Introduction
Myocardial infarction (MI) is caused by occlusion of
the coronary artery, and is pathologically characterized by severe
necrosis of cardiomyocytes due to persistent ischemia. MI is now a
common disease affecting human health and coronary atherosclerosis
is the major contributing factor for MI (1). In addition, hypertension, diabetes
and blood lipid abnormalities can also contribute to MI, and
smoking and alcohol abuse may be important inducers of blood
abnormalities (2). Investigations
using molecular methods have also suggested that the accumulation
of reactive oxygen species stress, upregulation of adhesive
molecules, and increases of serum homocysteine, blood fibrinogen
and coagulation, may lead to abnormalities of blood lipids,
resulting in MI (3).
Coronary atherosclerosis is elicited by a variety of
endogenous and exogenous factors through different intracellular
signaling pathways (4). MSC
transplantation can promote angiogenesis in the infarct area to
ameliorate cardiac function in MI (5). However, the clinical application of
MSC transplantation is restricted by low survival rates of MSCs
following transplantation (6).
Several signaling pathways are abnormally regulated in the process
of MI, including transforming growth factor-β (TGF-β)/small mothers
against decapentaplegic, Janus kinase-signal transducers and
activators of transcription, p38 mitogen-activated protein kinases
(p38MAPK), phosphoinositide-3-kinase (PI3K)/Akt-endothelial nitric
oxide synthase, PI3K/protein kinase C and Wnt (7–9).
Among these signaling pathways, p38MAPK is involved in regulating
the inflammatory process. Tumor necrosis factor (TNF)-α and
interleukin (IL)-6 are classical pro-inflammatory factors, which
are modulated by the p38MAPK pathway (8). The p38MAPK pathway is also important
in cell differentiation and development (10).
p38MAPK functions through phosphorylating nuclear
transcription factors and regulating gene expression. The
inhibition of p38MAPK has been reported to protect against MI
(4). However, the effects of the
inhibition of p38MAPK on MSC therapy in an MI model remain to be
elucidated. In the present study, an MI model was established in
rats by ligation of the left anterior descending branch of the
coronary artery. The MI model was exposed to the combined
application of p38MAPK inhibition and MSC transplantation. The aim
was to provide novel evidence for MI therapy.
Materials and methods
Isolation and culture of MSCs
Male Sprague-Dawley (SD) rats (4-week old; 250±50 g)
were obtained from the Shanghai Laboratory Animal Center (SLAC;
Shanghai, China). All experiments were approved by the ethics
committee of Jiangxi Provincial People's Hospital (Nanchang,
China). MSCs were isolated as previously described, with minor
modifications (11). Briefly, 50
SD rats were housed at room temperature with a standard 12-h
light/dark cycle and ad libitum access to food and water for
at least 7 days, following which the rats were sacrificed by
cervical dislocation and soaked in 75% alcohol for 2 min. The
bilateral tibial and femoral shafts were isolated and washed twice
in cold PBS. The tibia was cut from the middle, and washed in
low-glucose DMEM (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) containing 100 U/ml heparin without serum. According to
the Percoll lymphocyte separating method (cat. no. 17-0891-01;
Pharmacia; GE Healthcare Life Sciences, Piscataway, NJ, USA), the
suspension was centrifuged at a 500 g density gradient for 25 min
at room temperature and the mononuclear cell layer was collected.
The cells (5×103/ml) were resuspended in low-glucose
DMEM with 15% FBS (GE Healthcare Life Sciences), 100 U/ml
penicillin and streptomycin at 37°C and 5% CO2. The
cells were distinguished using flow cytometry with antibodies
against CD34 (1:100; cat. no. ab23830, Abcam, Shanghai, China),
CD44 (1:100; cat. no. ab46793, Abcam) and CD105 (1:100; cat. no.
