Introduction
There are five human RecQ-like proteins (RECQL1,
BLM, WRN, RECQL4 and RECQ5), each having 3′ to 5′ DNA helicase
activity but little sequence similarity outside the helicase motifs
(1). Three of these helicases
encode causative genes for Bloom syndrome (BLM), Werner syndrome
(WRN) and Rothmund-Thomson syndrome (RECQL4), respectively. These
three syndromes show genomic instability and cancer susceptibility,
but each also has distinctive features (2). RECQL4 is the causative gene for
Rothmund-Thomson syndrome (OMIM 266280) characterized by
poikiloderma and skeletal defects. Homozyogous mutations or
compound heterozygous mutations of the RECQL4 gene causes
Rothmund-Thomson syndrome (3,4).
However, mutations in the RECQL4 gene have
been associated with two other recessive disorders: One is
RAPADILINO syndrome (OMIM 266280) which is characterized by radial
hypoplasia, patella hypoplasia and arched plate, diarrhoea and
dislocated joints, little size and limb malformation, slender nose
and normal intelligence (4). The
other is Baller-Gerold syndrome (BGS) (OMIM 218600) characterized
by radial aplasia/hypoplasia and craniosynostosis (5). Three syndromes have overlapping
features, such as short stature and radial ray abnormalities
(6).
In Japan there have been no reports on BGS. We have
performed a nationwide questionnaire based study of BGS. Three BGS
patients were identified: one was the sporadic case with BGS, and
the remaining two were brothers. In this patient, we found for the
first time a homozygous large deletion in the RECQL4 gene in
Japan.
Materials and methods
Patients and questionnaire
From 2012 to 2014, a preliminary questionnaire for
soliciting information about BGS patients was sent to 1,407
Departments of 515 Pediatrics, 515 Dermatology Departments and 377
Cancer Hospitals in Japan. The response rate was 83, 68 and 36%,
respectively. Parents of the three patients suspected of having GBS
were requested to fill out more extensive questionnaire to obtain
detailed information on the three patients. The analyses were
approved by the ethics committee of Nagara Medical Center, and
written informed consent was obtained from the patients and/or
their parents.
Genetic analysis
Peripheral blood monocytes cells (PBMCs) were
separated using Ficoll-Paque (Amersham Bioscience, Uppsala,
Sweden). Genomic DNA from PBMCs of the patient and his parents were
prepared using a Sepa Gene kit (Sankyo Jyunyaku, Tokyo, Japan).
Amplification of the full-length RECQL4 gene region was performed
using PrimeSTAR GXL DNA polymerase (Takara Bio, Inc., Shiga,
Japan), using primers 5′-ATTGGCTGCTTGTCCGAG-3′ and
5′-GCCTGGAATATGTGATGTGC-3′. The PCR products were electrophoresed
on 0.7% agarose gel, and were also sequenced using Big Dye
Terminator v3.1 (Thermo Fisher Scientific, Waltham, MA, USA), using
primers 5′-GGTGAGCCATATGTGAACTGG-3′ and
5′-CACTGCATCCACAGAGCAAG-3′.
Results
Three patients in two families were identified to be
affected by BGS by the questionnaire-based survey in Japan. One
family had a 3-year-old older brother (case 1) and a 1-year-old
younger brother (case 2) with BGS. The older brother showed
craniosynostosis, thumb hypoplasia, radial ray defects and
imperforate anus and nasolacrimal duct malposition. Operations were
performed for craniosynostosis, imperforate anus and nasolacrimal
duct malposition. The younger brother showed left ptosis and was
diagnosed as having BGS on the basis of his clinical features and
the finding of the X-ray examination of the systemic bone. Their
RECQL4 genes were not analyzed because informed consents
were not obtained.
