Introduction
Hypophosphatasia (HP; Online Mendelian Inheritance
in Man no. 146300) is an inherited disease that affects the
development of bones and teeth by disrupting the mineralization
process. The incidence of serious HP is estimated to be 1/100,000
(1), while the incidence of mild
HP is higher and varies between racial groups (2). The clinical manifestations of HP are
highly variable. Severe forms of HP may result in mortality in
utero, whereas the mild form may result in early exfoliation of
deciduous teeth. HP is primarily caused by mutation of a gene
encoding tissue non-specific alkaline phosphatase, and is
characterized by hypocalcification of bones and teeth.
Dental deformity is common in HP, which contributes
to early diagnosis of the disease. Dental features in patients with
HP include tooth shape abnormalities (for example, small spherical
crown, constricted neck and enlargement of pulp chamber), structure
abnormalities (for example, maldevelopment of enamel, dentin and
cementum), color and tooth eruption abnormalities, and premature
loss of deciduous or permanent teeth (3). In addition, patients with HP may
display periodontal problems, as the collagen fiber of periodontal
ligaments fail to connect to the roots through Shapey's fiber
(4).
Regarding the vast clinical types of HP, elucidating
the underlying mechanisms may facilitate diagnosis, treatment and
prognosis determination. The serum level of alkaline phosphatase
liver/bone/kidney (ALPL) is an indicator of the severity of HP
(5). A study by Zurutuza et
al (6) suggested that the
clinical manifestations of HP are associated with ALPL activity.
ALPL, which consists of 524 amino acids, is a phosphomonoesterase
anchored to the cytomembrane via phosphatidylinositol glycan
(7). This protein catalyzes the
hydrolysis of phosphomonoester, and isolates inorganic phosphorus
Pi to generate hydroxyapatite crystals, which has an important role
in mineralization of the bone and teeth.
ALPL is encoded by the ALPL gene, which consists of
12 exons and is located on lp36.12 of chromosome 1 (location,
21,508,981–21,578,411 bp; http://ghr.nlm.nih.gov/gene/ALPL). According to the
ALPL gene mutation database (http://www.sesep.uvsq.fr/03_hypo_mutations.php)
established by Mornet and his colleagues (Versailles
Saint-Quentin-en-Yvelines University, France), 302 disease-causing
mutations have been reported until March 7th, 2016. The position
and type of mutation may affect the severity of HP (8).
Mutation screening of the ALPL gene has contributed
to diagnosis of the disease (9).
Furthermore, it may help to reveal the nature and underlying
mechanisms of HP. The present study identified that p.Trp29Arg, a
novel missense mutation in the ALPL gene, contributed to the unique
clinical features in a 5-year-old Chinese girl.
Materials and methods
Patient information
The patient was a 5-year-old girl with premature
loss of the deciduous teeth and severe mobility of the remaining
teeth. Medical history reported an absence of previous dental
treatment, recurrent infection, systemic disease, trauma or
surgery. No history of HP or consanguineous marriage was identified
in the family of the patient. X-ray examination, bone mineral
densitometry, pathological examinations and laboratory tests were
conducted. In addition, bidirectional sequencing of the ALPL gene
was performed. All the examinations were performed with permission
from the patient and parents in the form of full informed written
consent.
Bidirectional sequencing of the ALPL
gene
Blood samples (2 ml) were collected from the patient
and parents. Gene sequencing was conducted using an ABI
PRISM® 3730 DNA Analyzer (Shanghai South Gene
Technology, Co., Ltd., Shanghai, China). Sites of variation were
identified as described previously (10). The ALPL sequence derived from
GenBank (accession no. NM_000478.4) was referred to. Altered
nucleotides were confirmed by bidirectional sequencing. The novelty
of the two variants were determined from the National Center for
Biotechnology Information (NCBI) human SNP database (dbSNP,
https://www.ncbi.nlm.nih.gov/snp/), the
1000 Genomes Project database (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/),
and the Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/) (10).
Predicting the impact of the ALPL
mutation with Sorting Intolerant from Tolerant (SIFT) and
Polymorphism Phenotyping v2 (PolyPhen-2) tools
The ALPL sequences of other species were obtained
from GenBank, and conservation analysis was performed with
Clustalw2 software (ebi.ac.uk/Tools/msa/clustalw2/). The SIFT (sift.jcvi.org/) and PolyPhen-2 (genetics.bwh.harvard.edu/pph2/) algorithms were used
to predict whether the missense mutations may affect the protein
function.
Plasmid construction and site-directed
mutagenesis
The plasmid construction and site-directed
mutagenesis was performed by GenScript (Nanjing, China). Briefly, a
full-length cDNA for wild-type human ALPL (NM_000478.3) was
synthesized and confirmed by DNA sequencing using ABI PRISM 3700
DNA Sequencer (Perkin-Elmer Applied Biosystems, USA). Amplification
was performed using polymerase chain reaction (PCR) with primers
ALPL forward (F) 5′-AGCTCATGCATAACATCAGG-3′ and reverse (R)
5′-GTGGCAACTCTATCTTTGGT-3′. PCR was conducted using Phusion high
fidelity DNA polymerase (New England Biolabs, Inc., Ipswich, MA,
USA) according to the manufacturer's protocol.
Site directed mutagenesis was performed following
the overlap extension PCR method as previously described (11). Mutagenic primer W29R
5′-CCTTAGTGCCAGAGAAAGAGAAAGACCCCAAGTACCGGCGAGACCAAGCGCAAGAG-3′ was
designed containing the c.85T>C mutation. Primers for I395V F
5′-CGTGGCAACTCTGTCTTTGGTCTGGCCCCCATGCTGAGTGACACAGAC-3′ and R
5′-AGCATGGGGGCCAGACCAAAGACAGAGTTGCCACGGGGGGTGTATCCAC-3′ were used
to generate the c.1183A>G mutation. With amplified the ALPL gene
as templates, the fragments containing overlapping regions were
amplified through the first two PCR with mutagenic primers (W29R,
ALPL-mF, ALPL-mR for the c.85T>C mutation; I395V-F, I395V-R,
ALPL-mF, ALPL-mR for c.1183A>G mutation). Following purification
using a DNA purification kit (Beyotime Institute of Biotechnology,
Haimen, China), the products of the two reactions in the first PCR
were mixed and subjected to the second PCR using a 5′-end primer
ALPL-TBF
(5′-GCTAGCGCTACCGGACTCAGATCTCGAGATGATTTCACCATTCTTAGTACTGGCCATTGGCA-3′;
the XhoI site is underlined) with a XhoI restriction
site and a 3′ end primer ALPL-TBR
(5′-TTAGGGGGGGGGGAGGGAGAGGGGCGGTACCTCAGAACAGGACGCTCAGGGGGTAGA-3′;
the KpnI site is underlined) with a KpnI restriction
site to acquire full-length cDNAs encoding mutated ALPL.
For construction of ALPL expression vectors, we used
a CMV-driven plasmid pIRES2-EGFP (5.3 kb; BD Biosciences, San Jose,
CA, USA) containing enhanced green fluorescent protein (EGFP) gene.
In this vector, the gene of interest and the EGFP gene are
translated respectively, which means the ALPL would be expressed as
an unfused protein (Fig. 1).