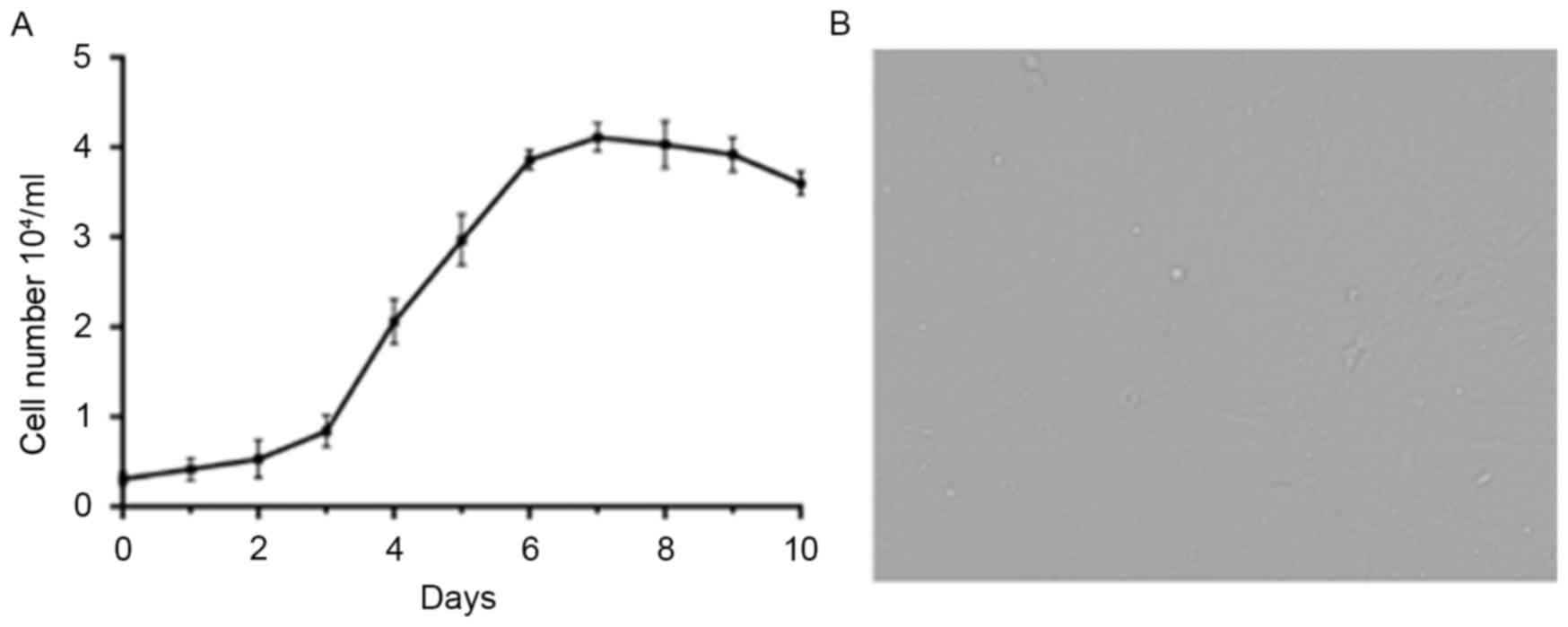
ab53318; Abcam). The cell growth curve is shown in Fig. 1.
MI model establishment
The specific pathogen-free rats (weight, 250±50 g)
were purchased from the SLAC and raised in an animal room for at
least 7 days prior to experiments. The acute MI model was
established by ligating the anterior descending branch of the
coronary artery. Following anesthesia via intraperitoneal injection
of 40 mg/kg pentobarbital with 0.l mg/kg atropine, the rats were
fixed on the bench in the supine position. A non-invasive method
was used to insert a tube (18G) into the mouth in order to expose
the throat. Positive pressure ventilation was applied using a
ventilator to assist with animal breathing. The tidal volume was
set at 9–12 ml and the breath rate was set at 1:1.5 with a
frequency of 60/min. Following exposure of the heart, the anterior
descending branch of the left coronary artery was identified and
ligated using 5–0 silk. The ST elevation was determined upon ECG
and the tissues downstream of the coronary artery ligation were
white or purple in color. Expansion of the left atrial appendage
was performed to determine successful modeling of the MI model. The
MI rats were then randomly divided into four groups: MSC therapy
group, P38MAPK inhibition group, MSC therapy with p38MAPK
inhibition group and saline control group. Subsequently, MSCs
(1.5×106/100 µ1) and/or SB203580 (0.2 mg/kg) were
injected twice each week for a consecutive 4-week period. HE
staining and a TUNEL assay were then performed to determine the
pathological changes and apoptosis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The heart tissues were collected for RT-qPCR
analysis. Total mRNA was isolated according to the manufacturer's
protocol of the kit (cat. no. 74134; Qiagen, Inc., Valencia, CA,
USA). The concentration and purity of the total RNA were determined
using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific,
Inc.). Subsequently, the mRNAs underwent RT-qPCR combined with SYBR
staining to amplify the expression (cat. no. RR430A; Takara
Biotechnology Co., Ltd., Dalian, China). The primers were
synthesized by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China).
The primers were as follows: p38MAPK, forward
5′-tcgagaccgtttcagtccat-3′ and reverse 5′-cca cgg acc aaa tatcca
ct-3′; transforming growth factor β-activated kinase 1 (TAK1),
forward 5′-cca tcc caa tgg cgt atc tta c-3′ and reverse 5′-tca tcc
tgg tcc aat tct gca a-3′; GAPDH, forward 5′-act tga agg gtg gag cca
aa-3′ and reverse 5′-cca gga aa tga gct tga ca-3′. The Cq value for
each gene was detected and the expression levels of target genes
were calculated using the 2−ΔΔCq method (12). RT-qPCR was performed on a real-time
PCR system (LightCycler 96; Roche Diagnostics, Basel, Switzerland)
with the following procedure: Initial denaturation at 95°C for 5
min, denaturation at 95°C for 30 sec, annealing at 60°C for 13 sec
and extension at 72°C for 30 sec, for 37 cycles. The reaction
sample consisted of 10 µl 2X SYBR Fast qPCR mix, 0.8 µl
forward/reverse primers (10 mM), 0.4 µl 50X ROX Reference Dye II
and 2 µl cDNA template.