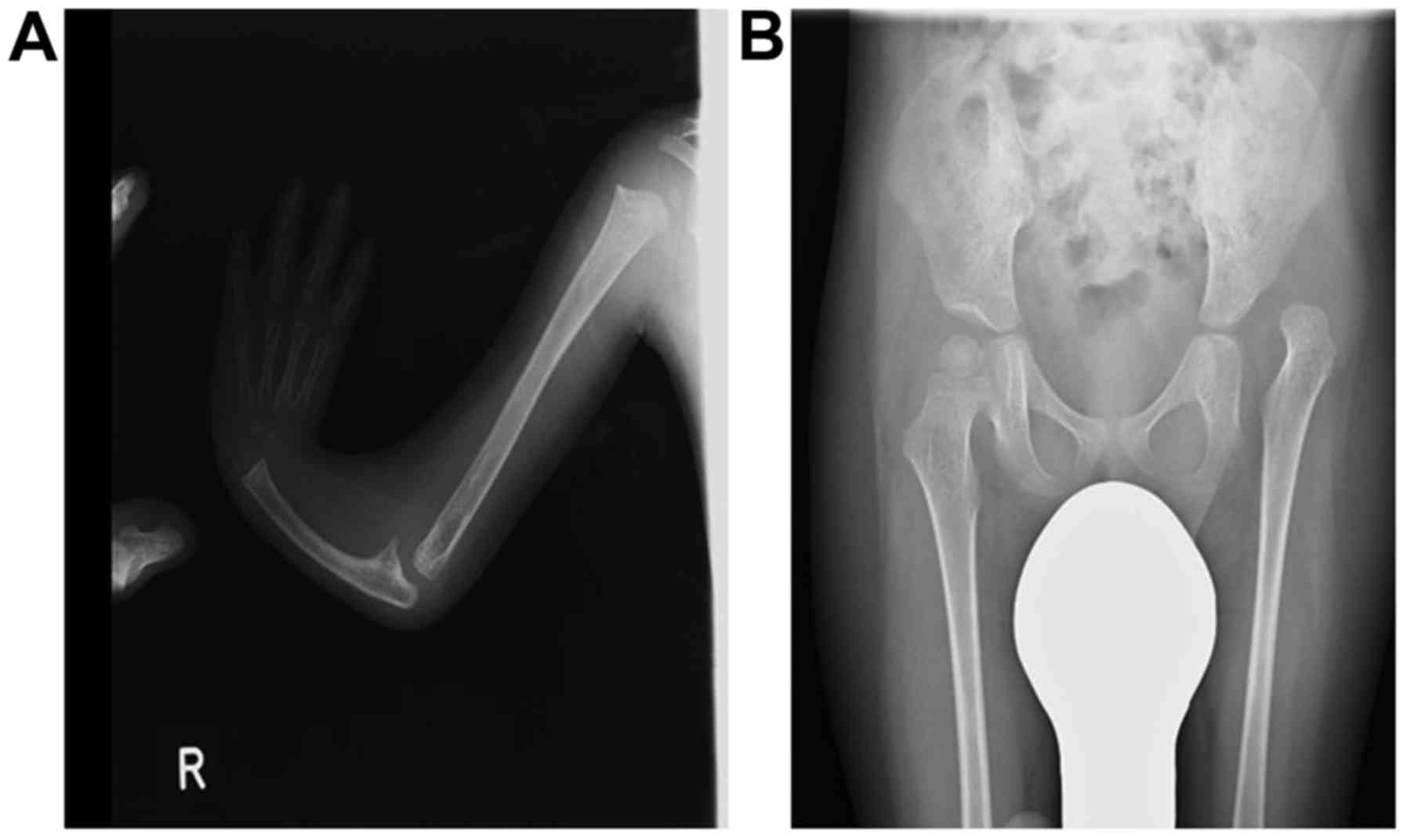
Case 3 was a 4-year-old boy. In addition to
craniosynostosis and thumb hypoplasia (Fig. 1), he showed left hip dislocation,
left knee joint dislocation, bent elbows, and excessive hand
abduction. His intelligence is within the normal range, as
determined by a new edition K-type development inspection. At 4
years of age he was still not ambulatory but could sit.