Recombinant of the plasmids were constructed as follows, after
purification using the DNA purification kit (Beyotime Institute of
Biotechnology), double restriction enzymes (XhoI and
KpnI) digested the wild type and mutated full-length ALPL
gene and the pIRES2-EGFP plasmid. Subsequently, the digested
products were isolated using 0.8% agarose gel electrophoresis and
purified with a DNA clear up kit (Beyotime Institute of
Biotechnology). The digested fragments were inserted into
pIRES2-EGFP plasmid according to the manufacturer's protocol of the
DNA Ligation kit (Takara Biotechnology Co., Ltd., Dalian, China).
Next, the recombinant vectors were transformed into competent
Escherichia coli cells. The transformation procedures and
colony PCR were performed as previously described (12). The recombinant plasmids were
extracted using Plasmid Preparation kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Primers
were designed using the Primer Designer™ Tool (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) (Table I). The insertion was verified by
sequencing and restriction-enzyme digestion.
 | Figure 1.Plasmid construction map, which is
based on a restriction map created by the Clontech Laboratories,
Inc. (vector information published 03 October 2002, PT3267-5). The
Homo sapiens ALPL, transcript variant 1, mRNA (NM_000478.3) was
cloned into pIRES2-EGFP by XhoI and KpnI. Unique restriction sites
are shown in bold. ALPL, alkaline phosphatase, liver/bone/kidney.
MCS, multiple cloning site; PCMV IE, immediate early
promoter of cytomegalovirus; IRES, internal ribosome entry site;
EGFP, enhanced green fluorescent protein; poly A, poly-adenosine;
ori, origin of replication; Neor, neomycin resistance
gene; Kanr, kanamycin resistance gene; HSV TK, herpes
simplex virus thymidine kinase. |
 | Table I.Primers used for ALPL gene
amplification and site directed mutagenesis. ALPL-mF and ALPL-mR
were designed from the conserved region in ALPL gene. W29R,
I395V-F, and I395V-R were designed to introduce point mutations
(underlined). ALPL-TBF and ALPL-TBR were designed to acquire full
length ALPL gene with restriction sites. |
Table I.
Primers used for ALPL gene
amplification and site directed mutagenesis. ALPL-mF and ALPL-mR
were designed from the conserved region in ALPL gene. W29R,
I395V-F, and I395V-R were designed to introduce point mutations
(underlined). ALPL-TBF and ALPL-TBR were designed to acquire full
length ALPL gene with restriction sites.
| Name | Primer Sequence |
|---|
| ALPL-F |
5′-AGCTCATGCATAACATCAGG-3′ |
| ALPL-R |
5′-GTGGCAACTCTATCTTTGGT-3′ |
| ALPL-mF |
5′-CCATTCTTAGTACTGGCCATTGGCACCTGCCTTACTAACTCCTTAGTGCCAGAGAAAGAG-3′ |
| ALPL-mR |
5′-TCAGAACAGGACGCTCAGGGGGTAGAGGGCCAGCGCGAGCAGCAGGGGGCCTGCAGCAA-3′ |
| W29R |
5′-CCTTAGTGCCAGAGAAAGAGAAAGACCCCAAGTACCGGCGAGACCAAGCGCAAGAG-3′ |
| I395V-F |
5′-CGTGGCAACTCTGTCTTTGGTCTGGCCCCCATGCTGAGTGACACAGAC-3′ |
| I395V-R |
5′-AGCATGGGGGCCAGACCAAAGACAGAGTTGCCACGGGGGGTGTATCCAC-3′ |
| ALPL-TBF |
5′-GCTAGCGCTACCGGACTCAGATCTCGAGATGATTTCACCATTCTTAGTACTGGCCATTGGCA-3′ |
| ALPL-TBR |
5′-TTAGGGGGGGGGGAGGGAGAGGGGCGGTACCTCAGAACAGGACGCTCAGGGGGTAGA-3′ |
Cell culture and transfection
The HEK293 human embryonic kidney cell line was
purchased from the Cell Bank of Chinese Academy of Sciences
(Shanghai, China). These cells were cultured as described
previously (13). Cells were
seeded into a 6-well plate at a density of 2×105 cells
per well for 24 h before transfection. A 0.6 µg aliquot of plasmid
of wild type and mutated ALPL were transiently transfected into
HEK293 cells. The Attractene Transfection Reagent (Qiagen GmbH,
Hilden, Germany) was used according to the manufacturer's protocol.
Cells were observed under a fluorescence microscope after 48 h.
Each assay was duplicated or triplicated in independent
experiments.
ALP activity assay
At 48 h after transfection, the medium was removed.
The cells were lysed with 0.2 ml lysis buffer (Beyotime Institute
of Biotechnology). A protease inhibitor mixture without any
phosphatase inhibitor was added to the cell lysate as described
previously (14). The protease
inhibitor mixture, which consisted of phenylmethylsulfonyl fluoride
(100 mM), aprotinin (15 µM), leupeptin (100 µM), bestatin (100 µM),
pepstatin A (100 µM) and E-64 (80 µM), was purchased from
Shanghaibocai (Shanghai, China). The lysate and media were
centrifuged at 15,000 × g for 10 min at 4°C to remove insoluble
material. ALP activity was determined using the Alkaline
Phosphatase Assay kit (Beyotime Institute of Biotechnology)
according to the manufacturer's protocol. In this kit,
para-nitrophenyl phosphatase is adopted as the substrate of ALP.
This reaction generates para-nitrophenol, which is yellow in an
alkaline environment. The absorbance at a wavelength of 405 nm is
proportional to the concentration of para-nitrophenol. A six-point
standard curve is recommended in the range from 0.02–0.2 mM. Sample
(1 µl) was added to each system. The absorbance at a wavelength of
405 nm was detected every minute automatically by a SpectraMax M3
spectrophotometer (Molecular Technologies, Sunnyvale, CA, USA).
Each ALP assay was repeated at least twice in independent
experiments. Protein concentrations were determined using a
Bicinchoninic Acid Protein assay kit (Beyotime Institute of
Biotechnology, China). ALP activities were calculated relative to
protein concentration.
ALPL protein expression
Western blotting was performed to detect the protein
expression levels of ALPL in transfected cells. Briefly, the
remaining cell lysate was prepared in 5X SDS loading buffer. Equal
amounts of protein (20 µg per lane) were separated by 4–12%
SDS-PAGE (GenScript, Nanjing, China) and transferred onto a
polyvinylidene difluoride membrane. Following blocking in 5% bovine
serum albumin (BSA, Beyotime Institute of Biotechnology, Haimen,
China) at room temperature for 2 h, the membrane was incubated
overnight at 4°C with primary antibody. ALPL antibody purchased
from Bioworld Technology, Inc., Nanjing, China (cat. no. BS6134),
was diluted to 1:500 in 5% BSA. GAPDH antibody (cat. no. AP0063),
which was also purchased from Bioworld Technology, was diluted at
1:10,000 in 5% BSA. The next day, the blots were washed 3 times
with TBS and incubated with a horseradish peroxidase-conjugated
goat anti-rabbit IgG antibody (cat. no. BS13278; Bioworld
Technology, Inc., Nanjing, China) at a dilution of 1:50,000 for 2 h
at room temperature. The blots were washed again and subsequently
visualized with an ImageQuant LAS 4000 mini system (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). This experiment was performed
three times.
