Introduction
Macrophages are essential components of innate
immunity that serve a role in inflammation and host defense via
production of pro- or anti-inflammatory mediators in response to
various stimuli (1). Macrophages
respond to stimulation in a polarized manner. The differentiation
of macrophages to the classic activation (M1) phenotype is
triggered by interferon (IFN)-γ, bacterial lipopolysaccharide
(LPS), interleukin (IL)-1β, or tumor necrosis factor α (TNF-α),
whereas IL-3 or IL-13 stimulate macrophage differentiation of the
alternative activation phenotype (M2) (2–5). The
M1 phenotype is characterized by the expression of high levels of
pro-inflammatory cytokines. Conversely, M2 macrophages express an
anti-inflammatory functional profile and are associated with wound
repair and angiogenesis (6).
Therefore, inflammatory associated diseases may result from a
sustained pro-inflammatory reaction and failure of
anti-inflammatory control mechanisms of macrophages.
Activating transcription factor 3 (ATF3), a member
of the mammalian activating transcription factor/cAMP responsive
element binding protein (ATF/CREB) family of transcription factors,
is induced in a variety of stressed tissues, including mechanically
and toxin-injured liver tissue, blood-deprived heart tissue and
injured peripheral nerves (7,8). The
transcriptional target of ATF3 varies in different cell types and
conditions, which therefore leads to diverse effects on cell
survival, proliferation and death (9). In neurons, ATF3 is frequently
reported to be a novel neuronal marker of nerve injury, and
induction of ATF3 expression enhances nerve regeneration (10,11).
In cardiac myocytes, ATF3 is a novel cytoprotective factor in
doxorubicin-induced apoptosis (12). Additionally, it is reported that
ATF3 protects renal cells and b-cells against oxidative
stress-induced cell death and apoptosis (13). Therefore, ATF3 is a protective
factor in numerous tissues.
Conversely, ATF3 is an inducible transcriptional
repressor in innate immune cells, which regulates the magnitude and
duration of inducible pro-inflammatory gene expression. Recently,
it was revealed that ATF3 is an important transcriptional regulator
that inhibits the inflammatory response by modulating the
expression of cytokines and chemokines and demonstrated that ATF3
is a negative regulator of Toll-like receptor 4 (TLR4) signaling in
macrophages (14). Activation of
TLR4 by LPS induces the expression of ATF3, which subsequently
inhibits the expression of various inflammatory genes induced by
TLR signaling, including IL-6, IL-12β, and TNF-α. Additionally,
ATF3 may modulate the expression levels of IFN-γ in macrophages by
controlling basal and inducible levels of IFN-β, and the expression
of genes downstream of IFN (15).
Therefore, ATF3 is able to negatively regulate transcription of
pro-inflammatory cytokines in macrophages. Understanding the exact
role of ATF3 in macrophages in the context of inflammation is of
primary concern, and may lead to the design of beneficial
therapeutics for inflammatory associated diseases. However, the
effects of ATF3 on recruitment and anti-inflammatory control
mechanisms of macrophages remain to be investigated.
The present study investigated whether ATF3 exerted
anti-inflammatory activities by modulating M1/M2 differentiation of
macrophages. Macrophage migration and markers of M1/M2 macrophages
were tested following overexpression of ATF3. Subsequently, the
underlying mechanism of how overexpression of ATF3 modulates
macrophage migration and M1/M2 polarization was investigated.
Materials and methods
Cell culture
Mouse macrophage RAW 264.7 cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagle's medium (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum,
100 U/ml streptomycin and 100 U/ml penicillin (Gibco; Thermo Fisher
Scientific, Inc.) in a humidified atmosphere at 37°C and 5%
CO2. Wntpalmitoyltransferase inhibitor
(IWP-2,N-(6-M
ethyl-2-benzothiazolyl)-2-[(3,4,6,7-tetrahydro-4-oxo-3-phenylthieno[3,2-d]pyrimidin-2-yl)thio]-acetamide)
was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany)
and 30 µM was used to treat the cells.
Plasmids and siRNA transfection
For overexpression of ATF3, ATF3 cDNA generated from
reverse transcription-quantitative PCR (RT-qPCR; The primers used
for the cloning were: 5′-AAAAAGCTTATGATGCTTCAACATCCAGG-3′ and
5′-TTTGAATTCTTAGCTCTGCAATGTTCCTT-3′) was subcloned into a pcDNA™3.1
plasmid (Invitrogen; Thermo Fisher Scientific, Inc.) between Hind3
and EcoR I sites to express ATF3 in abundance in E. coli
DH5α cells to generate an ATF3 expression plasmid. The third
generation of cells (5×105) were seeded into a 24-well
plate and transfected with the pcDNA-ATF3 or the empty
vector negative control plasmid, psDNA3.1(−), using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol at 37°C. After incubation for 48 h, cells
were harvested and ATF3 expression was determined. To knockdown
ATF3 and tenascin (TNC), The third generation of cells
(5×105) were seeded into a 24-well plate and ATF3
siRNA, its negative control (sham), or TNC siRNA and its negative
control siRNA (scramble) were transfected at 37°C using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.; 0.1
µM siRNA with 20 µl Lipofectamine). Following transfection for 48
h, the transfection medium was exchanged for normal medium and the
cells were used in the subsequent experiments or harvested for ATF3
and TNC expression measurement.
Migration assays
Migration assays were performed using 8 µm pore size
filters within 24-well transwell cell culture chambers with
polycarbonate filters (Corning Life Sciences, Glendale, Arizona,
USA) as previously described (16). A total of 5×105 cells
were transfected with pcDNA-ATF3 or the psDNA3.1(−), and
subsequently seeded into the upper chamber of the transwell.
Transwells were either uncoated (5 µm pore size) or coated with
Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA) diluted 1:50 (8
µm pore size). As a chemoattractant, monocyte chemoattractant
protein-1 (MCP-1; 100 ng/ml; R&D Systems, Inc., Minneapolis,
MN, USA) was present in the lower wells. After incubation for 18 h
at 37°C in 5% CO2, cells in the lower chambers that
passed through the filter were counted under a Carl Zeiss Primo
Vert microscope (Carl Zeiss AG, Oberkochen, Germany).