Western blot analysis
Myocardial tissues were collected for protein
isolation (ReadyPrep; GE Healthcare Life Sciences). The protein
concentrations were determined using a BCA protein assay kit
(Thermo Fisher Scientific, Inc.). 25 µg proteins from each group
were obtained for sodium dodecyl sulfate polyacrylamide gel
electrophoresis (12%) and transferred onto nitrocellulose membranes
for western blot analysis. The membranes were incubated with the
following primary antibodies: p38MAPK (1:2,000; cat. no.
sc-17852-R, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), TAK1
(1:2,000; cat. no. sc-7967, Santa Cruz Biotechnology, Inc.) and
actin (1:3,000; cat. no. sc-58673, Santa Cruz Biotechnology, Inc.).
Following incubation with the primary antibodies at 4°C overnight,
the nitrocellulose membranes were washed three times and incubated
with secondary antibody (HRP-labeled goat anti-rabbit IgG; cat. no.
A16104SAMPLE, Thermo Fisher Scientific, Inc.) at 4°C for 2 h. The
staining of the blots was enhanced using an ECL kit (Thermo Fisher
Scientific, Inc.). The densities of the blots were quantified using
Quantity One software (v4.62; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Statistical analyses
Data are presented as the mean ± standard error of
the mean. Statistical analyses were performed using one-way
analysis of variance with GraphPad Prism 6.0 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibition of p38MAPK potentiates the
protective effect of MSC therapy against acute MI injury
An acute MI model was produced using rats by
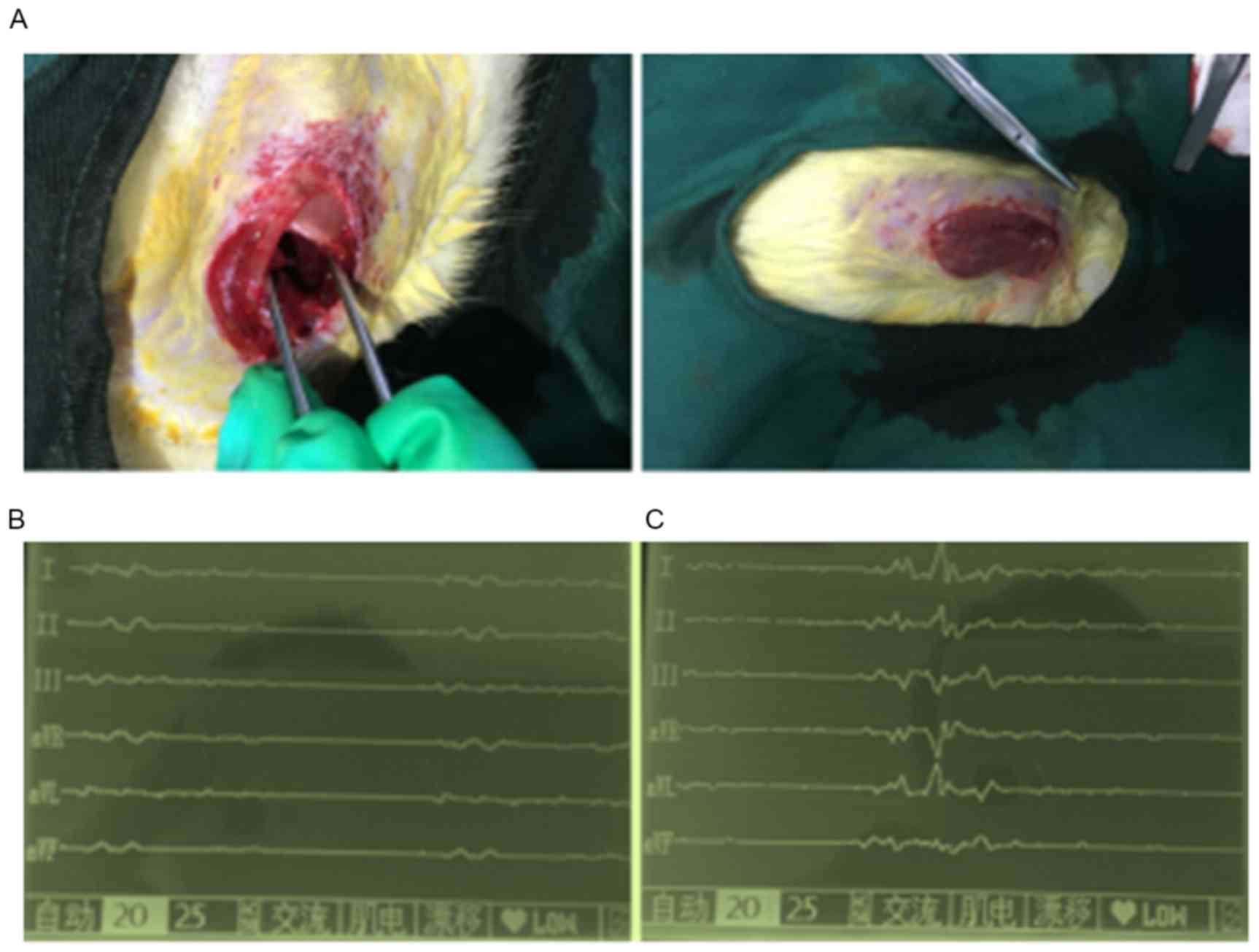
ligating the anterior descending branch of the coronary artery. The
modeling process is shown in Fig.