All-atom molecular dynamics (MD)
simulation
To investigate the effect of the two mutations
p.Trp29Arg (c.85T>C) and p.Ile395Val (c.1183A>G) on ALPL
dynamics, all-atom MD simulations were performed on wild type and
mutated ALPL in monomer form. The Protein Data Bank structure
(1EW2) (15) was selected as the
initial structure of wild-type ALPL. The mutated structure was
built by Modeller version 9.13 software (16). All the MD simulations were carried
out using Gromacs package software version 5.0.4 (17) with Amber99sb-ildn force field
(18). The TIP3P water model was
used for the solvate (19). The
monomer was solvated in a 9.893×9.893×9.893 nm water box. The
Zn2+ and Mg2+ in the crystal structure were
explicitly included in the simulations but without consideration of
the effects of charge transfer and protonation/deprotonation, since
the current simulations do not involve the binding/unbinding of
Zn2+ and Mg2+ ions. To achieve charge neutral
and an equivalent metal ion concentration of 150 mM/l, 85
Na+ and 85 Cl− were added to the water box of
wild-type ALPL, and 85 Na+ and 86 Cl− to the
water box of mutated ALPL. The electrostatic interaction was
treated using Particle Mesh Ewald with a cutoff of 10 nm. The same
cutoff was used in the calculation of the van der Waals
interactions. All bonds were constrained using the LINCS algorithm
and the MD time step was set to 2 fsec (20). V-rescale and Parrinello-Rahman
algorithms were used for temperature and pressure coupling
(21,22). The initial structure of each system
was first subjected to a minimization of 50,000 steps. Following
minimization, a 500 fsec constant number of atoms, volume and
temperature equilibration at 300 K was performed. Subsequently, a
500 fsec number of particles, pressure and temperature (NPT)
equilibration at 1 atm and 300 K was conducted, followed by a 100
nsec production simulation under NPT conditions at 1 atm and 300
K.
Statistical analysis
Data were statistically analyzed using GraphPad
Prism version 5.0 (GraphPad Software Inc., La Jolla, CA, USA).
Results are presented as the mean ± standard derivation. A one-way
analysis of variance and Student-Newman-Keuls test were conducted
to determine the significant differences in mean values among
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical examination and auxiliary
examination
A physical examination demonstrated that the body
weight and height of the patient was just above the norm. She had a
waddling gait without limb or skin abnormalities. Extensive
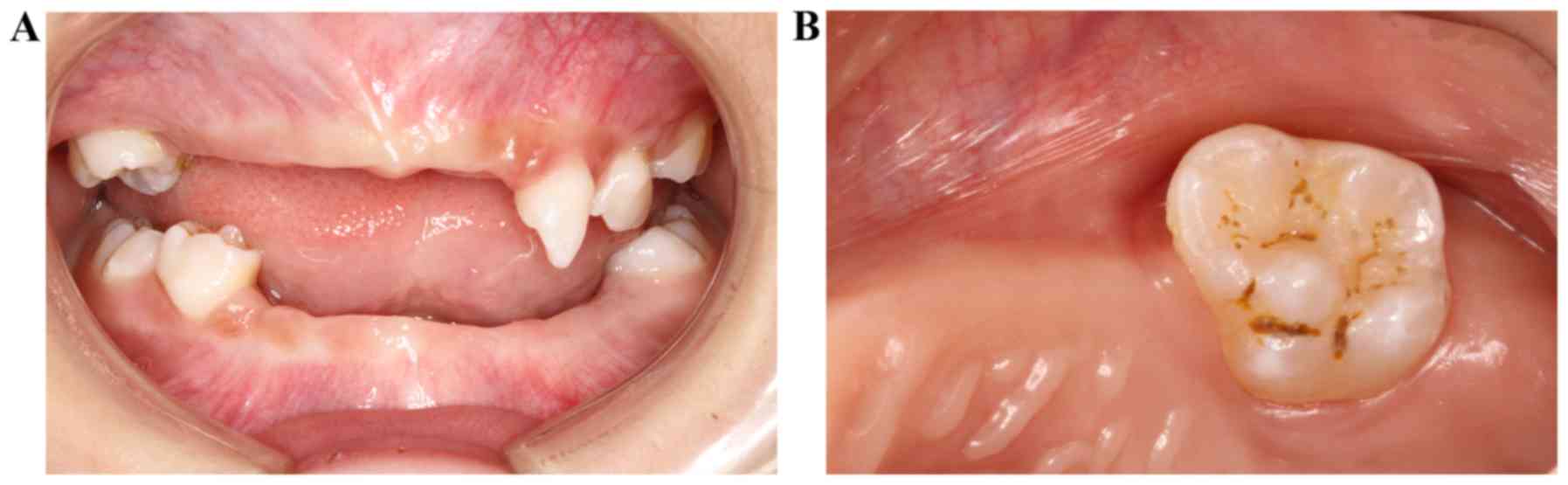
premature loss of deciduous teeth was observed, with only 55, 63,
64, 65, 75, 84 and 85 remaining (Fig.