From his clinical features and bone X-ray
examination findings he was suspected of having BGS. Therefore, his
RECQL4 gene was analyzed. His RECQL4 gene showed a
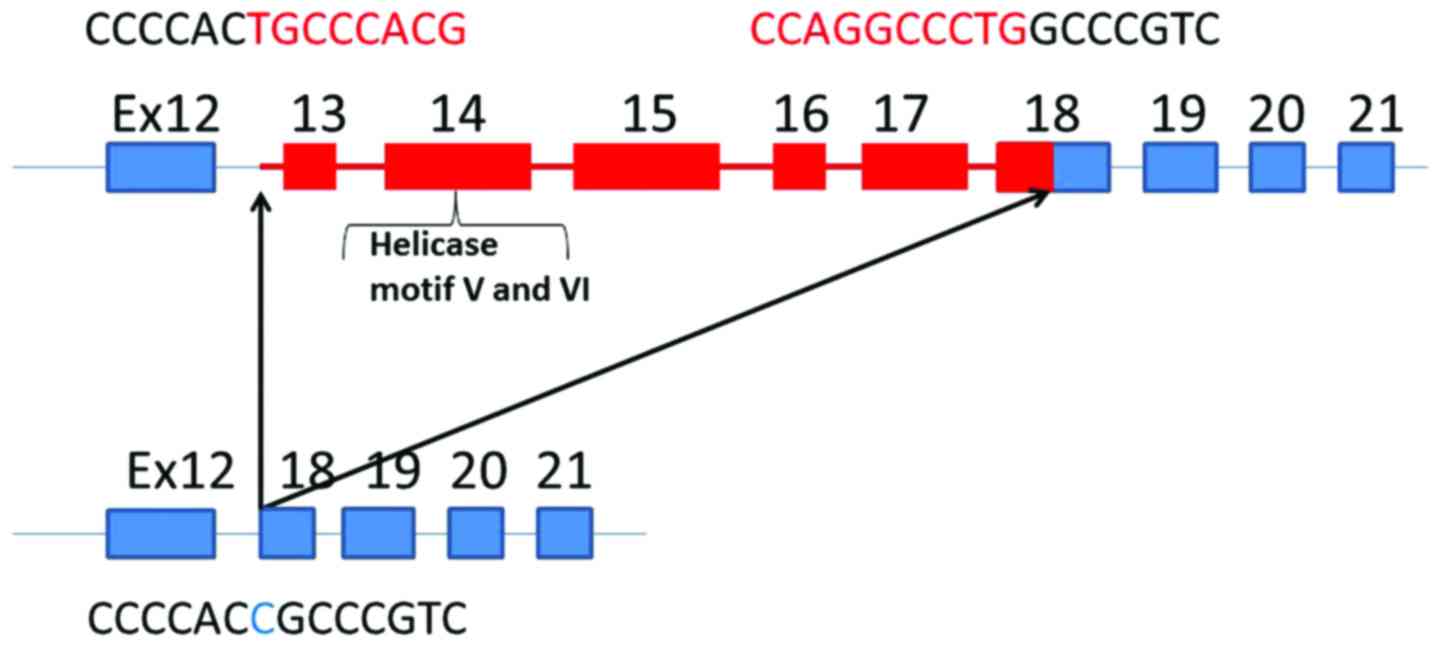
1,614 bp homozygous deletion and 1 bp (G) insertion
(NC_000008.10:g.145737562_145739175delinsC) (Fig. 2A). Deleted DNA spanned from intron
12 to the former part of exon 18, which contained the helicase
motif (Fig. 3). Agarose gel
analysis (Fig. 2B) showed that his
father and mother carried the wild type and mutant RECQL4
genes, respectively.
