian species (Fig. 2).
HA-SSCP was also used to screen the 96 unaffected control
individuals (Fig. 3); however, no
mutations were identified.
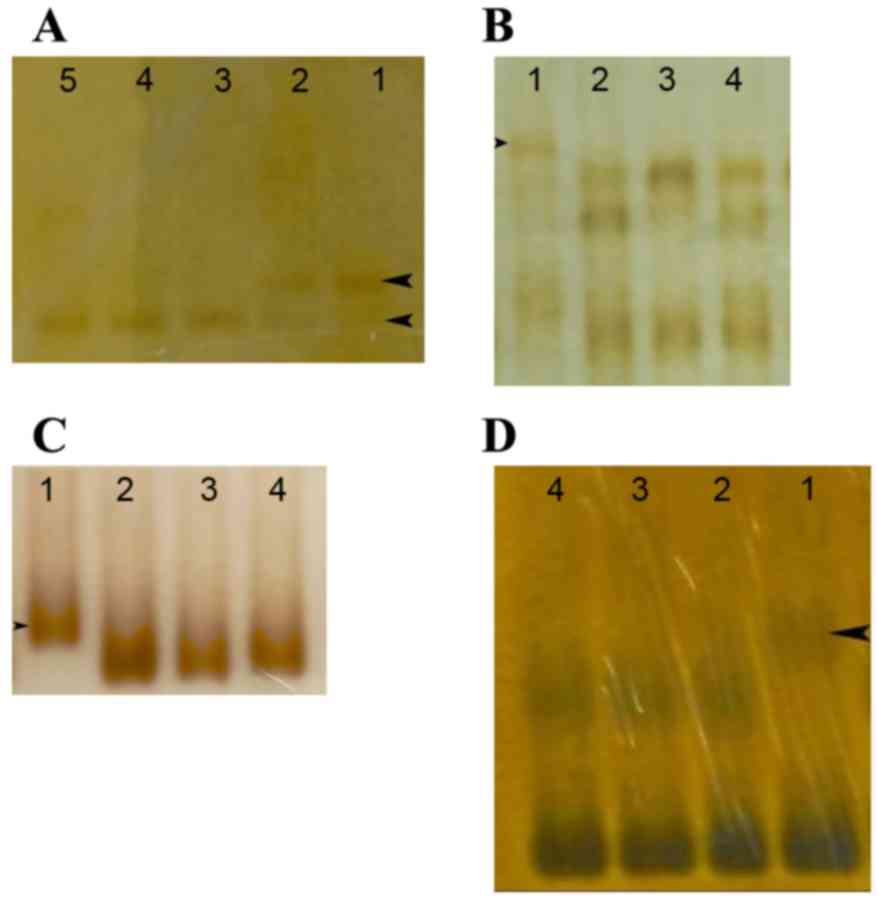 | Figure 3.HA-SSCP analysis of the GPR143 genetic
mutations in patients with OA1, arrows indicate the differential
bands. (A) Lane 1, band from a patient; lane 2, the bands from a
carrier with the c.333G>A (p.W111X) mutation. Lanes 3–5, bands
from normal controls. (B) Lane 1, band from a patient with the
c.658+2T>G mutation. Lanes 2–4, bands from normal controls. (C)
HA-SSCP analysis of the GPR143 genetic mutation in a patient with
OA1. Lane 1, band from a patient with the c.215_216 int CGCTGC
(p.71-72intAA) mutation. Lanes 2–4, bands from normal controls. (D)
Lane 1, band from a patient with the c.17T>C (p. L6P) mutation.
Lanes 2–4, bands from normal controls. HP-SSCP, heteroduplex-single
strand conformation polymorphism; OA1, ocular albinism type 1;
GPR143, G protein-coupled receptor 143. |
PolyPhen and Blosum 62 analyses were used to
determine if whether the missense mutations identified in the
present study resulted in changes in the residue weight at the
protein level. Blosum 62 was used to evaluate the missense
mutations and PolyPhen was used to determine whether an amino acid
change had a ‘probable damaging’ effect. It was determined that the
p.G118E and p.L6P mutations may be damaging with a score 0.999 by
PolyPhen and they led to a change in the residue weighting from 6
to-2 and from 4 to-3 by Blosum 62, respectively.