2A). In addition, constricted neck and denudation of dentin on
the occlusal surface was observed in several teeth (Fig. 2B). Furthermore, although abnormal
mobility was detected, the patient had a good oral hygiene,
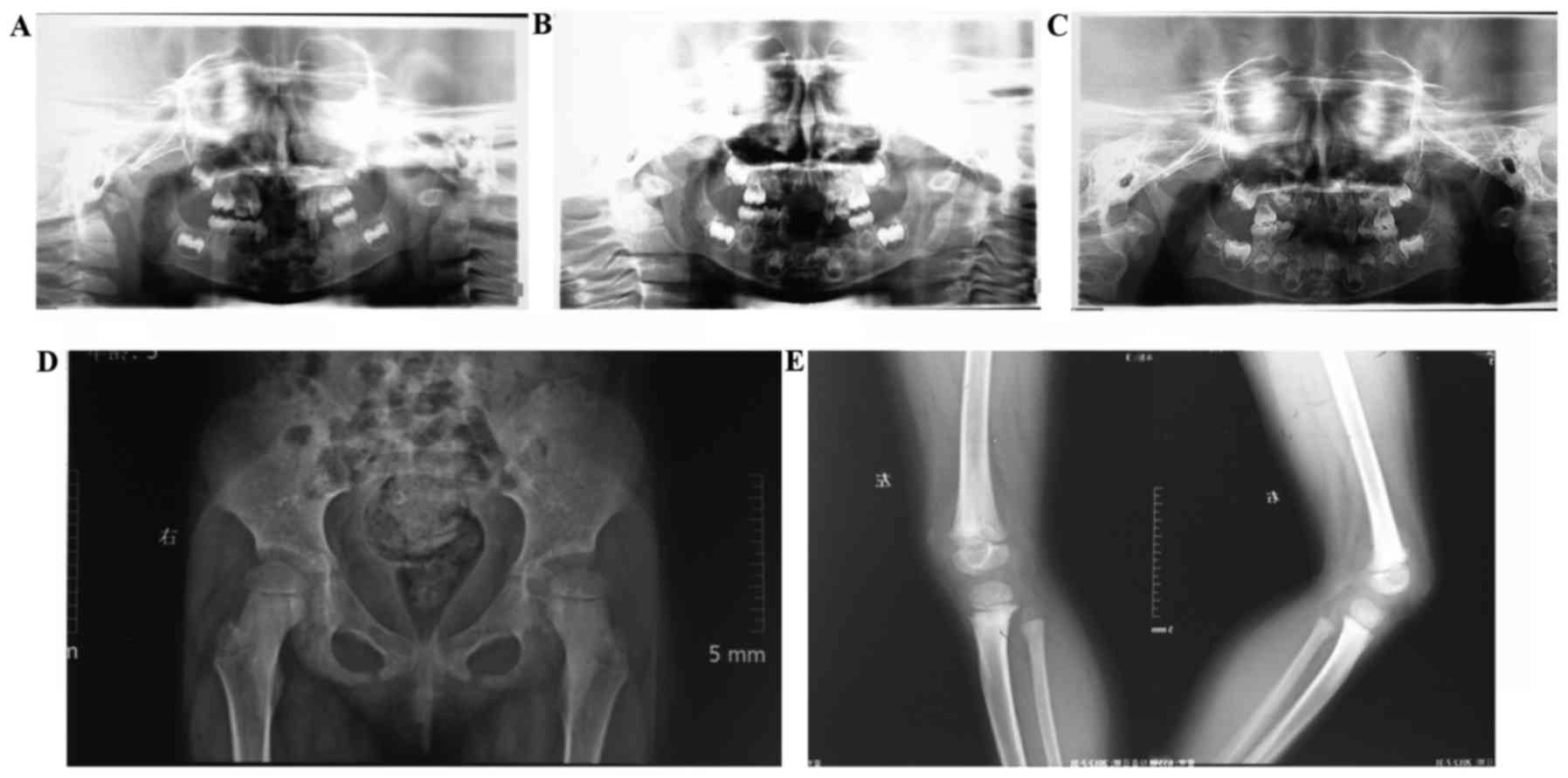
demonstrating no sign of attachment loss. Panoramic radiographs
demonstrated malformed deciduous teeth without congenital loss of
permanent teeth. No considerable alveolar bone resorption was
observed during the 3-year follow-up (Fig. 3A-C).
Slight dislocation of the hip and disrupted
Shenton's lines were observed with mild osteodystrophy in the knee.
In addition, a wide epiphyseal plate with rough surface was
observed (Fig. 3D and E). Bone
mineral densitometry velocity of sound was 3698 m/s, and the
Z-value was 1.96 m with a relative risk of fracture of 1.000.
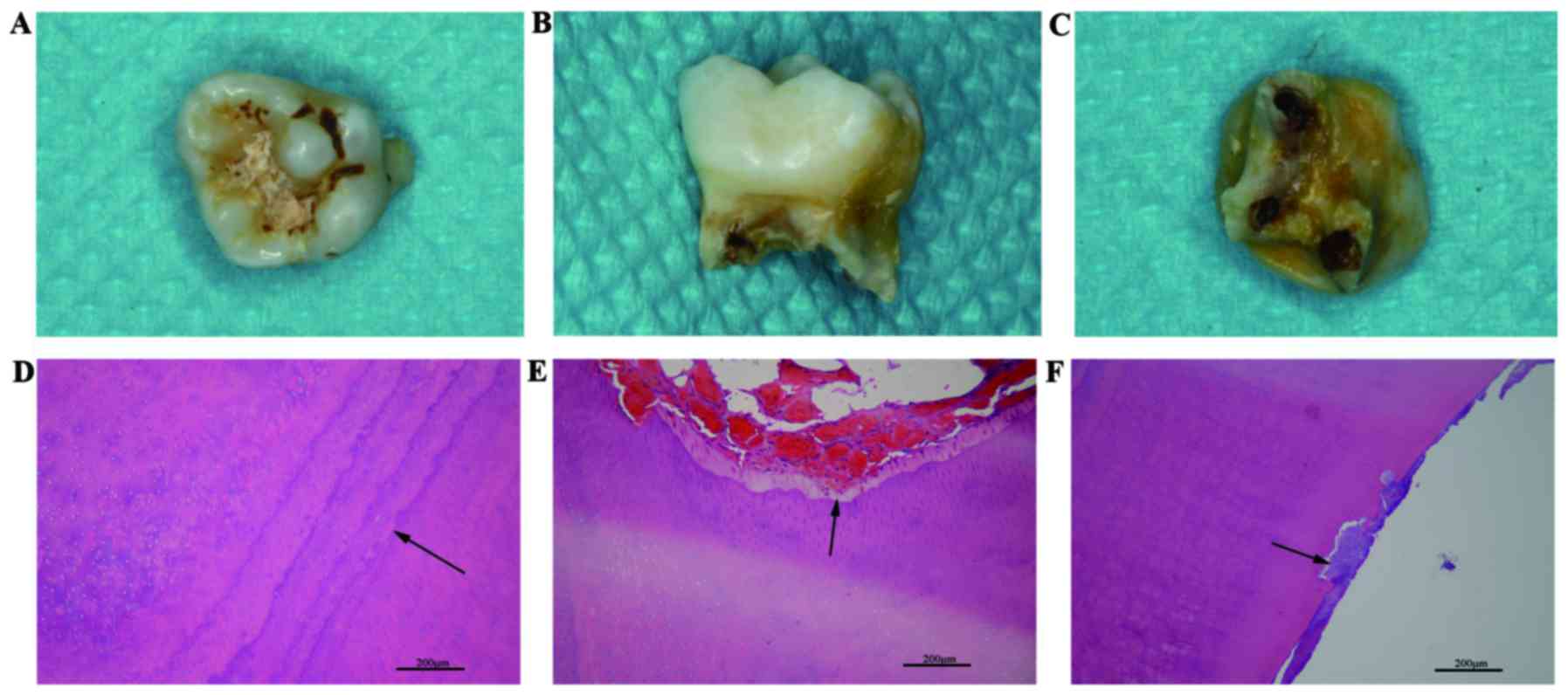
The naturally lost primary tooth exhibited enamel
defects, peculiar short roots and widened root canals (Fig. 4A-C). Hematoxylin and eosin staining
of the histological slices demonstrated that calcification of the
dentin was abnormal. An unusually distinct incremental line of
dentin and an uneven width of the predentin were observed (Fig. 4D and E) and patches of cementum
were found to be embedded in dentin (Fig. 4F).
The patient (5-years-old) had decreased serum ALP,
parathyroid hormone and serum osteocalcin compared with the normal
levels at 5-years-old (Table II).
Additionally, the level of ALP was also decreased compared with
earlier time points: 83 U/l at 3 years old, and 74 U/l at 4 years
old.
| Table II.Examination of the 5-year-old
patient. |
Table II.
