Introduction
The prevalence of diabetes has increased worldwide.
Previous studies have demonstrated that patients with diabetes are
more vulnerable to myocardial ischemia reperfusion (IR) injury, and
the risk of post-myocardial infarction death is increased by
200–400% in patients with diabetes compared with non-diabetic
individuals (1). Ischemic
post-conditioning (IPO), administered at the onset of reperfusion,
has been demonstrated to be an effective method to combat
myocardial IR injury (2,3). IPO may be a more promising approach
compared with ischemic preconditioning (IPC) due to the
difficulties associated with predicting the onset of myocardial
ischemia in clinical practice. However, previous studies have
demonstrated that diabetic hearts were unresponsive to IPO, and the
underlying mechanisms remain unclear (4–7).
AMP-activated protein kinase (AMPK), an
evolutionarily conserved serine/threonine kinase, is a principal
regulator of adenosine 5′-triphosphate homeostasis and energy
metabolism in the body (8). AMPK
serves a role in cell survival under stress conditions, including
oxidative stress, starvation, ischemia and hypoxia (9–11).
The beneficial effects of AMPK activation are mediated by
maintaining the homeostasis of reduced nicotinamide adenine
dinucleotide phosphate (NADPH), phosphorylating the tuberous
sclerosis complex to inhibit mammalian target of rapamycin (mTOR),
thereby promoting cytoprotective autophagy though direct
(serine/threonine protein kinase ULK1 phosphorylation) or indirect
(mTOR inhibition) mechanisms (10,12).
Autophagy is a conserved intracellular self-digestion process for
long-lived cytoplasmic proteins, organelles and macromolecules, and
is essential for maintaining cellular homeostasis under normal
conditions and affording protective responses to adverse conditions
(13).
Previous studies have observed that AMPK activation
is able to induce autophagy and, subsequently, provide protective
effects against IR injury in heart (14), brain (15), liver (16), kidney (17) and muscular tissues (18). IPC and IPO have been demonstrated
to combat IR injury by triggering AMPK-regulated autophagy
(15,19). In the diabetic myocardium, AMPK was
observed to be inhibited in combination with decreased cardiac
autophagy, and further studies demonstrated that cardiac function
was improved in diabetes by promoting AMPK-regulated autophagy
(20). These previous experimental
data suggested that AMPK-regulated autophagy may serve a role in
protecting the myocardium against IR injury and hyperglycemic
insult. However, whether AMPK-regulated autophagy is associated
with the pathophysiological process of myocardial IR injury in
diabetes, and its underlying mechanisms, remains to be
elucidated.
The aims of the present study were to investigate
whether hyperglycemia-induced AMPK inhibition is responsible for
the ineffectiveness of IPO by impairing autophagy in diabetic
hearts, and if so, whether activation of AMPK is able to restore
the sensitivity of diabetic hearts to IPO-induced cardioprotection
through autophagy activation.
Materials and methods
Experimental animals
A total of 120 male Sprague-Dawley rats of specific
pathogen-free level, weighing 250±10 g (age, 6–8 weeks) were
provided by Hunan SLAC JD Laboratory Animal Co., Ltd. (Hunan,
China). All rats were housed at 24°C, with a fixed light/dark cycle
(12 h light/12 h dark) and with ad libitum access to food
and water. All of the experimental protocols were in accordance
with the principles of Animal Care of Wuhan University (Wuhan,
China), and approved by the Committee for the Use of Live Animals
in Teaching and Research. Diabetic rats were induced by a single
intraperitoneal (i.p.) injection of streptozotocin (60 mg/kg;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) as previously
described, and rats exhibiting hyperglycemia (blood glucose ≥16.7
mmol/l) were considered to be diabetic (4,5). The
body weight, blood glucose and food and water intake of all rats
were observed and recorded.