Clinical phenotype
In the present study, all patients with OA1
exhibited different degrees of horizontal nystagmus; however, they
typically manifested a severe decline in visual acuity. Uncorrected
visual acuity ranged from 0.1 to 0.3, and corrected visual acuity
ranged from 0.1 to 0.4 (Table
II).
| Table II.Visual acuity of patients with
OA1. |
Table II.
Visual acuity of patients with
OA1.
| Patient ID | Gender | Visit age
(years) | Naked vision | Corrected vision |
|---|
| QT299 | Male | 6 months | Perception of
light | N/A |
| QT418 | Male | 7 | 0.2/0.2 | 0.2/0.2 |
| QT593 | Male | 43 | 0.1/0.1 | 0.1/0.1 |
| QT638 | Female | 10 | NA | 0.4/0.4 |
| QT646 | Male | 8 months | Perception of
light | N/A |
| QT676 | Male | 2 months | N/A | N/A |
| QT720 | Male | 10 |
0.1/0.3− |
0.3/0.3+ |
| QT751 | Male | 7 | 0.1/0.1 | 0.2/0.2 |
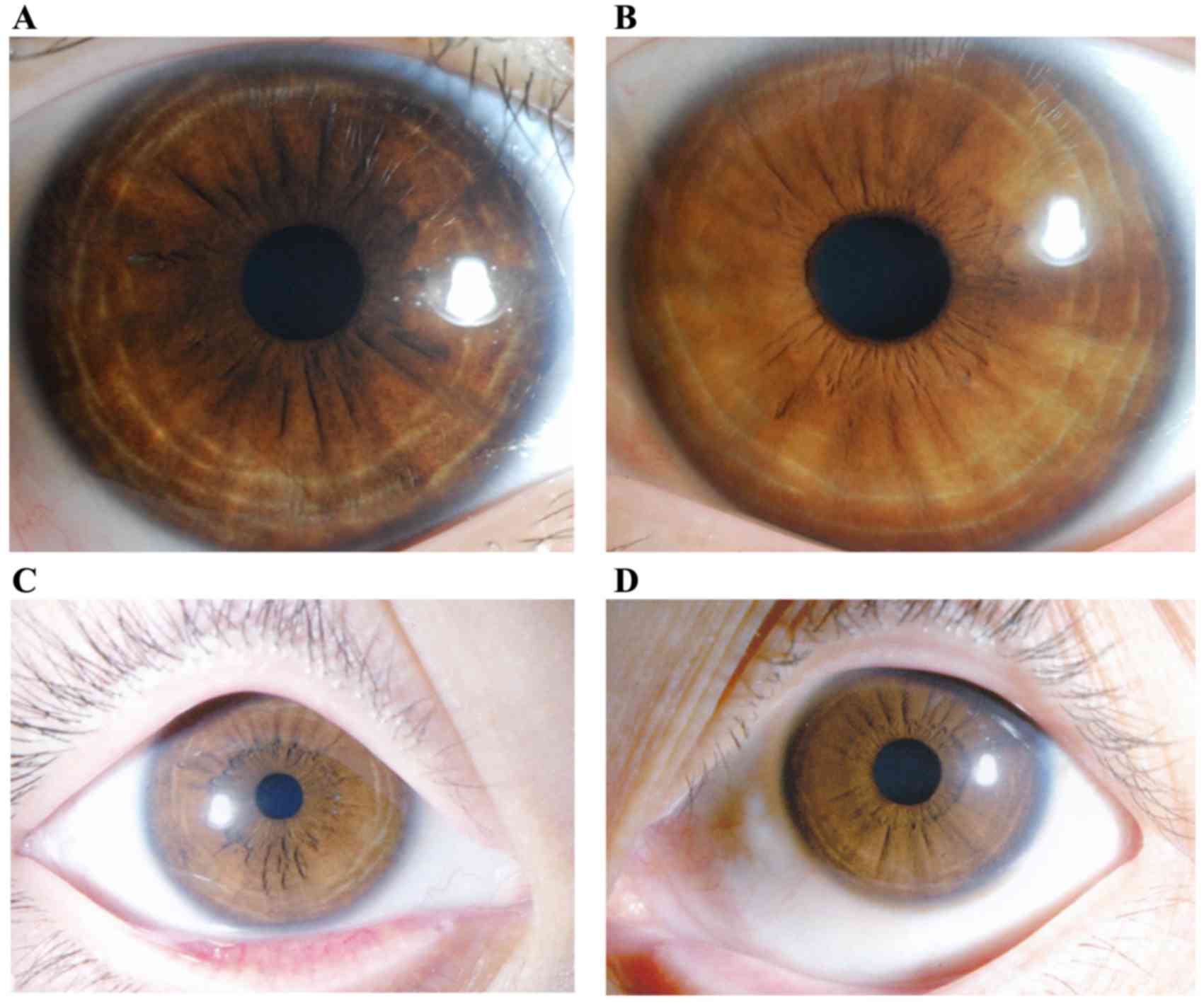
Upon slit lamp examination, different degrees of
abnormal pigmentation were observed in the irises of patients.