2A. During the present study, four rats died due to
infection-induced heart failure. The ECG examination showed that
the model rats exhibited acute anterior ST elevation (Fig. 2B and C). At 4 weeks post-treatment,
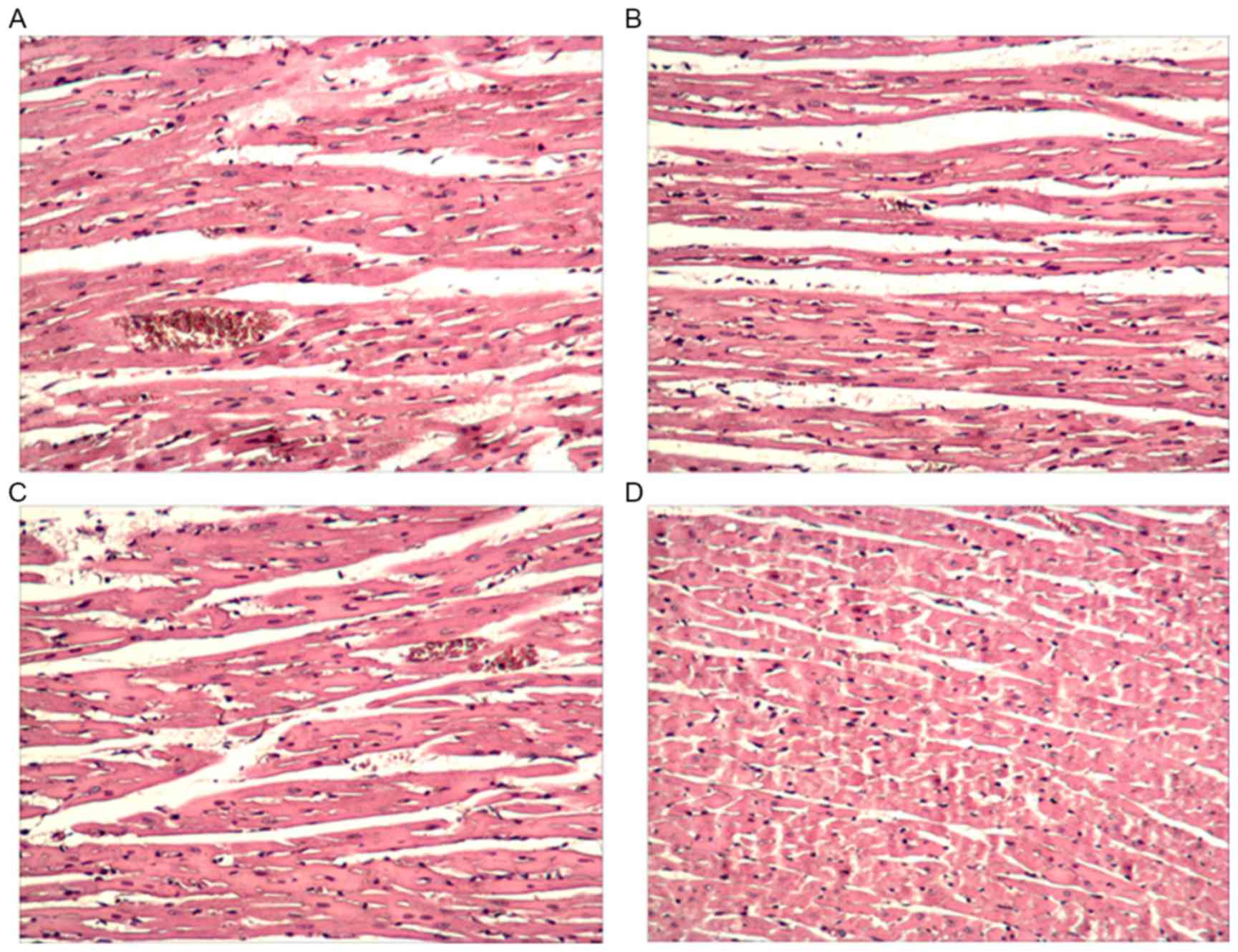
the heart tissues were collected for HE staining. As shown in
Fig. 3A, the cells in the model
control group were loosely arranged with numerous bubbles,
inflammatory cell infiltration and tissue injury. Following
treatment with MSCs or SB203580, the cells were tightly arranged
with few bubbles (Fig. 3B and C).