Myocardial IR injury model
A well-established myocardial IR injury model was
used in the present study (4). All
rats were anesthetized (sodium pentobarbital; 50 mg/kg i.p.;
Sigma-Aldrich; Merck KGaA) with tracheotomy and ventilation. The IR
injury model was achieved by occluding the left anterior descending
artery for 30 min followed by 120 min of reperfusion. IPO was
established by 3 cycles of 10 sec of reperfusion and ischemia at
the onset of reperfusion. Sham-operated rats were subjected to the
same surgical procedures without ligation. Ischemia was confirmed
by elevation of the ST segment with limb lead II and discoloration
of the ischemic zone.
Experimental protocols
A total of 8 weeks subsequent to the onset of
diabetes, diabetic (D) and age-matched non-diabetic (N) rats were
randomly divided into 10 groups (n=12/group) as follows: 1, N+sham
(S); 2, N+IR; 3, N+IPO; 4, D+S; 5, D+IR; 6, D+IPO; 7,
D+IR+A-769662; 8, D+IPO+A-769662; 9, D+IPO+3-MA+A-769662; and 10,
D+IR+3-MA. A-769662 (6 mg/kg; catalogue no. S2697) (21) and 3-MA (catalogue no. S2767; 15
mg/kg) (both from Selleck Chemicals, Houston, TX, USA) (22) were given as i.p. injections 30 min
prior to myocardial ischemia.
Cardiac function assessment
Invasive hemodynamic monitoring was performed to
evaluate cardiac function. Left ventricular systolic pressure
(LVSP), maximal rates of increase and decrease in LVSP (±
dP/dtmax), and heart rate (HR) were intermittently monitored using
an electrophysiolograph (MH150; BioPAC Systems, Inc., Goleta, CA,
USA) and the data were analyzed using AcqKnowledge software
(version 5.0; BioPAC Systems, Inc.).
Infarct size determination
Myocardial infarct size was measured using 3% Evans
blue dye and 1% 2,3,5-triphenyltetrazolium chloride (both from
Sigma-Aldrich; Merck KGaA) staining, and scanning (v30; Seiko Epson
Corporation, Nagano, Japan) and image analysis using Image-Pro Plus
software (version 3.0, Media Cybernetics, Inc., Rockville, MD,
USA), as described previously (4).
The risk areas were stained red, while the infarct areas remained
pale.
Creatine kinase-MB (CK-MB) assay
Blood samples were centrifuged (1,200 × g for 10 min
at 4°C), and the serum was collected to measure CK-MB using
commercial kits (catalogue no. 1327c; Elabscience Biotechnology
Co., Ltd., Wuhan, China), according to the manufacturer's
protocol.
Oxidative stress detection
Myocardial tissue and H9c2 cells were homogenized
and centrifuged (2,400 × g for 15 min at 4°C) to obtain the
supernatants. The activity of superoxide dismutase (SOD) was
detected using a SOD assay kit, which employed the hydroxylamine
method (catalogue no. A001-1; Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). The expression of malondialdehyde (MDA)
was determined using an ELISA assay kit (catalogue no. 0060c;
Elabscience Biotechnology Co., Ltd.), according to the
manufacturer's protocol.
Electron microscopy
Observation of the number of autophagosomes under a
transmission electron microscope (TEM) is a direct qualitative
measure of autophagy (23).
Ischemic heart tissue samples of ~1 mm3 were removed and
pre-fixed in a solution of 2.5% glutaraldehyde at 4°C for 24 h, and
subsequently post-fixed in 1% OsO4 at 4°C for 30 min,
dehydrated in an ascending series of alcohol, and embedded in epoxy
resin. The slide was stained by uranyl acetate and lead citrate at
4°C for 0.5–1 h, and observed under a TEM (HT7700; Hitachi, Ltd.,
Tokyo, Japan).