Peripheral iris depigmentation, such as a ring (which was irregular
in some patients) or fan, was observed, although the pigmentation
of the iris around the pupil area appeared normal and the overall
iris color appeared darker than normal (Fig. 4A). However, some patients did not
have obvious depigmentation of the iris; therefore, determining
whether the patient had OA1 or was healthy was difficult in such
cases (Fig. 4B). Iris
depigmentation did not lead to iris transillumination in the
present study (Fig. 4).
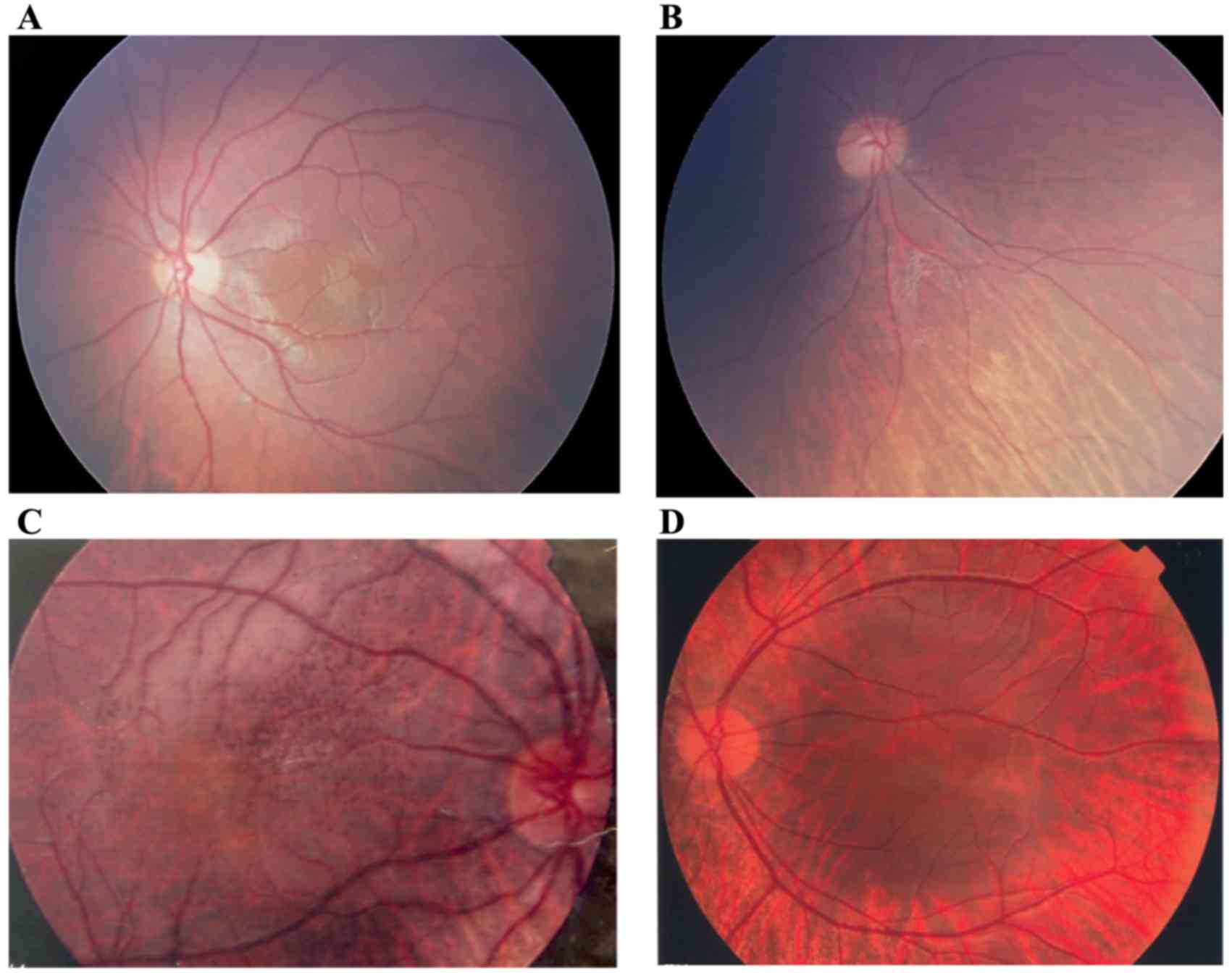
Based on the findings of the present study, patients
with OA1 have different degrees of retinal depigmentation as
determined by fundus examination. Hypopigmentation of the retina
was universal and the choroidal blood vessels were clearly visible,
which is different from the phenotype of oculocutaneous albinism.