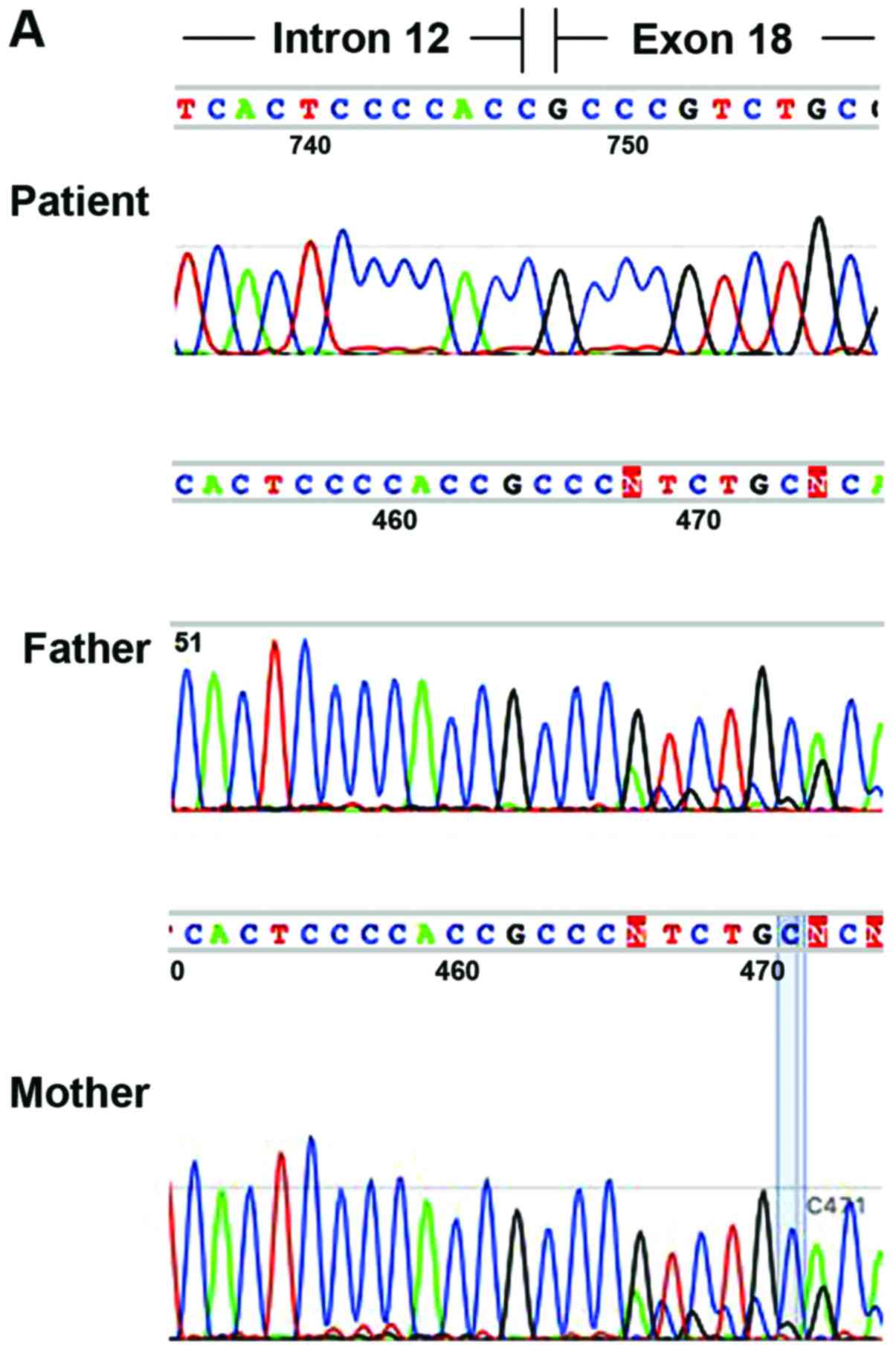 | Figure 2.(A) Chromatogram of DNA sequences at
DNA break point (NC_000008.10:g.145737562_145739175delinsC). (B)
Large homozygous deletion of RECQL4 gene in case 3 detected
by agarose gel electrophoresis. λ/H, HindIII digested DNA
marker, M, DNA marker, Co, control, Pt, case 3, Mo, Mother, Fa,
Father. |
Discussion
The prevalence of BGS is unknown; it is probably
less than 1:1,000,000 (7). This
rarity of BGS makes its diagnosis difficult in some cases. Piard
et al (8). Showed that the
mean age at referral for BGS was 7 years. In our nationwide survey
only two families were identified to have members affected by BGS.