Western blotting
Total protein was lysed using
radioimmunoprecipitation assay (RIPA) buffer (Beyotime Institute of
Biotechnology, Nantong, China). After determining the protein
concentration with a Bicinchoninic acid assay kit (Beyotime
Institute of Biotechnology, Haimen, China), ~30 µg of proteins were
loaded onto 10% gels and subjected to SDS-PAGE, prior to transfer
onto polyvinylidene difluoride membranes (Invitrogen; Thermo Fisher
Scientific, Inc.). Subsequently, membranes were blocked for 1 h in
a blocking solution (5% skimmed milk, 0.05% Tween 20) at 37°C and
probed with the following primary antibodies: Mouse anti-β-catenin
(2698; 1:1,000), rabbit anti-c-myc 9402; 1:1,000) and anti-cyclin
D1 (2292; 1:1,000; all from Cell Signaling Technology, Inc.,
Danvers, MA, USA), and mouse anti-ATF3 (sc81189; 1:500), rabbit
anti-TNC (sc20932; 1:1,000), and mouse anti-β-actin (sc130300;
1:1,000; all from Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
at 4°C overnight. Horseradish peroxidase-conjugated rabbit
anti-mouse (sc358917; 1:3,000; Santa Cruz Biotechnology) or goat
anti-rabbit (RPN4301; 1:5,000; GE Healthcare Life Sciences,
Chalfont, UK) secondary antibodies were added to the membranes for
1 h at room temperature. Protein bands were detected using the
Enhanced Chemiluminescence substrate detection system (Amersham
Biosciences Corporation, Piscataway, USA). The intensities of the
resulting bands were quantified using Carestream Molecular Imaging
software version 5.0.2.30 (Carestream Health, Woodbridge, CT, USA)
on a Gel Logic 2000 imaging system (Kodak, Rochester, NY, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Reverse transcription PCR was performed on an
Applied Biosystems® 7500 fast sequence detection system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Briefly,
total RNA was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed using the MMLV Reverse Transcriptase kit (Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's protocol. qPCR was performed using SYBR Green
reagent (Qiagen, Inc., Valencia, CA, USA). Cycling conditions were
as follows: An initial predenaturation step at 95°C for 5 min,
followed by 40 cycles of denaturation at 95°C for 15 sec, annealing
at 58°C for 30 sec and extension at 72°C for 20 sec. The experiment
was performed three times. The relative expression levels of the
target genes were calculated using the 2−∆∆Cq method
(17) and normalized to GAPDH.
Forward and reverse sequences of the primers used for all target
genes and GAPDH are listed in Table
I.
 | Table I.Primers used. |
Table I.
Primers used.
| Gene | Primer
sequences |
|---|
| MCP-1 | F:
5′-TCAGCCAGATGCAGTTAACGC-3′ |
|
| R:
5′-TGGATGCATTAGCTTCAGATTTACG-3′ |
| CD16 | F:
5′-GACAGTGTGACTCTGAAG-3′ |
|
| R:
5′-GCACCTGTACTCTCCAC-3′ |
| iNOS | F:
5′-CCCTTCCGAATTTCTGGCAGCAGC-3′ |
|
| R:
5′-GGCTGTCAGAGCCTCGTGGCTTTGG-3′ |
| TNF-19α | F:
5′-TTGACCTCAGCGCTGAGTTG-3′ |
|
| R:
5′-CCTGTAGCCCACGTCGTAGC-3′ |
| Arg-1 | F:
5′-CAGAAGAATGGAAGAGTCAG-3′ |
|
| R:
5′-CAGATATGCAGGGAGTCACC-3′ |
| CD163 | F:
5′-ATGGGTGGACACAGAATGGTT-3′ |
|
| R:
5′-CAGGAGCGTTAGTGACAGCAG-3′ |
| Mrc-1 | F:
5′-TCTTTTACGAGAAGTTGGGGTCAG-3′ |
|
| F:
5′-ATCATTCCGTTCACAGAGGG-3′ |
| PPARγ | R:
5′-GGAGATCTCCAGTGATATCGACCA-3′ |
|
| F:
5′-ACGGCTTCTACGGATCGAAACT-3′ |
| TNC | R:
5′-GTTTGGAGACCGCAGAGAAGAA-3′ |
|
| F:
5′-TGTCCCCATATCTGCCCATCA-3′ |
| GAPDH | R:
5′-AGGTCGGTGTGAACGGATTTC-3′ |
|
| F:
5′-TGTAGACCATGTAGTTGAGGTCA-3′ |
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparisons between two groups were analyzed by unpaired Student's
t-test. Experiments were repeated three times. P<0.05 was
considered to indicate a statistically significant difference
analyzed using SPSS version 18.0 (SPSS, Inc., Chicago, IL,
USA).