Diffuse reduction of pigmentation in the retina, but not in the
choroidal vessels, was clearly observed, as the color of the retina
was slightly redder than the healthy tissue. The entire retina
exhibited features similar to highly myopic eyes, such as irregular
retinal depigmentation and hyperpigmentation in some regions. In
particular, the retina exhibited regional depigmentation around the
optic disc and peripheral retina, where the choroidal vessels may
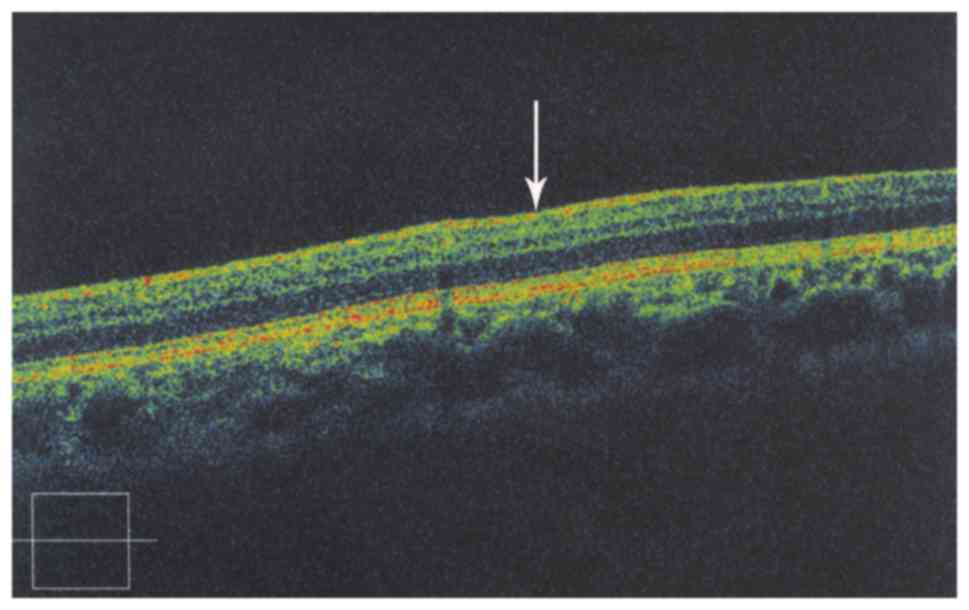
be observed (Fig. 5). The macular
foveal structure was not visible in OCT examination (Fig. 6).
Discussion
The GPR143 gene encodes a pigment cell-specific G
protein-coupled receptor protein (GPCR) that is localized
intracellularly in melanosomes. OA1 has been reported to regulate
melanosome formation in the biogenesis stage, as well as the
motility of melanosomes (14).
Genetic analysis has detected >60 mutations in
the GPR143 gene in sporadic and familial cases. Previous studies
have determined that the GPR143 mutations are deletion mutations
and that most of these deletion mutations are point mutations
(1,15–17).