All of the affected members had craniosynostosis and radial ray
defects. On the other hand Rothmund-Thomson syndrome and RAPADILINO
syndrome are two recessively inherited syndromes whose clinical
features overlap those of BGS. Rothmund-Thomson syndrome is
characterized by poikiloderma. Radial ray hypoplasia or absent
thumbs occur in a minority of cases. RAPADILINO syndrome is
characterized by radial ray defects. BGS is characterized by
craniosynostosis in association with radial hypoplasia. Our cases
showed radial ray defect with carniosyonostosis without
poikiloderma which is consistent with the characteristic of
BGS.
The RECQL4 gene in our genetically analyzed
patient showed homozygous deletion from intron 12 to the former
part of exon 18 resulting in the deletion of amino acids after
Ala687. To our knowledge this large deletion of RECQL4 gene
has not been reported in other BGS patients. This deletion spans
the RecQ helicase motif from IV to VI. The deleted RECQL4 protein
in the patient might not be functional. To the best of our
knowledge, this deletion in the RECQL4 gene has not been
reported. Piard et al reported that no RECQL4 mutations were
found in their BGS group without poikiloderma. However, our patient
had café-au-lait-like spots but not poikiloderma (8). The relationship between poikiloderma
and RECQL4 gene mutation should be further examined.
In our patient the parents had heterozygous
RECQL4 gene mutation without evident consanguinity. A
molecular study-based diagnosis is powerful tool for genetic
counselling of individuals affected by BGS. Cao et al
Reported the case of BGS prenatally diagnosed (9). The cases of BGS diagnosed on the
basis of molecular genetics should be accumulated.
Acknowledgements
This study was supported in part by Health and Labor
Science Research Grants for Research on Intractable Diseases from
The Ministry of Health, Labour and Welfare of Japan (H23-075).
References
|
1
|
Nakayama K: RecQ family helicases: Roles
as tumor suppressor proteins. Oncogene. 21:9008–9021. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaneko H and Kondo N: Clinical features of
Bloom syndrome and function of the causative gene, BLM helicase.
Expert Rev Mol Diagn. 4:393–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kitao S, Shimamoto A, Goto M, Miller RW,
Smithson WA, Lindor NM and Furuichi Y: Mutations in RECQL4 cause a
subset of cases of Rothmund-Thomson syndrome. Nat Genet. 22:82–84.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larizza L, Roversi G and Volpi L:
Rothmund-Thomson syndrome. Orphanet J Rare Dis. 5:22010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Maldergem L, Verloes A, Lejeune L and
Gillerot Y: The Baller-Gerold syndrome. J Med Genet. 29:266–268.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Maldergem L, Siitonen HA, Jalkh N,
Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL,
et al: Revisiting the craniosynostosis-radial ray hypoplasia
association: Baller-Gerold syndrome caused by mutations in the
RECQL4 gene. J Med Genet. 43:148–152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van Maldergem L; Pagon RA, Adam MP,
Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT,
Mefford HC, Smith RJH and Stephens K: Baller-Gerold Syndrome.
Seattle (WA): University of Washington, Seattle; pp. 1993–2016.
2007
|
|
8
|
Piard J, Aral B, Vabres P,
Holder-Espinasse M, Mégarbané A, Gauthier S, Capra V, Pierquin G,
Callier P, Baumann C, et al: Search for RECQL4 mutations in 39
patients genotyped for suspected Rothmund-Thomson/Baller-Gerold
syndromes. Clin Genet. 87:244–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cao DH, Mu K, Liu DN, Sun JL, Bai XZ,
Zhang N, Qiu GB and Ma XW: Identification of novel compound
heterozygous RECQL4 mutations and prenatal diagnosis of
Baller-Gerold syndrome: A case report. Genet Mol Res. 14:4757–4766.
2015. View Article : Google Scholar : PubMed/NCBI
|