Results
Overexpression of ATF3 promotes the
migration of macrophages
To examine whether ATF3 regulates macrophage
migration, the pcDNA-ATF3 plasmid and ATF3 siRNA were
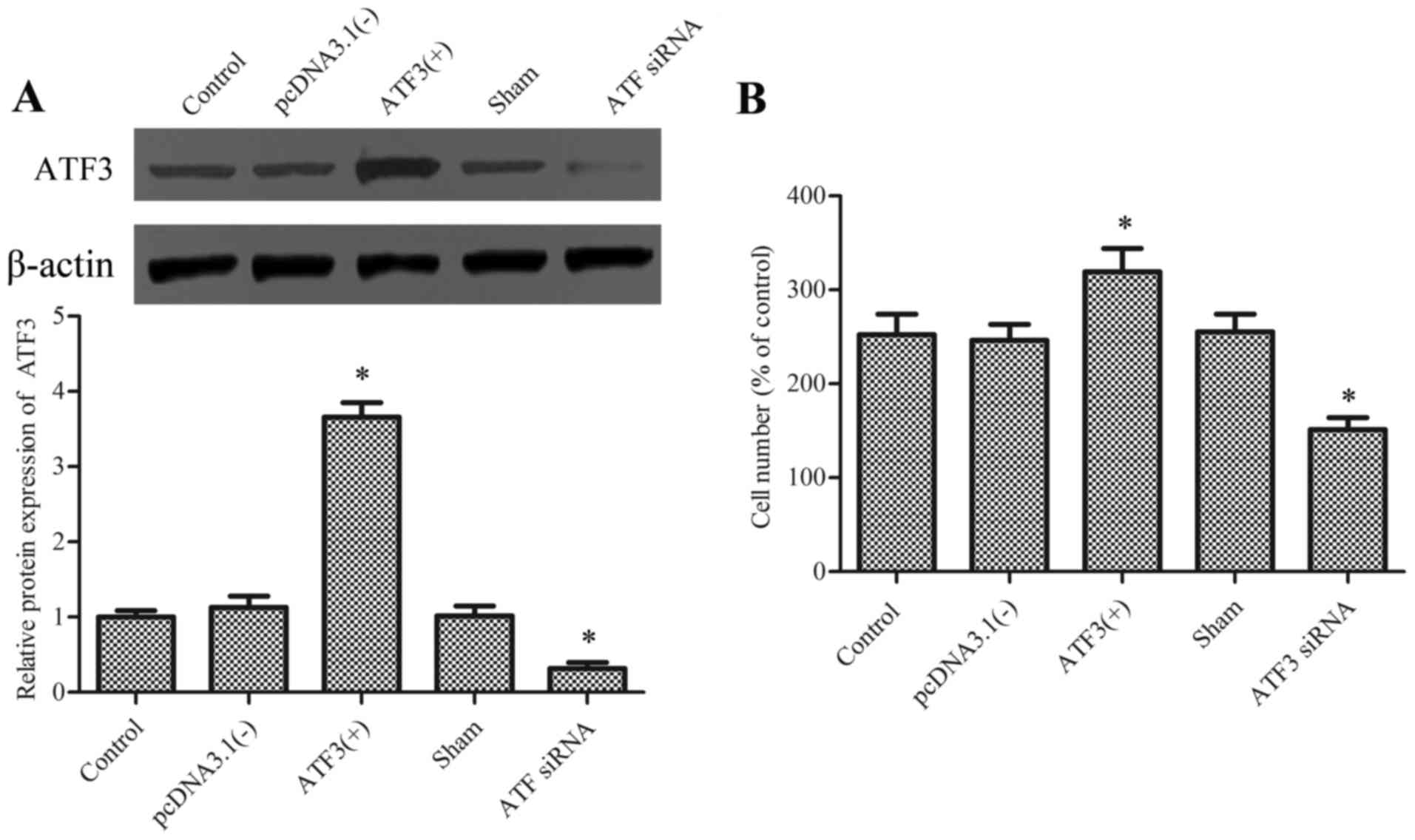
utilized to overexpress and knockdown the ATF3 protein,
respectively (Fig. 1A). Cell
viability was tested using an MTT assay and the ATF3 siRNA had no
effect on cellular viability (data not show).
Subsequently, migration of macrophages was evaluated
in the presence of chemotaxis-inducing agent MCP-1. Results
demonstrated that overexpression of ATF3 significantly promoted the
migration of macrophages compared with the empty vector
[pcDNA3.1(−)], whereas knockdown of ATF3 significantly reduced
migration compared with the sham group and the difference between
the control and sham group were not significant (Fig. 1B).
Overexpression of ATF3 promotes
macrophage differentiation of the M2 phenotype
To determine the function of ATF3 in the process of
macrophage polarization, markers of the M1 phenotype [MCP-1,
inducible nitric oxide synthase (iNOS), cluster of differentiation
(CD) 16 and TNF-α] and the M2 phenotype [CD163, mannose receptor C
type 1 (Mrc-1), arginase 1 (Arg-1) and peroxisome
proliferator-activated receptor γ (PPARγ)] were measured. Results
revealed that the mRNA expression levels of M1-associated genes
encoding MCP-1, iNOS, CD16 and TNF-α, were reduced following
transfection with pcDNA-ATF3 compared with the empty vector
control group, and were enhanced following transfection with
ATF3 siRNA compared with the sham group (P<0.05; Fig. 2A). In addition, overexpression of
ATF3 in RAW 264.7 cells enhanced the expression levels of
the genes encoding CD163, Mrc-1, Arg-1 and PPARγ compared with the
empty vector control group, and these levels were then reduced by
knockdown of ATF3 compared with the sham group (P<0.05;
Fig. 2B). These results suggested
that ATF3 promotes polarization of M2 in macrophages.