In these mutants, large fragment deletion mutations were not
uncommon. However, the present study identified one case of an
insertion mutation. A potential explanation may be due to ethnic
differences and the biased selection of the volunteers in the
present study. A previous study demonstrated that the OA1 protein
was a GPCR protein that spans the integral membrane seven times,
with its C terminus oriented towards the cytoplasm, and this
protein may function as an intracellular protein localized in
endolysosomal organelles (18).
In the present study, the GPR143 mutations
c.333G>A (p.W111X), c.353G>A (p.G118E) and c.17T>C (p.L6P)
were located in the loop regions within the lumen of melanosomes or
lysosomes. The missense mutations, p.G118E and p.L6P, were
relatively highly conserved in seven different mammalian species
and had high pathogenicity, as determined by Blosum 62 and
PolyPhen. It is possible that the missense mutations c.353G>A
(p.G118E) and c.17T>C (p.L6P) may alter genetic receptor
binding, which may lead to functional changes; however, further
investigation is required to confirm this conclusion as the
predictions of the current study are based only on bioinformatics
data, requiring experimental support. The insertion mutation,
c.215_216 ins CGCTGC (p.71-72insAA), is located in transmembrane
region (TM) 2. Systematic and experimental topology analyses may be
used in future studies to identify the function of the TM domains
in terms of GPCR functionality, screen ligands and identify
proteins located in OA1 signaling pathways.
The GPR143 gene has been previously reported as
being characterized by a high mutation rate of up to 95% (19). The present study identified a
mutation rate of 62.5%, which may be primarily due to the small
sample size, probands from sporadic cases, or bias in the selection
of volunteers. GPR143 is the only identified pathogenic gene that
may lead to the development of OA1 and, as it has few axons, this
decreases the cost of genetic testing.
Furthermore, the positive screening rate of the
GPR143 gene is relatively high. All of the above contribute to the
relative ease of genetic testing for this gene and disease. The
results from a genetic test may be used to further diagnose
patients with existing clinical criteria and also aid in prenatal
diagnosis.
The present study identified different degrees of
hypopigmentation in the iris and retina, which was different from
the sclera. Iris and fundus differences observed between the
pigmented and white regions may be due to ethnic differences
(20,21). Therefore, according to the
diagnostic criteria for OA1 in China, the use of clinical features
based solely on the conditions of the sclera may lead to a
misdiagnosis or missed diagnosis.
In conclusion, the findings of the present study
expand the genetic mutation spectrum for the GPR143 gene and
provide additional evidence for the clinical diagnosis,
differential diagnosis and genetic counseling for OA1. Further
investigation of the functional properties of the GPR143 protein,
particularly by generating animal models, will provide information
regarding how genetic mutations alter the molecular interactions
involved in the generation of motor nystagmus. The analysis of
these interactions may help to elucidate the underlying mechanisms
of the disease.
Acknowledgments
The authors would like to thank the patients and
family members for their participation. The present study was
supported in part by the National Natural Science Foundation of
China (grant nos. 81170847 and 81270972).
References
|
1
|
Schnur RE, Gao M, Wick PA, Keller M, Benke
PJ, Edwards MJ, Grix AW, Hockey A, Jung JH, Kidd KK, et al: OA1
mutations and deletions in X-linked ocular albinism. Am J Hum
Genet. 62:800–809. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosenberg T and Schwartz M: X-linked
ocular albinism: Prevalence and mutations-a national study. Eur J
Hum Genet. 6:570–577. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Donnell FE Jr, King RA, Green WR and
Witkop CJ Jr: Autosomal recessively inherited ocular albinism. A
new form of ocular albinism affecting females as severely as males.