The combined application of MSCs with SB203580 exhibited optimal
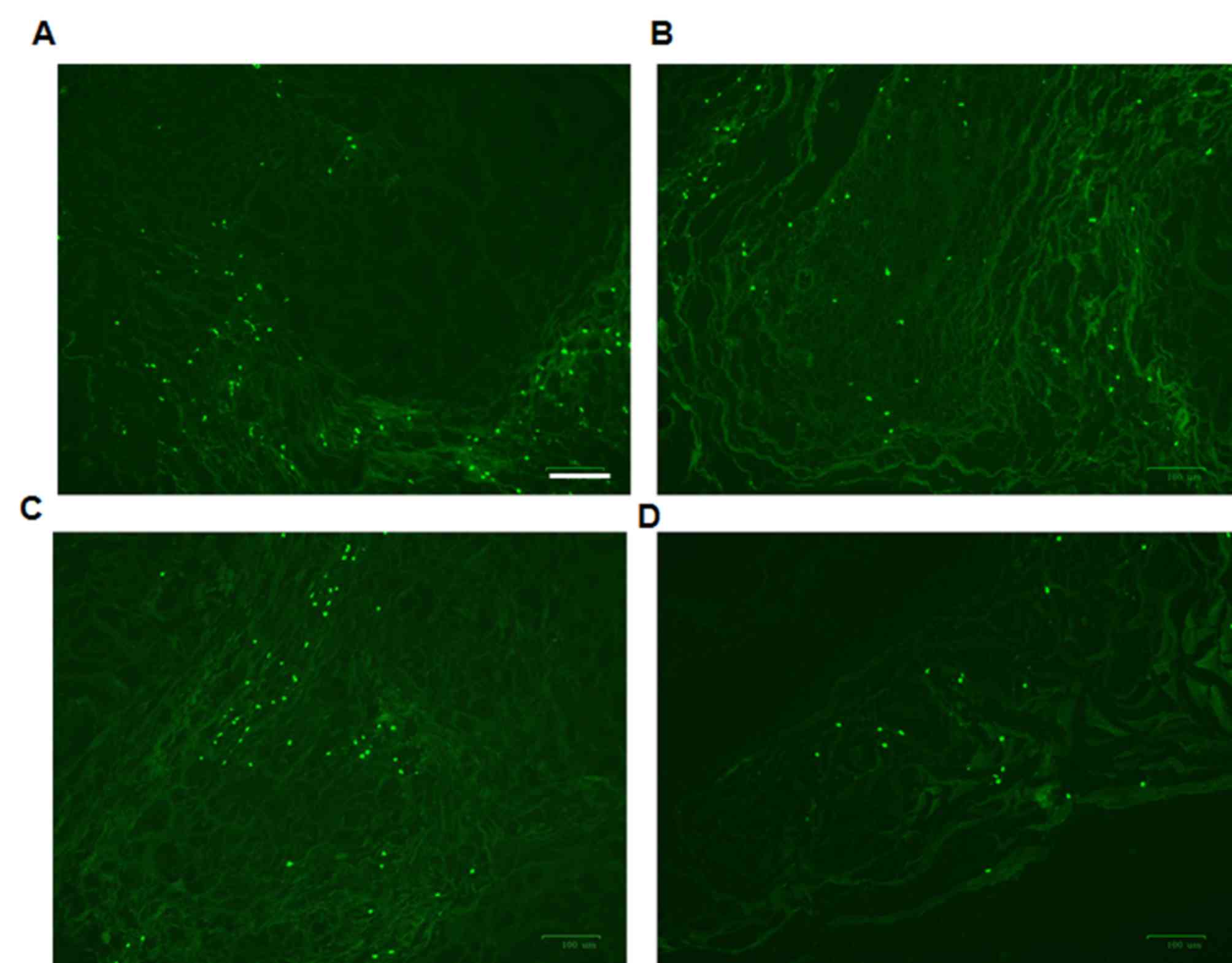
effects (Fig. 3D). A TUNEL assay
was also performed to detect apoptosis. Apoptotic cells in the
control group were apparent (Fig.
4A). Following treatment with MSCs or SB203580, the number of
apoptotic cells was significantly decreased (Fig. 4B and C). Consistent with the HE
staining, the combined application of MSCs with SB203580 (Fig. 4D) showed optimal effects in
decreasing apoptosis.