Examination of the 5-year-old
patient.
| Component | Measurement | Normal range (5 years
old) |
|---|
| Serum ALP
(U/l) | 61 | 150–370 |
| Blood calcium
(mmol/l) | 2.47 | 2.25–2.75 |
| Serum phosphorus
(mmol/l) | 1.96 | 1.29–1.94 |
| Parathyroid hormone
(pg/ml) | 9.67 | 15.00–65.00 |
| Serum osteocalcin
measure (ng/ml) | 73 | 11–43 |
| 25-hydroxy vitamin
D (ng/ml) | 26.64 | 30–100 |
ALPL gene analysis
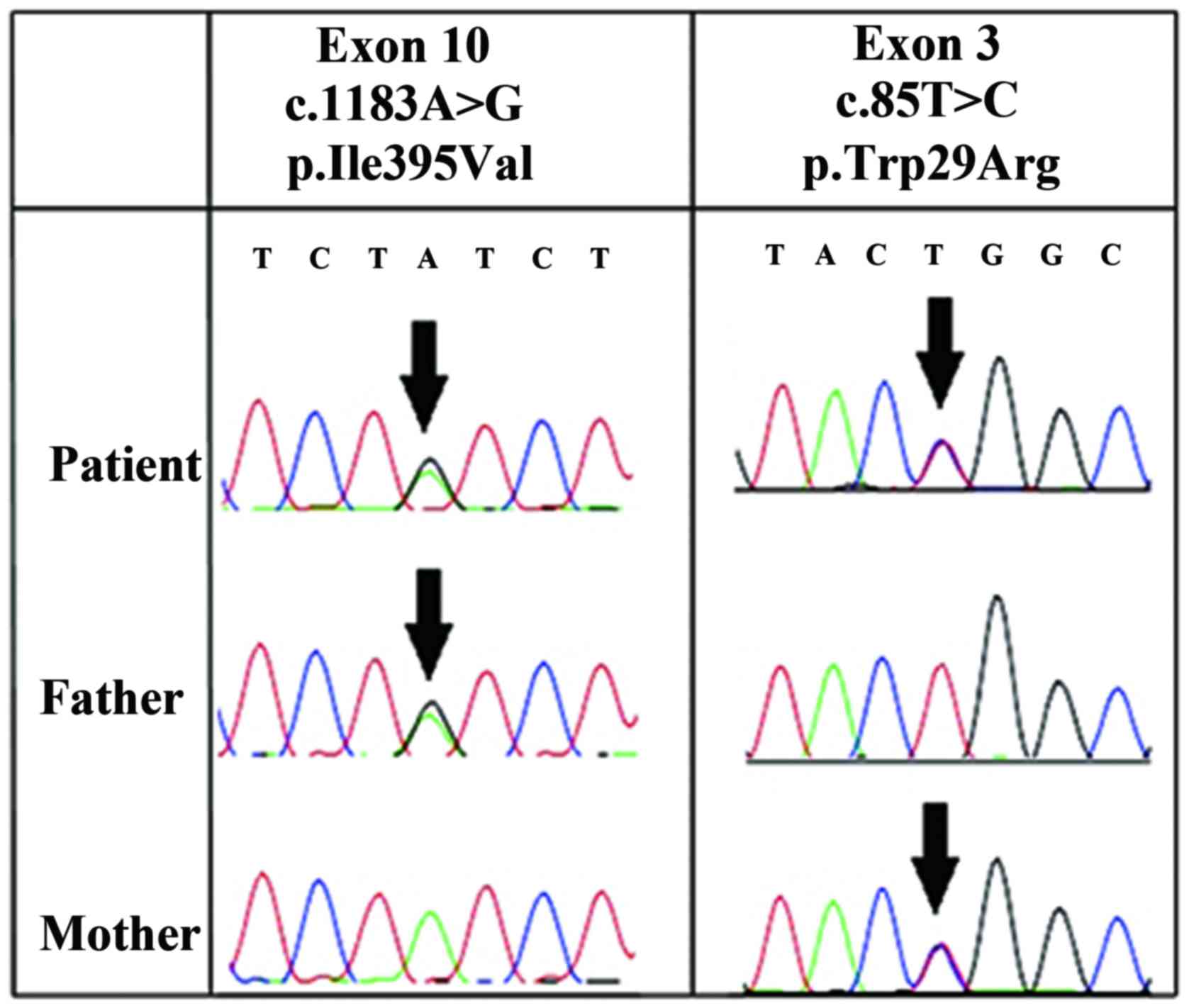
Direct sequencing of the 12 exons of ALPL
demonstrated that the patient was a compound heterozygote with two
missense mutations in the ALPL gene: p.Trp29Arg (c.85T>C) and
p.Ile395Val (c.1183A>G). p.Trp29Arg (c.85T>C) is located on
exon 3 and p.Ile395Val (c.1183A>G) is on exon 10. The p.Trp29Arg
(c.85T>C) mutation was also present on exon 3 of the mother's
ALPL gene, while the p.Ile395Val (c.1183A>G) mutation was
present on exon 10 of the father's ALPL gene (Fig. 5).
Predicting the impact of the mutations
with SIFT and PolyPhen-2
SIFT and PolyPhen-2 were used to predict the impact
of the substitution on ALPL function. The prediction by SIFT
indicated that p.Trp29Arg and p.Ile395Val are ‘tolerable’. The
prediction conducted by PolyPhen-2 demonstrated that p.Trp29Arg and
p.Ile395Val are ‘possibly damaging’ in terms of conservation, with
scores of 0.554 and 0.817, respectively. However, p.Trp29Arg and
p.Ile395Val were ‘benign’ in term of variation, with scores of
0.238 and 0.422, respectively.