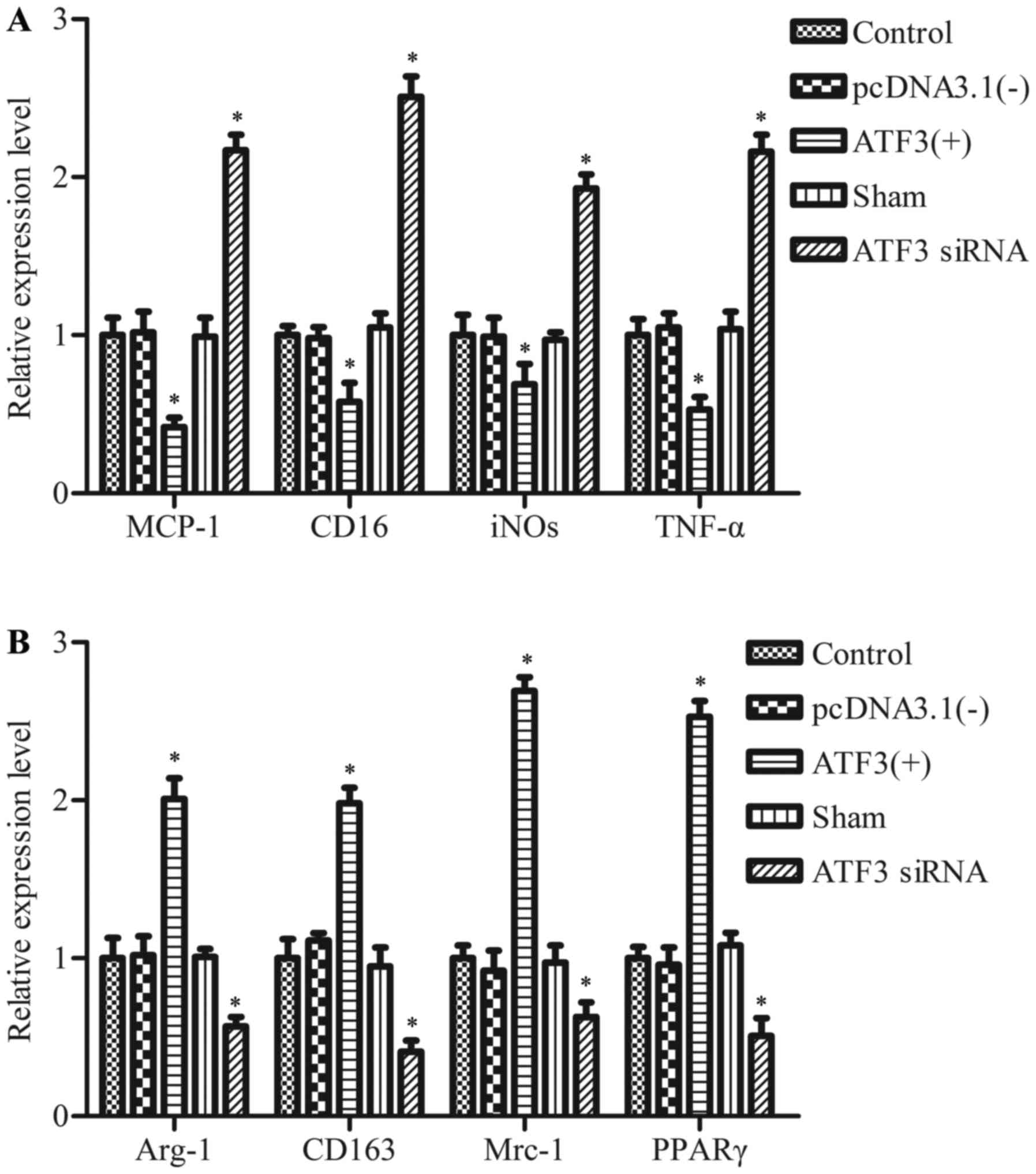 | Figure 2.Effect of ATF3 overexpression on the
characterization of M1 or M2 macrophage polarization. RAW 264.7
cells transfected with pcDNA3.1 (−), pcDNA-ATF3 (ATF3 (+)),
ATF3 siRNA or negative control siRNA (sham) for 48 h. mRNA
expression levels of markers of the (A) M1 state and (B) M2 state
were determined using reverse transcription-quantitative polymerase
chain reaction. Data are expressed as the mean ± standard deviation
of the three independent experiments. *P<0.05 vs. sham or
pcDNA3.1 (−). ATF3, activating transcription factor 3; siRNA, short
interfering RNA; pcDNA3.1(−), empty vector; MCP-1, monocyte
chemoattractant protein-1; iNOS inducible nitric oxide synthase;
CD, cluster of differentiation; TNF-α, tumor necrosis factor;
Mrc-1, mannose receptor C type 1; Arg-1, arginase 1; PPARγ,
peroxisome proliferator-activated receptor γ. |
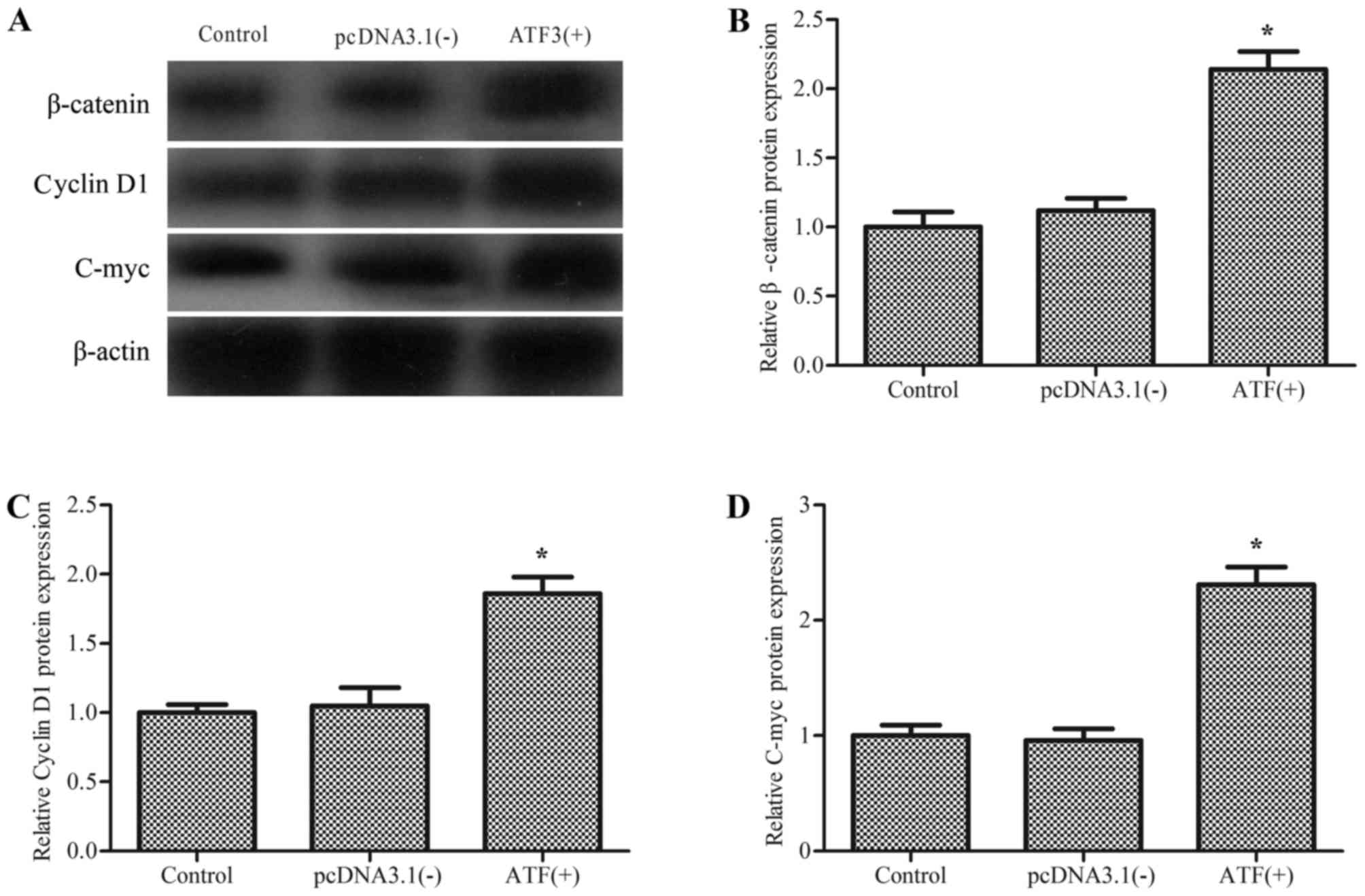