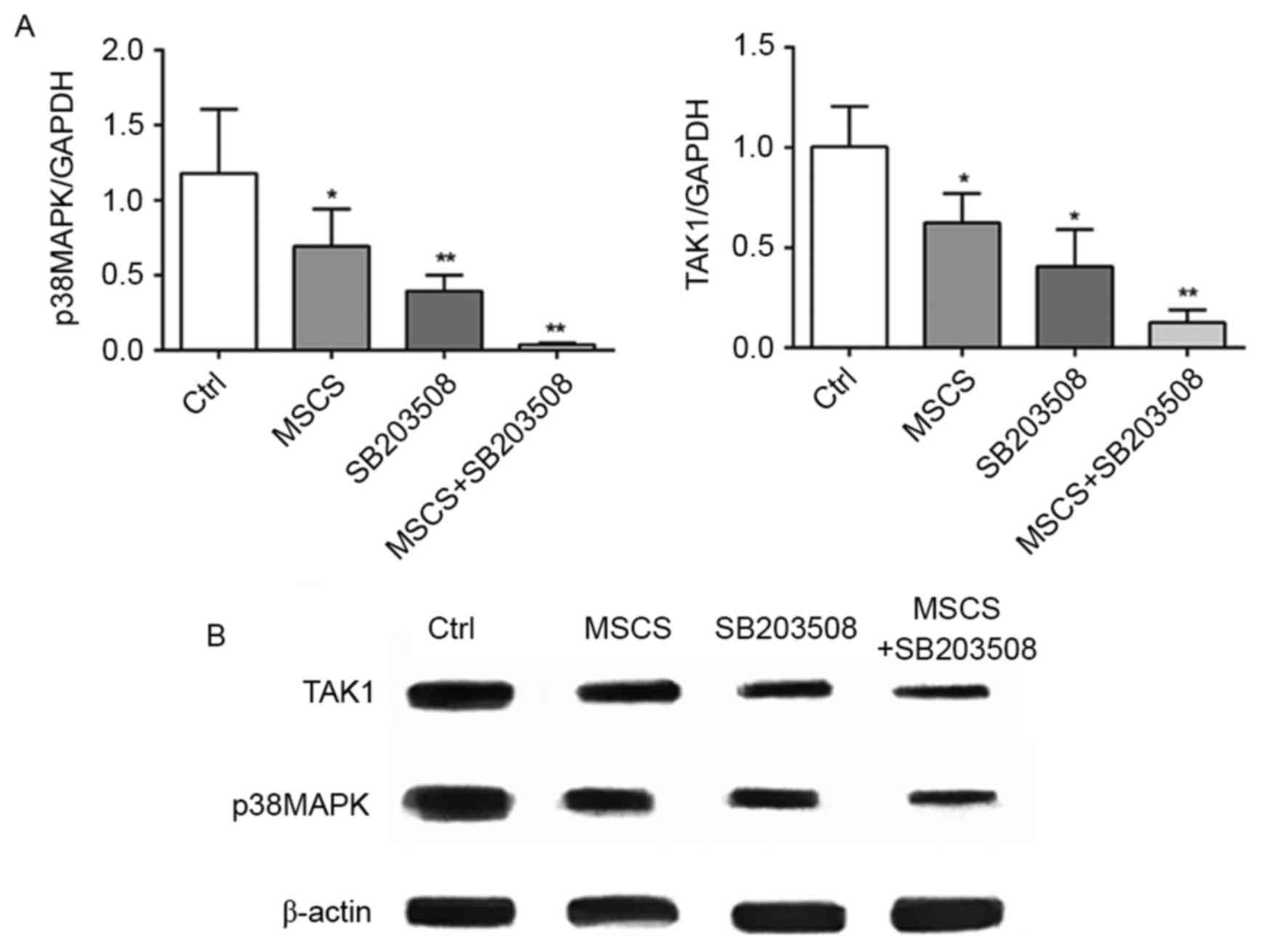
Inhibition of p38MAPK and MSC therapy
functions through the p38MAPK-TAK1 pathway
mRNA and proteins were isolated to examine the
underlying mechanisms. The expression levels of p38MAPK and TAK1
were determined. Compared with the control group, MSC therapy or
SB203580 injection significantly decreased the expression of
p38MAPK at the mRNA (Fig. 5A) and
protein (Fig. 5B) levels. In
addition, the combined application of MSC therapy with SB203580 led
to a further decrease in the expression of p38MAPK. The expression
of TAK1 was also detected, which is upstream of the p38MAPK
signaling pathway in the different groups. A consistent trend was
observed in the groups as shown for p38MAPK (Fig. 5A and B).
Discussion
The MAPK family is a family of serine/threonine
protein kinases, which can transfer signal transduction from the
extracellular environment to the nucleus through the conservative
three-cascade reaction (MAPKKK-MAPKK-MAPK) (13). There are four classical MAPK
signaling pathways, including the extracellular signal-regulated
kinase (ERK) signaling pathway, c-Jun N-terminal
kinase/stress-activated protein kinase, p38 MAPK signaling pathway
and the ERK5/big MAPK 1 signaling pathway. These signaling pathways
have functions in several biological processes, including
regulating cell migration, apoptosis, differentiation and
proliferation (14). In the
present study, it was found that the p38MAPK signaling pathway was
critical in potentiating MSC therapy in MI injury.