In vitro analysis
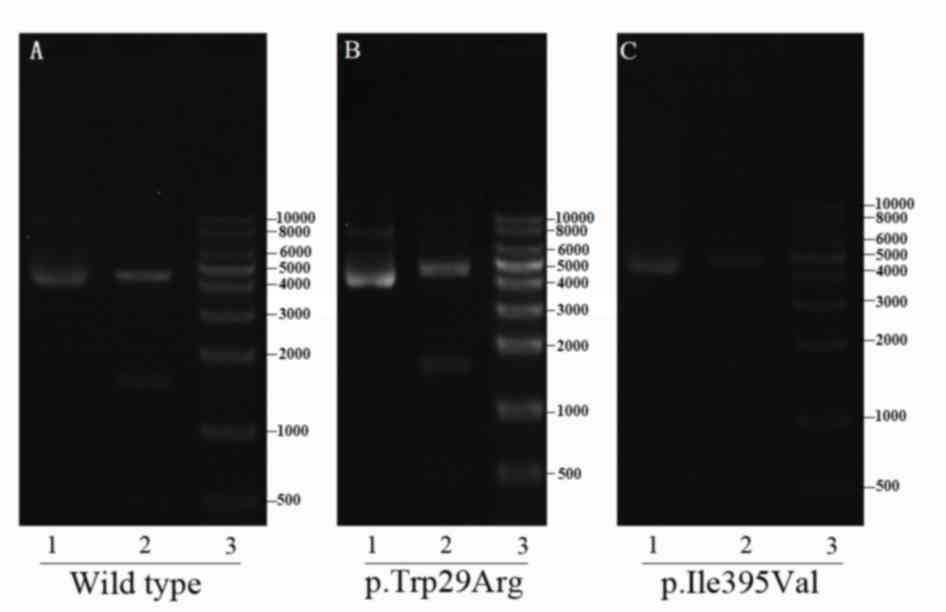
The correct construction of the plasmids was
verified through sequencing and restriction-enzyme digestion
(Fig. 6). All six groups
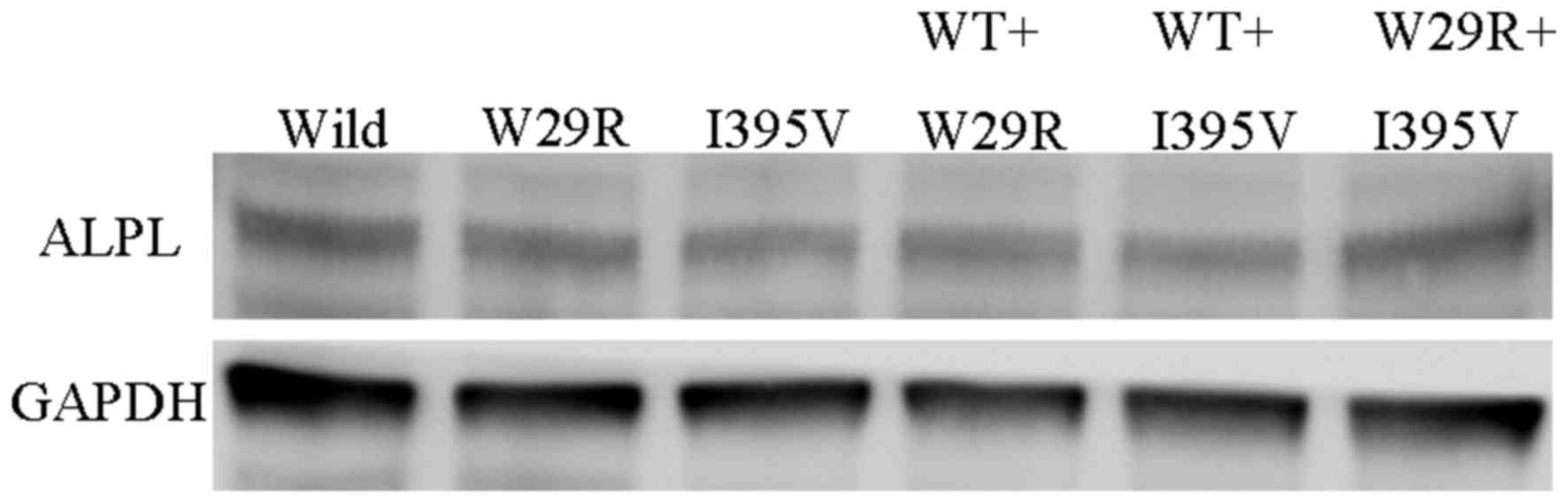
transfected with wild type and/or mutated ALPL demonstrated similar
ALPL protein expression levels (Fig.
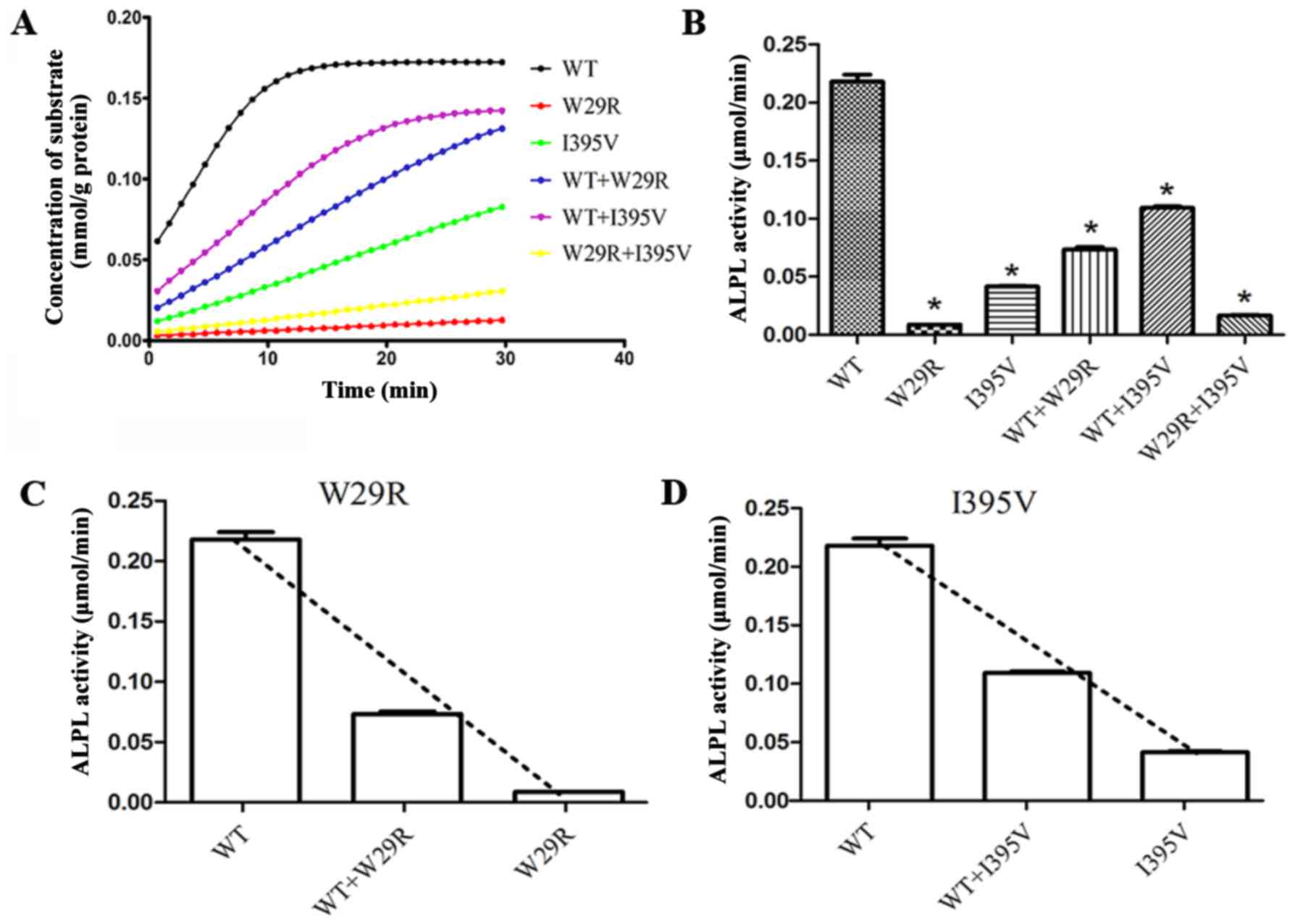
7). Loss of protein function was observed in p.Trp29Arg and
p.Ile395Val. When Trp29 of all the monomers were substituted by
Arg, only 4.1% ALP activity remained relative to wild type ALP.
When half of the monomers were wild type, the ALP activity
increased to 33.7% relative to wild type ALP. The corresponding
data for p.Ile395Val, wild type + p.Ile395Val and p.Trp29Arg +
p.Ile395Val were 19.1, 50.1 and 7.6%, respectively (Fig. 8A and B). Furthermore, the activity
of wild type ALP was reduced when co-expressed with p.Trp29Arg and
reduced to a lesser degree by p.Ile395Val, suggesting a dominant
negative effect of these two mutants on wild type ALPL (Fig. 8C and D).