Overexpression of ATF3 activates the
Wnt/β-catenin signaling pathway
β-catenin, encoded by the CTNNB1 gene, is a
transcriptional co-activator and serves a role in the inflammatory
response (18,19). To explore the mechanism of ATF3
regulation of macrophage migration and M2 polarization, the effect
of ATF3 on Wnt/β-catenin signaling was investigated. As
demonstrated in Fig. 3,
overexpression of ATF3 resulted in enhanced expression levels of
β-catenin and its target genes cyclin D1 and c-myc compared with
cells transfected with the empty vector control. This suggested
that overexpression of ATF3 induced the activation of the
Wnt/β-catenin signaling pathway in macrophages.
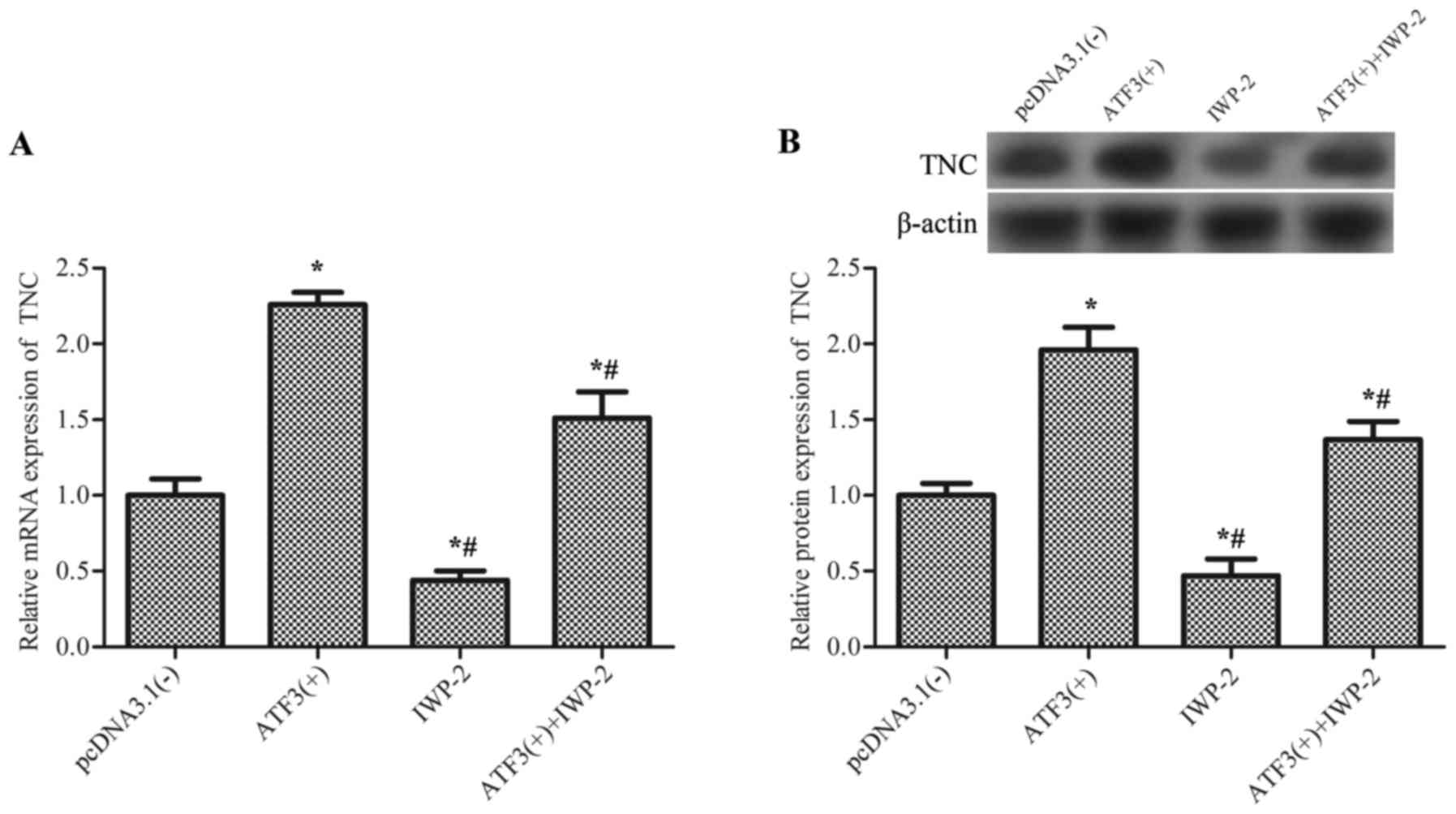
TNC is activated by ATF3 via the
Wnt/β-catenin signaling pathway
The gene encoding TNC is a canonical Wnt target
(20), and serves a role in
macrophage behavior and function (21,22).
Therefore, the association between ATF3 and TNC was investigated.