The p38MAPK signaling pathway is one of the MAPK
pathways, which can be activated by stress, cytokine stimulation,
insulin and growth factor stimulation. In normal immune and
inflammatory reactiona, the p38MAPK signaling pathway is activated
to phosphorylate transcription factors and regulate gene
expression, being involved in a variety of intracellular biological
activities (15). Upstream of the
p38MAPK signal transduction pathway, TAK1 regulates the
phosphorylation of p38MAPK through the activation of TGF-β.
Therefore, the expression or activation of TAK1 is an important
indicator of the activity of the TGF-β-TAK1-p38MAPK signaling
pathway. Several studies have suggested that the p38MAPK pathway is
activated in the MI model (5,16)
and p38MAPK has become a therapeutic target for heart diseases
(17,18). However, other studies have
demonstrated the pro-survival function of p38MAPK in MI (19,20).
In the present study, it was demonstrated that the inhibition of
p38MAPK alone ameliorated the injury induced by MI. The expression
of p38MAPK was also decreased following treatment with p38MAPK
inhibitor. The inhibition of p38MAPK potentiated the protective
effect of MSC therapy in MI.
Acute coronary artery occlusion causes blood flow
interruption and leads to necrosis of heart tissue. Following MI,
inflammatory cell infiltration, and serum levels of IL-6 and TNF-α
are upregulated, suggesting that inflammation is closely associated
with MI (21,22). In the present study, ligation of
the left coronary anterior descending branch was performed to
produce MI, and the MI model was confirmed by ST elevation and
pathological changes. This animal model is widely used to examine
the mechanisms and enable screening of therapeutic agents. In the
present study, the MI model was treated with p38MAPK inhibitor
and/or MSCs twice each week for 4 weeks. MSCs are a type of
non-hematopoietic stem cell found in bone marrow, which has the
potential to differentiate into osteoblasts, fibroblasts, reticular
cells, fat cells and endothelial cells. MSCs are characterized by
their abundance, biodegradability and regeneration (22,23).
Previously, MSC therapy has been used in the treatment of several
diseases, including acute MI (24). In the present study, a Percoll
lymphocyte separating system was applied to obtain MSCs. This
separation system has the advantage of obtaining high purity MSCs.
In the present study, the MI rats were randomly divided into four
groups. Following MSC therapy or injection with the p38MAPK
inhibitor, the pathological changes observed in the heart tissue
were ameliorated. In addition, the combined application of MSCs
with the p38MAPK inhibitor showed additive effects. The
ameliorative effects were reflected by a decrease in inflammation
and morphological changes. The results of the TUNEL assay also
indicated that that number of apoptotic cells following MI was
significantly attenuated by MSC therapy or the inhibition of
p38MAPK.
In conclusion, the present study evaluated the
effects of inhibiting p38MAPK on MSC therapy against MI injury.
Although the inhibition of p38MAPK or MSC therapy alone exerted
mild protective effects, their combined application had a superior
effect. These findings provide novel implications for the clinical
application of MSC therapy for MI.
References
|
1
|
Pitts R, Daugherty SL, Tang F, Jones P, Ho
PM, Tsai TT, Spertus J and Maddox TM: Optimal secondary prevention
medication use in acute myocardial infarction patients with
nonobstructive coronary artery disease is modified by management
strategy: Insights from the TRIUMPH Registry. Clin Cardiol. Apr
7–2017.(Epub ahead of print) doi: 10.1002/clc.22686. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shibutani H, Akita Y, Yutaka K, Yamamoto
S, Matsui Y, Yoshinaga M, Karakawa M and Mori Y: Acute myocardial
infarction with ‘wrap around’ right coronary artery mimicking
Takotsubo cardiomyopathy: A case report. BMC Cardiovasc Disord.
16:712016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun HJ, Zhao MX, Ren XS, Liu TY, Chen Q,
Li YH, Kang YM, Wang JJ and Zhu GQ: Salusin-β promotes vascular
smooth muscle cell migration and intimal hyperplasia after vascular
injury via ROS/NFκB/MMP-9 pathway. Antioxid Redox Signal.
24:1045–1057. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma SF, Luo Y, Ding YJ, Chen Y, Pu SX, Wu
HJ, Wang ZF, Tao BB, Wang WW and Zhu YC: Hydrogen sulfide targets
the Cys320/Cys529 motif in Kv4.2 to inhibit the Ito potassium
channels in cardiomyocytes and regularizes fatal arrhythmia in
myocardial infarction. Antioxid Redox Signal. 23:129–147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li G, Qian W and Zhao C: Analyzing the
anti-ischemia-reperfusion injury effects of ginsenoside Rb1
mediated through the inhibition of p38α MAPK. Can J Physiol
Pharmacol. 94:97–103. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang F, Zhou H, Du Z, Chen X, Zhu F, Wang
Z, Zhang Y, Lin L, Qian M, Zhang X, et al: Cytoprotective effect of
melatonin against hypoxia/serum deprivation-induced cell death of
bone marrow mesenchymal stem cells in vitro. Eur J Pharmacol.