Study in H9c2 cell lines
Rat cardiomyocyte-derived H9c2 cells (American Type
Culture Collection, Manassas, VA, USA) were maintained in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 100 µg/ml
penicillin/streptomycin in an atmosphere containing 5%
CO2 at 37°C. The cells were randomly divided into 10
groups: 1, low glucose (5.5 mM) medium (LG); 2,
LG+hypoxia/reoxygenation (HR); 3, LG+hypoxia post-conditioning
(HPO); 4, high glucose (30 mM) medium (HG); 5, HG+HR; 6, HG+HPO; 7,
HG+HR+A-769662; 8, HG+HPO+A-769662; 9, HG+HPO+A-769662+3-MA; 10,
HG+HR+3-MA. A-769662 (100 mM) and 3-MA (10 nM) (24) was given 1 h prior to hypoxia, and
the cells underwent 4 h of hypoxia followed by 2 h of
reoxygenation. HPO was performed by 3 cycles of 5 min reoxygenation
and hypoxia. Hypoxic conditions were obtained using a gas incubator
(5% CO2 and 95% N2). Each experiment was performed ≥3
times independently in triplicate. Cells and supernatants were
collected for further analysis.
Cell viability and lactate
dehydrogenase (LDH) release assay
Cell viability was determined using a Cell Counting
Kit-8 (CCK-8) assay kit at a wavelength of 450 nm (catalogue no.
04-11; Dojindo Molecular Technologies, Inc., Kumamoto, Japan), and
LDH was measured using a cytotoxicity assay kit a wavelength of 490
nm (catalogue no. 0218c; Elabscience Biotechnology Co., Ltd.),
according to the manufacturer's protocols.
Western blot analysis
Western blotting was performed as described
previously (4). Tissues or cells
were homogenized with radioimmunoprecipitation assay lysis buffer.
Equivalent proteins were separated using SDS-PAGE on a 5–15% gel
and electro-transferred onto a polyvinylidene fluoride membrane.
The membranes were incubated with anti-GAPDH (catalogue no. 2118),
anti-microtubule associated protein 1 light chain 3 β/α (LC3B/A,
catalogue no. 12741), anti-nuclear pore glycoprotein p62 (p62,
catalogue no. 5114), anti-mTOR (catalogue no. 2983),
anti-phosphorylated (p) mTOR (ser2448, catalogue no. 5536) (all
from Cell Signaling Technology, Inc., Danvers, MA, USA), AMPKα
(catalogue no. sc25792) and p-AMPKα (Thr172, catalogue no.
sc101630) (both from Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) primary antibodies (1:500-1,000 dilution) overnight at 4°C,
followed by Alexa Fluor secondary antibody (1:10,000 dilution,
catalogue no. A-21210; Thermo Fisher Scientific, Inc.) for 1 h at
room temperature. Signals were detected using an Odyssey
fluorescence imaging scanner and quantified using Odyssey software
v3.0.29 (both from LI-COR Biosciences, Lincoln, NE, USA).
Statistical analysis
Rats and H9c2 cell culture dishes were randomly
assigned to treated or control groups. Western blot analysis was
conducted blindly, with samples separated into numbered groups at
random. Data are presented as the mean ± standard deviation. An
unpaired Student's t-test was used to detect the differences in
characteristics between non-diabetic and diabetic rats. Two-way
repeated-measures analysis of variance (ANOVA) followed by
Bonferroni's post-hoc test was used to analyze the differences in
left ventricular function data between the groups. All other data
were evaluated using one-way ANOVA followed by Bonferroni's
post-hoc test. Analysis was performed using Prism software (version
5.0.7; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Characteristics of experimental
diabetic rats
No significant difference was observed in body
weight and blood glucose prior to diabetes induction. Following 8
weeks of STZ-induced diabetes, the rats were characterized by a
decreased body weight, increased blood glucose, and increased food
and water intake compared with the age-matched non-diabetic rats
(data not presented).