All-atom MD simulation
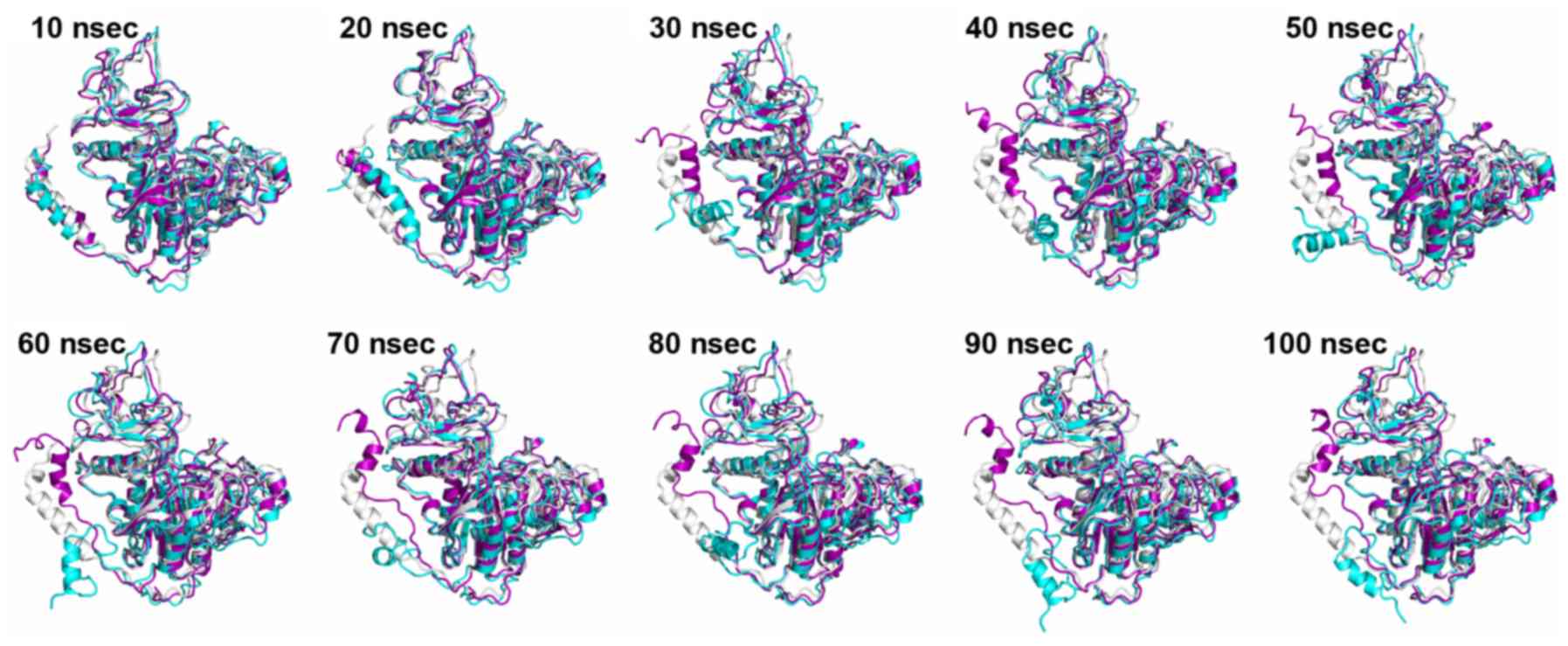
The simulations revealed that the N-terminal helix
of wild type and mutated ALPL exhibit varying behaviors. Several
snapshots were extracted at different stages along the simulations
of wild type and mutated monomer ALPL, and these were aligned to
the crystal structure of monomer ALPL (Fig. 9). After 30 nsec, the N-terminal
helix of mutated ALPL separated from the main body of the protein,
and moved freely. However, in the wild type ALPL, this part
remained close to the rest of monomer at all times. Except for the
N-terminal helix, the overall structure of the remaining part of
ALPL was minimally affected by the mutation. Taking into
consideration that Trp29 is located at the N-terminal helix, it was
speculated that the mutation (p.Trp29Arg) may affect the dynamics
of ALPL significantly, whereas the substitution (p.Ile395Val) may
have little effect on ALPL.
Discussion
It has been widely reported that HP is caused by
mutations in the ALPL gene. As the position and type of mutation
determines the severity of HP, it is important to elucidate the
exact impact of a specific mutation. The patient assessed in the
present study has two mutations: p.Trp29Arg and p.Ile395Val.
p.Trp29Arg is a novel mutation that has not been previously
reported. p.Ile395Val has been reported (23); however, the exact impact of the
mutation was not studied.
Online forecasting tools are widely used to predict
the severity of a substitution. However, in the case of these
mutations, results from SIFT and PolyPhen-2 were not fully
consistent with each other. Furthermore, the results were not in
complete agreement with the phenotype of the patient. Recently,
Silvent et al (24)
reported that the 47 substitutes predicted by PolyPhen-2 were found
to be false positives, which counted for 9% of the 524 residues of
ALPL. Therefore, these methods should not be solely relied on to
clarify the impact of the mutations on protein function.
Trp29 of ALPL is highly conserved throughout many
species. Located at the N-terminal α-helix, Trp29 is a crucial site
interacting with the monomer (25). Ile395 is also a conserved site
located at the crown domain, and associates with the process of
dimerization. Amino acid alterations at this site may disturb the
homodimer interface and thus, induce dysfunction of the protein.
All-atom MD simulation suggested that p.Trp29Arg resulted in
misfolding of the N-terminal helix, which may affect the structure
integration in the process of assembly (25), thus ultimately affecting enzyme
function. No abnormal alterations in dynamics around Ile395 were
detected in p.Ile395Val via all-atom MD simulation. However, in
vitro cell culture experiments indicated that p.Ile395Val
significantly reduced the function of ALPL. Therefore, p.Ile395Val
may affect the protein function in some way other than dynamic
disturbance.
Results from the cell experiment demonstrated that
p.Trp29Arg and p.Ile395Val significantly affected the protein
function. p.Trp29Arg+p.Ile395Val mutations markedly reduced ALP
activity, which was consistent with the phenotype of the patient to
a certain extent. Cells expressing wild type + p.Trp29Arg
(represents the genotype of the mother) and wild type+p.Ile395Val
(represents the genotype of the father) exhibited lower ALP
activity compared with wild type, but markedly increased ALP
activity compared with p.Trp29Arg+p.Ile395Val-expressing cells.
This is consistent with the hereditary mode of childhood HP, which
is primarily reported as autosomal recessive (26). Therefore, if the parents plan to
have a second child, they should be informed of the risk of HP.
Notably, although p.Trp29Arg+p.Ile395Val-expressing
cells demonstrated only 7.6% function in vitro, the patient
exhibited mild clinical symptoms. Similar paradoxical cases may be
observed in other reports (14). A
limitation of in vitro studies is that they demonstrate the
impact of a mutation on the protein in a relatively simple
environment, which may not be fully representative of the human
phenotype. Therefore, the underlying mechanisms require further
investigation.