Results revealed that overexpression of AFT3 significantly
upregulated the mRNA expression levels of TNC (Fig. 4A) and the TNC protein (Fig. 4B) compared with the empty vector
control, and this effect was partially inhibited by IWP-2, an
inhibitor of Wnt/β-catenin signaling, which suggested that ATF3
activates TNC via the Wnt/β-catenin signaling pathway.
ATF3 regulates the migration and
polarization of M2 macrophages by upregulating TNC expression
levels
To further investigate the role of TNC in
ATF3-mediated macrophage migration and polarization, cells were
transfected with pcDNA-ATF3 and knockdown of TNC using
TNC siRNA (Fig. 5A), prior
to determining cell viability and the expression levels of
M2-associated genes. Results revealed that transfection of
pcDNA-ATF3 significantly promoted macrophage migration
compared with the empty vector control, whereas transfection with
TNC siRNA reduced the migration induced by pcDNA-ATF3
compared with the scrambled control group (Fig. 5B). In addition, the expression
levels of genes associated with the M1 phenotype that were
downregulated by pcDNA-ATF3 were enhanced by transfection
with TNC siRNA (Fig. 5C),
whereas M2 gene expression levels that were upregulated by
pcDNA-ATF3 were inhibited by TNC siRNA (Fig. 5D).
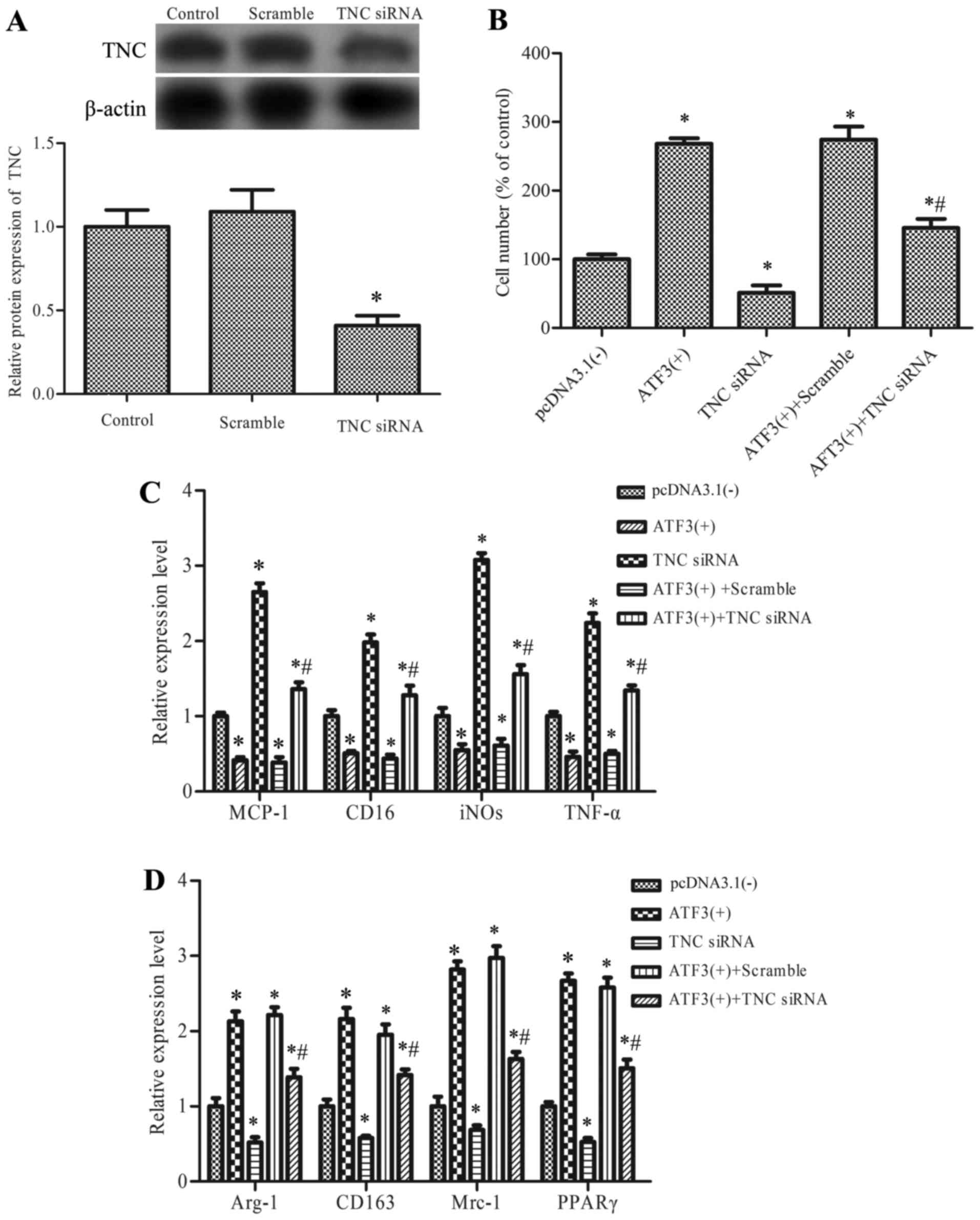 | Figure 5.The role of TNC in the regulation of
ATF3 on macrophage migration and M1/M2 polarization. (A) RAW 264.7
cells were transfected with TNC siRNA or negative control
siRNA (scramble). Cells without transfect vector were defined as
the control group. TNC protein expression levels were determined
using western blot and densitometric analysis. Cells were treated
with pcDNA-ATF (ATF3 (+)), TNC siRNA, ATF3(+)
+scramble or ATF3(+) +TNC siRNA. Cells without transfect
vector were defined as the control group. (B) macrophage migration,
(C) mRNA expression of markers of M1 state and (D) mRNA expression
of markers of the M2 state, were measured. Data are expressed as
the mean ± standard deviation of the three independent experiments.