IPO provides cardioprotection in
non-diabetic animals
No significant difference was observed in the area
at risk as a percentage of the left ventricle (AAR/LV) in all
groups (data not presented). As presented in Fig. 1A, diabetic rats exhibited increased
myocardial infarct size compared with non-diabetic rats, following
IR insult. IPO significantly decreased the infarct size in
non-diabetic rats, and not in diabetic rats. Biochemical markers of
myocardial injury and oxidative stress were additionally examined.
Diabetic hearts exhibited an increased level of MDA and decreased
activity of SOD. Compared with non-diabetes, IR significantly
increased CK-MB and MDA level and decreased SOD activity in
diabetes. IPO caused a significant reversion in non-diabetes, and
not in diabetes (Fig. 1B-D).
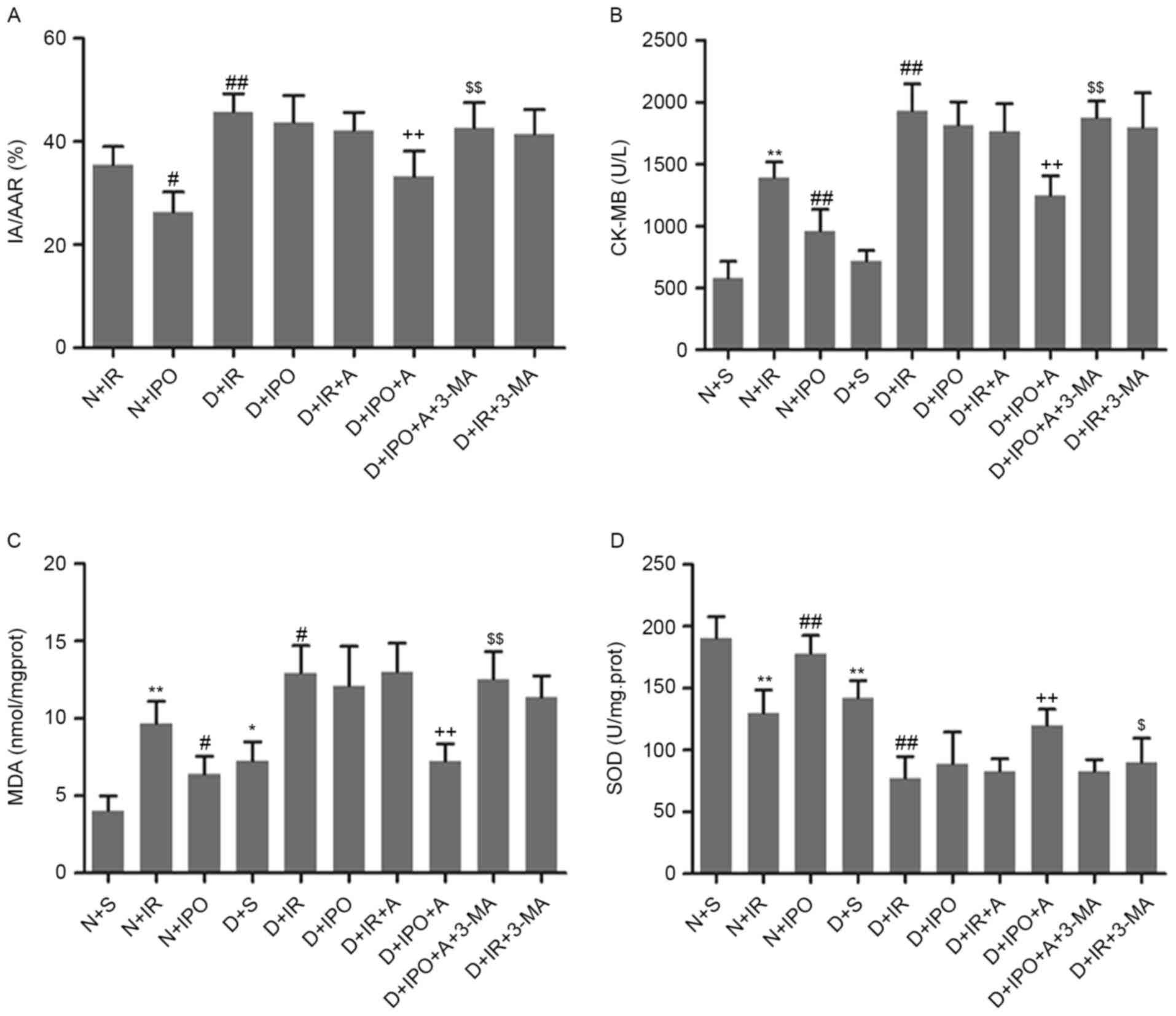 | Figure 1.Effects of IPO on myocardial infarct
size and biomarkers following 30 min ischemia followed by 2 h
reperfusion, in non-diabetic and diabetic rats. (A) Percentage of
area at risk vs left ventricle. Biomarkers of the degree of injury
were (B) CK-MB (C) MDA and (D) SOD. n=6/group. **P<0.01 vs. N+S
group; #P<0.05 and ##P<0.01 vs. N+IR
group; ++P<0.01 vs. D+S group; $P<0.05
and $$P<0.01 vs. D+IPO+A group. N, non-diabetes; D,
diabetes; S, sham; IR, ischemia reperfusion; IPO, ischemic
post-conditioning; A, A-769662; IA/AAR, infarct area/area at risk;