In conclusion, the present study reported a novel
missense mutation in the ALPL gene, p.Trp29Arg, which was located
in the N-terminal helix and significantly reduced enzyme activity
of ALPL. Evidence on how a substitution in the N-terminal helix may
influence the structure and function of ALPL was rather. Hoylaerts
et al (25) demonstrated
that the correct folding of the N-terminus is essential for the
overall structural integrity and the intramolecular transitions
during enzyme catalysis of ALP. They made it through deleting amino
acid in N-terminus rather than substitution. In terms of the
missense mutation, it is still unclear whether a substitution in
N-terminus may influence the folding or result in dysfunction of
the protein. The present study determined that the p.Trp29Arg may
lead to misfolding of the N-terminal helix, which may undermine the
structural integrity and the intramolecular transitions of ALPL and
significantly reduce the enzyme activity of ALPL. The p.Trp29Arg
substitution may lead to HP. Further investigations are required to
determine which residues participate in this transformation and
effect of the p.Trp29Arg substitution on the enzyme activity of the
wild-type ALPL, and the association between the phenotype and the
genotype of HP due to the substitution in the N-terminus.
Acknowledgements
The authors would like to thank Mr Haijian Fan from
Nanjing Gulou Hospital, Medical School of Nanjing University
(Nanjing, China) for his contribution on the imaging diagnosis, and
Professor Xiaofeng Huang and Mr Sheng Chen from Nanjing
Stomatological Hospital, Medical School of Nanjing University for
their contribution on the pathology diagnosis. Finally, the authors
would like to thank Ms Qian Zhang from Nanjing Stomatological
Hospital, Medical School of Nanjing University for her kind
information on dental embryology.
The present study was supported by the Key Project
of Science and Technology Bureau of Jiangsu Province (grant no.
BL2013002) and the Fundamental Research Funds for the Central
Universities (grant no. 021414310017).
References
|
1
|
Fraser D: Hypophosphatasia. Am J Med.
22:730–746. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mornet E, Hofmann C, Bloch-Zupan A,
Girschick H and Le Merrer M: Clinical utility gene card for:
Hypophosphatasia - update 2013. Eur J Hum Genet. 22:2014.doi:
10.1038/ejhg.2013.177. View Article : Google Scholar :
|
|
3
|
Reibel A, Manière MC, Clauss F, Droz D,
Alembik Y, Mornet E and Bloch-Zupan A: Orodental phenotype and
genotype findings in all subtypes of hypophosphatasia. Orphanet J
Rare Dis. 4:62009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van den Bos T, Handoko G, Niehof A, Ryan
LM, Coburn SP, Whyte MP and Beertsen W: Cementum and dentin in
hypophosphatasia. J Dent Res. 84:1021–1025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whyte MP: Hypophosphatasia and the role of
alkaline phosphatase in skeletal mineralization. Endocr Rev.
15:439–461. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zurutuza L, Muller F, Gibrat JF,
Taillandier A, Simon Bouy-B, Serre JL and Mornet E: Correlations of
genotype and phenotype in hypophosphatasia. Hum Mol Genet.
8:1039–1046. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jemmerson R and Low MG:
Phosphatidylinositol anchor of HeLa cell alkaline phosphatase.
Biochemistry. 26:5703–5709. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim EE and Wyckoff HW: Structure of
alkaline phosphatases. Clin Chim Acta. 186:175–187. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mornet E: Hypophosphatasia. Best Pract Res
Clin Rheumatol. 22:113–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang W, Shen L, Deng Z, Ding Y, Mo X, Xu
Z, Gao Q and Yi L: Novel missense variants of ZFPM2/FOG2 identified
in conotruncal heart defect patients do not impair interaction with
GATA4. PLoS One. 9:e1023792014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ho SN, Hunt HD, Horton RM, Pullen JK and
Pease LR: Site-directed mutagenesis by overlap extension using the
polymerase chain reaction. Gene. 77:51–59. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li W, Yang JG, Ren FX, Kang CL and Zhang
SY: PCR site-directed mutagenesis of long QT syndrome KCNQ1 gene in
vitro. Yi Chuan. 26:589–593. 2004.(In Chinese). PubMed/NCBI
|
|
13
|
Witzigmann D, Wu D, Schenk SH,
Balasubramanian V, Meier W and Huwyler J: Biocompatible
polymer-Peptide hybrid-based DNA nanoparticles for gene delivery.
ACS Appl Mater Interfaces. 7:10446–10456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang H, Wang L, Geng J, Yu T, Yao RE, Shen
Y, Yin L, Ying D, Huang R, Zhou Y, et al: Characterization of six
missense mutations in the tissue-nonspecific alkaline phosphatase
(TNSALP) gene in Chinese children with hypophosphatasia. Cell
Physiol Biochem. 32:635–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le Du MH, Stigbrand T, Taussig MJ, Menez A
and Stura EA: Crystal structure of alkaline phosphatase from human
placenta at 1.8 A resolution. Implication for a substrate
specificity. J Biol Chem. 276:9158–9165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sánchez R and Sali A: Comparative protein
structure modeling. Introduction and practical examples with
modeller. Methods Mol Biol. 143:97–129. 2000.PubMed/NCBI
|
|
17
|
Hess B, Kutzner C, van der Spoel D and
Lindahl E: GROMACS 4: Algorithms for highly efficient,
load-balanced, and scalable molecular simulation. J Chem Theory
Comput. 4:435–447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lindorff-Larsen K, Piana S, Palmo K,
Maragakis P, Klepeis JL, Dror RO and Shaw DE: Improved side-chain
torsion potentials for the Amber ff99SB protein force field.
Proteins. 78:1950–1958. 2010.PubMed/NCBI
|
|
19
|
Jorgensen WL, Chandrasekhar J and Madura
JD: Comparison of simple potential functions for simulating liquid
water. J Chem Phys. 79:9261983. View
Article : Google Scholar
|
|
20
|
Hess B, Bekker H, Berendsen HJC and
Fraaije JG: LINCS: A linear constraint solver for molecular
simulations. J Computation Chem. 18:1463–1472. 1998. View Article : Google Scholar
|
|
21
|
Bussi G, Donadio D and Parrinello M:
Canonical sampling through velocity rescaling. J Chem Phys.
126:0141012007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parrinello M and Rahman A: Crystal
Structure and Pair Potentials: A Molecular-Dynamics Study. Phys Rev
Lett. 45:1196–1199. 1980. View Article : Google Scholar
|
|
23
|
Wenkert D, McAlister WH, Coburn SP, Zerega
JA, Ryan LM, Ericson KL, Hersh JH, Mumm S and Whyte MP:
Hypophosphatasia: Nonlethal disease despite skeletal presentation
in utero (17 new cases and literature review). J Bone Miner Res.
26:2389–2398. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Silvent J, Gasse B, Mornet E and Sire JY:
Molecular evolution of the tissue-nonspecific alkaline phosphatase
allows prediction and validation of missense mutations responsible
for hypophosphatasia. J Biol Chem. 289:24168–24179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoylaerts MF, Ding L, Narisawa S, Van
Kerckhoven S and Millan JL: Mammalian alkaline phosphatase
catalysis requires active site structure stabilization via the
N-terminal amino acid microenvironment. Biochemistry. 45:9756–9766.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silva I, Castelao W, Mateus M and Branco
JC: Childhood hypophosphatasia with myopathy: Clinical report with
recent update. Acta Reumatol Port. 37:92–96. 2012.PubMed/NCBI
|