Arch Ophthalmol. 96:1621–1625. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cortin P, Tremblay M and Lemagne JM:
X-linked ocular albinism: Relative value of skin biopsy, iris
transillumination and funduscopy in identifying affected males and
carriers. Can J Ophthalmol. 16:121–123. 1981.PubMed/NCBI
|
|
5
|
Lam BL, Fingert JH, Shutt BC, Singleton
EM, Merin LM, Brown HH, Sheffield VC and Stone EM: Clinical and
molecular characterization of a family affected with X-linked
ocular albinism (OA1). Ophthalmic Genet. 18:175–184. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sallmann GB, Bray PJ, Rogers S, Quince A,
Cotton RG and Carden SM: Scanning the ocular albinism 1 (OA1) gene
for polymorphisms in congenital nystagmus by DHPLC. Ophthalmic
Genet. 27:43–49. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schiaffino MV, d'Addio M, Alloni A,
Baschirotto C, Valetti C, Cortese K, Puri C, Bassi MT, Colla C, De
Luca M, et al: Ocular albinism: Evidence for a defect in an
intracellular signal transduction system. Nat Genet. 23:108–112.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Samaraweera P, Donatien PD, Qazi S,
Kobayashi T, Hearing VJ, Panthier JJ and Orlow SJ: Identification
and characterization of a melanocyte-specific novel 65-kDa
peripheral membrane protein. Eur J Biochem. 266:924–934. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
d'Addio M, Pizzigoni A, Bassi MT,
Baschirotto C, Valetti C, Incerti B, Clementi M, De Luca M,
Ballabio A and Schiaffino MV: Defective intracellular transport and
processing of OA1 is a major cause of ocular albinism type 1. Hum
Mol Genet. 9:3011–3018. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan J and Dennin RH: High homologous
nucleotide to GBV-C was amplified from DNA of MT2 and HeLa cells
and PBMC of human and chimpanzee. Acta Pharmacol Sin. 22:320–326.
2001.PubMed/NCBI
|
|
11
|
Zhang Q and Minoda K: Detection of
congenital color vision defects using heteroduplex-SSCP analysis.
Jpn J Ophthalmol. 40:79–85. 1996.PubMed/NCBI
|
|
12
|
Stone EM: Leber congenital amaurosis-a
model for efficient genetic testing of heterogeneous disorders:
LXIV edward jackson memorial lecture. Am J Ophthalmol. 144:791–811.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sale MM, Craig JE, Charlesworth JC,
FitzGerald LM, Hanson IM, Dickinson JL, Matthews SJ, Heyningen Vv,
Fingert JH and Mackey DA: Broad phenotypic variability in a single
pedigree with a novel 1410delC mutation in the PST domain of the
PAX6 gene. Hum Mutat. 20:3222002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giordano F, Bonetti C, Surace EM, Marigo V
and Raposo G: The ocular albinism type 1 (OA1) G-protein-coupled
receptor functions with MART-1 at early stages of melanogenesis to
control melanosome identity and composition. Hum Mol Genet.
18:4530–4545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bassi MT, Bergen AA, Bitoun P, Charles SJ,
Clementi M, Gosselin R, Hurst J, Lewis RA, Lorenz B, Meitinger T,
et al: Diverse prevalence of large deletions within the OA1 gene in
ocular albinism type 1 patients from Europe and North America. Hum
Genet. 108:51–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schiaffino MV, Bassi MT, Galli L, Renieri
A, Bruttini M, De Nigris F, Bergen AA, Charles SJ, Yates JR, Meindl
A, et al: Analysis of the OA1 gene reveals mutations in only
one-third of patients with X-linked ocular albinism. Hum Mol Genet.
4:2319–2325. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rudolph G, Meindl A, Bechmann M, Schworm
HD, Achatz H, Boergen KP, Kampik A, Berninger T and Meitinger T:
X-linked ocular albinism (Nettleship-Falls): A novel 29-bp deletion
in exon 1. Carrier detection by ophthalmic examination and DNA
analysis. Graefes Arch Clin Exp Ophthalmol. 239:167–172. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sone M and Orlow SJ: The ocular albinism
type 1 gene product, OA1, spans intracellular membranes 7 times.
Exp Eye Res. 85:806–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Camand O, Boutboul S, Arbogast L, Roche O,
Sternberg C, Sutherland J, Levin A, Héon E, Menasche M, Dufier J
and Abitbol M: Mutational analysis of the OA1 gene in ocular
albinism. Ophthalmic Genet. 24:167–173. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Donnell FE Jr, Green WR, Fleischman JA
and Hambrick GW: X-linked ocular albinism in Blacks. Ocular
albinism cum pigmento. Arch Ophthalmol. 96:1189–1192. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shiono T, Tsunoda M, Chida Y, Nakazawa M
and Tamai M: X linked ocular albinism in Japanese patients. Br J
Ophthalmol. 79:139–143. 1995. View Article : Google Scholar : PubMed/NCBI
|