CK-MB, creatine kinase MB; MDA, malondialdehyde; SOD, superoxide
dismutase. |
Effects of IPO on myocardial autophagy
status, and AMPK and mTOR expression in diabetes and
non-diabetes
In the present study, myocardial autophagy status,
and AMPK and mTOR expression and phosphorylation, were observed
following IR injury or IPO treatment. As presented in Fig. 2A-D, compared with non-diabetes, a
decreased autophagosome number and LC3B/A ratio, in combination
with increased p62 expression, were observed in diabetes. IR insult
significantly increased the autophagosome number and LC3B/A ratio,
and decreased p62 expression, in non-diabetes; these alterations
were further increased by IPO. However, IR and IPO did not
significantly alter myocardial autophagosome number, LC3B/A ratio
and p62 expression in diabetic hearts. No significant difference
was detected in the total expression of AMPK and mTOR among all the
groups (data not presented). As presented in Fig. 2E and F, in non-diabetes, IR
increased AMPK phosphorylation and decreased mTOR phosphorylation,
which was further increased by IPO. Compared with non-diabetes, a
decrease in phosphorylated AMPK with an increase in phosphorylated
mTOR were detected in the diabetic myocardium. IR and IPO were
observed to increase AMPK phosphorylation and decrease mTOR
phosphorylation.