*P<0.05 vs. pcDNA3.1(−) and #P<0.05 vs. ATF3(+) +
scramble. ATF3, activating transcription factor 3; TNC, tenascin;
siRNA, short interfering RNA; MCP-1, monocyte chemoattractant
protein-1; iNOS, inducible nitric oxide synthase; CD, cluster of
differentiation; TNF-α, tumor necrosis factor; Mrc-1, mannose
receptor C type 1, Arg-1; arginase 1; PPARγ, peroxisome
proliferator-activated receptor γ. |
These results suggested that ATF3 regulates
macrophage migration and M2 polarization, in part, by upregulation
of TNC.
Discussion
Macrophages are primary producers of
pro-inflammatory mediators and the migration of macrophages from
the circulation into injured tissues serves a crucial role in wound
healing. Macrophage polarization is closely associated with
homeostatic tissue remodeling, resolution of inflammation,
remodeling and tissue repair (23). ATF3 is a transcriptional modulator,
induced by LPS and the TLR-dependent injury response, that
negatively regulates numerous pro-inflammatory cytokines and
chemokines in macrophages (24).
Previous reviews have reported that ATF3 modulates the expression
levels of a number of inflammatory genes (25). Therefore, the present study
investigated the effect of ATF3 on macrophage migration and
polarization.
Migration of macrophages serves a role in the onset
and course of inflammation. Chen et al (26) revealed that the epithelium-derived
exosomal ATF3 inhibited the expression of monocyte chemoattractant
protein 1 and macrophage migration, and Zmuda et al
(27) suggested that ATF3 knockout
islets inhibited macrophage recruitment in vivo. The present
study revealed that overexpression of ATF3 promoted M2 marker
expression and suppressed the expression levels of M1-associated
markers. This suggested that ATF3 may reverse M1-polarized
macrophages to M2 phenotypes, and that the ATF3-mediated
anti-inflammatory function is closely associated with macrophage
phenotype. It is evident that ATF3 serves an important role in
injury in numerous tissues, and it was revealed that ATF3 may
protect against acute kidney and lung injury (28,29).
M2 macrophages exhibit immunoregulatory functions including defense
against infection, promotion of angiogenesis and wound healing
(30). The results of the present
study suggested that ATF3 may be a protective regulator for injured
tissues by promoting the polarization of M2 macrophages.
ATF3 serves a role in the cellular adaptive-response
network in response to signals perturbing homeostasis (25). Previous data has suggested that
ATF3 activates the Wnt/β-catenin signaling pathway in human breast
cancer cells (31), which concurs
with the results of the present study, as overexpression of ATF3
activated the Wnt/β-catenin pathway in macrophages. Various
components of the Wnt/β-catenin signaling pathway are involved in
the inflammatory response, including in inflammatory conditions in
humans and in the LPS-treated macrophage cell line (32). Wnt upregulates the expression
levels of TNC (20), which is a
large hexameric extracellular matrix glycoprotein that is highly
expressed during embryonic development, cancer invasion and wound
healing. It has been reported that TNC may be expressed in
macrophages and regulates their behavior and function (21,22).
TNC has been suggested to act as pro-inflammatory modulator in
various diseases (33,34) and accelerates macrophage migration
(35). The results of the present
study demonstrated that overexpression of ATF3 upregulated TNC
expression levels via the Wnt/β-catenin signaling pathway. In
addition, the present study demonstrated that TNC was an effector
for ATF3 in modulating macrophage migration and M2
polarization.
In conclusion, the present study revealed that
overexpression of ATF3 in macrophage cells promoted their migration
to chemotaxis-inducing agent MCP-1, and influenced the M1/M2
phenotype. These results emphasize that ATF3 expression levels
affect macrophages, partially by upregulating TNC via the
Wnt/β-catenin signaling pathway. The present study may provide an
insight into the positive regulation of ATF3 on macrophage
migration, and tissue regeneration via modulation of the M2
macrophage.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81001196) and
Shaanxi Social Development for Science and Technology Project
(grant no. 2016SF-330).
Glossary
Abbreviations
Abbreviations:
|
ATF3
|
activating transcription factor 3
|
|
TNC
|
tenascin
|
|
IFN-γ
|
interferon-γ
|
|
TNF-α
|
tumor necrosis factor-α
|
|
ATF/CREB
|
activating transcription factor/cAMP
responsive element binding protein
|
|
TLR4
|
Toll-like receptor 4
|
References
|
1
|
Gordon S and Martinez FO: Alternative
activation of macrophages: Mechanism and functions. Immunity.
32:593–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martinez FO, Sica A, Mantovani A and
Locati M: Macrophage activation and polarization. Front Biosci.
13:453–461. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuroda E, Ho V, Ruschmann J, Antignano F,
Hamilton M, Rauh MJ, Antov A, Flavell RA, Sly LM and Krystal G:
SHIP represses the generation of IL-3-induced M2 macrophages by
inhibiting IL-4 production from basophils. J Immunol.
183:3652–3660. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Odegaard JI and Chawla A: Mechanisms of
macrophage activation in obesity-induced insulin resistance. Nat
Clin Pract Endocrinol Metab. 4:619–626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Porcheray F, Viaud S, Rimaniol AC, Léone
C, Samah B, Dereuddre-Bosquet N, Dormont D and Gras G: Macrophage
activation switching: An asset for the resolution of inflammation.
Clin Exp Immunol. 142:481–489. 2005.PubMed/NCBI
|
|
6
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hai T and Hartman MG: The molecular
biology and nomenclature of the activating transcription
factor/cAMP responsive element binding family of transcription
factors: Activating transcription factor proteins and homeostasis.
Gene. 273:1–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hai T, Wolfgang CD, Marsee DK, Allen AE
and Sivaprasad U: ATF3 and stress responses. Gene Expr. 7:321–335.
1999.PubMed/NCBI
|
|
9
|
Thompson MR, Xu D and Williams BR: ATF3
transcription factor and its emerging roles in immunity and cancer.