748:157–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei N, Zhang C, He H, Wang T, Liu Z, Liu
G, Sun Z, Zhou Z, Bai C and Yuan D: Protective effect of saponins
extract from Panax japonicus on myocardial infarction: Involvement
of NF-κB, Sirt1 and mitogen-activated protein kinase signalling
pathways and inhibition of inflammation. J Pharm Pharmacol.
66:1641–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: Physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao H, Xu YN, Luo H, Chen Y, Zhang YY,
Tao L, Jiang Y and Shen XC: OMT inhibited TGF-β1-induced cardiac
fibroblast proliferation via down-regulating p38MAPK
phosphorylation in vitro. Zhongguo Zhong Yao Za Zhi. 40:2168–2173.
2015.(In Chinese). PubMed/NCBI
|
|
10
|
Blanc A, Pandey NR and Srivastava AK:
Synchronous activation of ERK 1/2, p38mapk and PKB/Akt signaling by
H2O2 in vascular smooth muscle cells:
Potential involvement in vascular disease (review). Int J Mol Med.
11:229–234. 2003.PubMed/NCBI
|
|
11
|
Bourzac C, Smith LC, Vincent P, Beauchamp
G, Lavoie JP and Laverty S: Isolation of equine bone marrow-derived
mesenchymal stem cells: A comparison between three protocols.
Equine Vet J. 42:519–527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu G, Li J, He L, Wang X and Hong X:
MPTP-induced changes in hippocampal synaptic plasticity and memory
are prevented by memantine through the BDNF-TrkB pathway. Br J
Pharmacol. 172:2354–2368. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Osaki LH and Gama P: MAPKs and signal
transduction in the control of gastrointestinal epithelial cell
proliferation and differentiation. Int J Mol Sci. 14:10143–10161.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
del Barco Barrantes I and Nebreda AR:
Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans.
40:79–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miloso M, Scuteri A, Foudah D and Tredici
G: MAPKs as mediators of cell fate determination: An approach to
neurodegenerative diseases. Curr Med Chem. 15:538–548. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arabacilar P and Marber M: The case for
inhibiting p38 mitogen-activated protein kinase in heart failure.
Front Pharmacol. 6:1022015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Donoghue ML, Glaser R, Aylward PE,
Cavender MA, Crisp A, Fox KA, Laws I, Lopez-Sendon JL, Steg PG,
Theroux P, et al: Rationale and design of the LosmApimod To Inhibit
p38 MAP kinase as a TherapeUtic target and moDify outcomes after an
acute coronary syndromE trial. Am Heart J. 169:622–630.e6. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang M, Li Z, Zhang X, Xie X, Zhang Y,
Wang X and Hou Y: Rosuvastatin attenuates atrial structural
remodelling in rats with myocardial infarction through the
inhibition of the p38 MAPK signalling pathway. Heart Lung Circ.
24:386–394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitra A, Ray A, Datta R, Sengupta S and
Sarkar S: Cardioprotective role of P38 MAPK during myocardial
infarction via parallel activation of α-crystallin B and Nrf2. J
Cell Physiol. 229:1272–1282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li C, He J, Gao Y, Xing Y, Hou J and Tian
J: Preventive effect of total flavones of Choerospondias axillaries
on ischemia/reperfusion-induced myocardial infarction-related MAPK
signaling pathway. Cardiovasc Toxicol. 14:145–152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang M, Yang D, Xiang M and Wang J: Role
of interleukin-6 in regulation of immune responses to remodeling
after myocardial infarction. Heart Fail Rev. 20:25–38. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Somasuntharam I, Yehl K, Carroll SL,
Maxwell JT, Martinez MD, Che PL, Brown ME, Salaita K and Davis ME:
Knockdown of TNF-α by DNAzyme gold nanoparticles as an
anti-inflammatory therapy for myocardial infarction. Biomaterials.
83:12–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sekar D, Saravanan S, Karikalan K,
Thirugnanasambantham K, Lalitha P and Islam VI: Role of microRNA 21
in mesenchymal stem cell (MSC) differentiation: A powerful
biomarker in MSCs derived cells. Curr Pharm Biotechnol. 16:43–48.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu X, Chen P, Wu Y, Wang K, Xu Y, Chen H,
Zhang L, Wu R, Webster KA, Yu H, et al: MiR-211/STAT5A signaling
modulates migration of mesenchymal stem cells to improve its
therapeutic efficacy. Stem Cells. 34:1846–1858. 2016. View Article : Google Scholar : PubMed/NCBI
|