J Mol Med (Berl). 87:1053–1060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsujino H, Kondo E, Fukuoka T, Dai Y,
Tokunaga A, Miki K, Yonenobu K, Ochi T and Noguchi K: Activating
transcription factor 3 (ATF3) induction by axotomy in sensory and
motoneurons: A novel neuronal marker of nerve injury. Mol Cell
Neurosci. 15:170–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Averill S, Michael GJ, Shortland PJ,
Leavesley RC, King VR, Bradbury EJ, McMahon SB and Priestley JV:
NGF and GDNF ameliorate the increase in ATF3 expression which
occurs in dorsal root ganglion cells in response to peripheral
nerve injury. Eur J Neurosci. 19:1437–1445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nobori K, Ito H, Tamamori-Adachi M, Adachi
S, Ono Y, Kawauchi J, Kitajima S, Marumo F and Isobe M: ATF3
inhibits doxorubicin-induced apoptosis in cardiac myocytes: A novel
cardioprotective role of ATF3. J Mol Cell Cardiol. 34:1387–1397.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida T, Sugiura H, Mitobe M, Tsuchiya
K, Shirota S, Nishimura S, Shiohira S, Ito H, Nobori K, Gullans SR,
et al: ATF3 protects against renal ischemia-reperfusion injury. J
Am Soc Nephrol. 19:217–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nguyen CT, Kim EH, Luong TT, Pyo S and
Rhee DK: TLR4 mediates pneumolysin-induced ATF3 expression through
the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7
cells. Mol Cells. 38:58–64. 2015.PubMed/NCBI
|
|
15
|
Labzin LI, Schmidt SV, Masters SL, Beyer
M, Krebs W, Klee K, Stahl R, Lütjohann D, Schultze JL, Latz E and
De Nardo D: ATF3 is a key regulator of macrophage IFN responses. J
Immunol. 195:4446–4455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lighvani S, Baik N, Diggs JE, Khaldoyanidi
S, Parmer RJ and Miles LA: Regulation of macrophage migration by a
novel plasminogen receptor Plg-R KT. Blood. 118:5622–5630. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JS, Yeo S, Shin DG, Bae YS, Lee JJ,
Chin BR, Lee CH and Baek SH: Glycogen synthase kinase 3beta and
beta-catenin pathway is involved in toll-like receptor 4-mediated
NADPH oxidase 1 expression in macrophages. FEBS J. 277:2830–2837.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kraus C, Liehr T, Hülsken J, Behrens J,
Birchmeier W, Grzeschik KH and Ballhausen WG: Localization of the
human beta-catenin gene (CTNNB1) to 3p21: A region implicated in
tumor development. Genomics. 23:272–274. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pedersen EA, Scannell CA, Menon R and
Lawlor ER: Tenascin C is a canonical Wnt target gene in Ewing
sarcoma and its expression is potentiated by R-spondin. Cancer Res.
74:3085. 2014. View Article : Google Scholar
|
|
21
|
Kimura T, Tajiri K, Hlroe M, et al:
Tenascin-C regulates macrophage behavior during tissue repair after
myocardial infarction in mouse model. Molecular Biology Of The
CellAmer Soc Cell Biology. Bethesda, MD: pp. 20814–2755. 2014
|
|
22
|
Shimojo N, Hashizume R, Kanayama K, Hara
M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T and Imanaka-Yoshida K:
Tenascin-C may accelerate cardiac fibrosis by activating
macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6
axis. Hypertension. 66:757–766. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mantovani A, Biswas SK, Galdiero MR, Sica
A and Locati M: Macrophage plasticity and polarization in tissue
repair and remodelling. J Pathol. 229:176–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gilchrist M, Thorsson V, Li B, Rust AG,
Korb M, Roach JC, Kennedy K, Hai T, Bolouri H and Aderem A: Systems
biology approaches identify ATF3 as a negative regulator of
Toll-like receptor 4. Nature. 441:173–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hai T, Wolford CC and Chang YS: ATF3, a
hub of the cellular adaptive-response network, in the pathogenesis
of diseases: Is modulation of inflammation a unifying component?
Gene Expr. 15:1–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen HH, Lai PF, Lan YF, Cheng CF, Zhong
WB, Lin YF, Chen TW and Lin H: Exosomal ATF3 RNA attenuates
pro-inflammatory gene MCP-1 transcription in renal
ischemia-reperfusion. J Cell Physiol. 229:1202–1211. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zmuda EJ, Qi L, Zhu MX, Mirmira RG,
Montminy MR and Hai T: The roles of ATF3, an adaptive-response
gene, in high-fat-diet-induced diabetes and pancreatic beta-cell
dysfunction. Mol Endocrinol. 24:1423–1433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li HF, Cheng CF, Liao WJ, Lin H and Yang
RB: ATF3-mediated epigenetic regulation protects against acute
kidney injury. J Am Soc Nephrol. 21:1003–1013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shan Y, Akram A, Amatullah H, Zhou DY,
Gali PL, Maron-Gutierrez T, González-López A, Zhou L, Rocco PR,
Hwang D, et al: ATF3 protects pulmonary resident cells from acute
and ventilator-induced lung injury by preventing Nrf2 degradation.
Antioxid Redox Signal. 22:651–668. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan L, Della Coletta L, Powell KL, Shen J,
Thames H, Aldaz CM and MacLeod MC: Activation of the canonical
Wnt/β-catenin pathway in ATF3-induced mammary tumors. PLoS One.
6:e165152011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee H, Bae S, Choi BW and Yoon Y:
WNT/β-catenin pathway is modulated in asthma patients and
LPS-stimulated RAW264.7 macrophage cell line. Immunopharmacol
Immunotoxicol. 34:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carey WA, Taylor GD, Dean WB and Bristow
JD: Tenascin-C deficiency attenuates TGF-ß-mediated fibrosis
following murine lung injury. Am J Physiol Lung Cell Mol Physiol.
299:L785–L793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Midwood K, Sacre S, Piccinini AM, Inglis
J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et
al: Tenascin-C is an endogenous activator of Toll-like receptor 4
that is essential for maintaining inflammation in arthritic joint
disease. Nature Med. 15:774–780. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shimojo N, Hashizume R, Kanayama K, Suzuki
Y, Hara M, Nishioka T, Yoshida T and Yoshida KI: A functional role
of tenascin-C in angiotensin II-induced cardiac fibrosis.
Circulation. 130:A96322014.
|