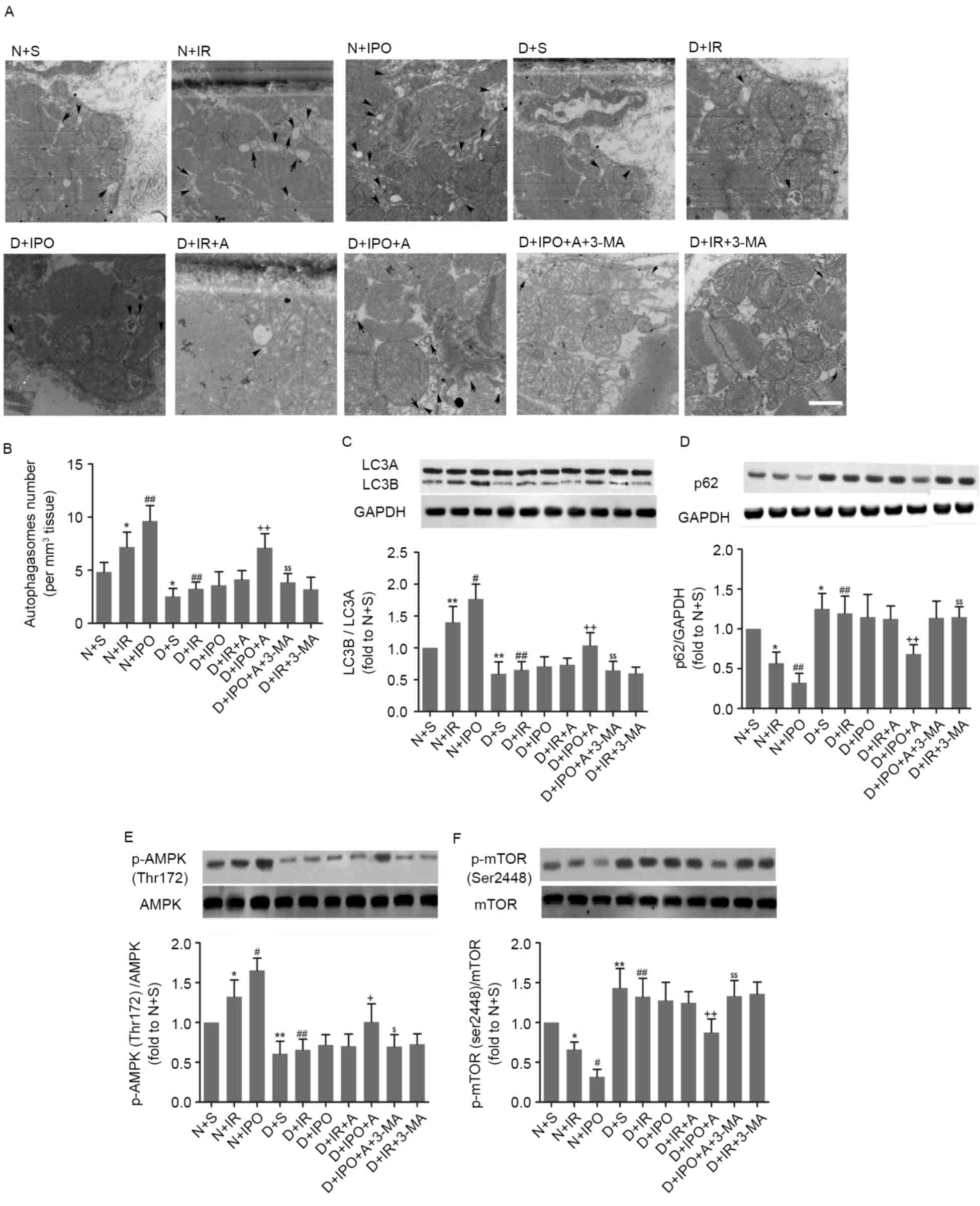 | Figure 2.Effects of IPO with or without A on
myocardial autophagy and the AMPK/mTOR signaling pathway in
non-diabetic and diabetic rats, with 30 min ischemia followed by 2
h reperfusion. (A) Myocardial transmission electron microscopy
analysis. (B) Autophagosome number. (C) Ratio of LC3B/A. (D)
Expression of p62. (E) AMPK phosphorylation. (F) mTOR
phosphorylation. Scale bar=1 µm. n=6/group. *P<0.05 and
**P<0.01 vs. N+S group; #P<0.05 and
##P<0.01 vs. N+IR group; +P<0.05 and
++P<0.01 vs. D+IR group; and $P<0.05
and $$P<0.01 vs. D+IPO+A group. N, non-diabetes; D,
diabetes; S, sham; IR, ischemia reperfusion; IPO, ischemic
post-conditioning; A, A-769662; LC3, microtubule associated protein
1 light chain 3; AMPK, 5′-AMP-activated protein kinase catalytic
subunit α-1; mTOR, mammalian target of rapamycin; p62, nuclear pore
glycoprotein p62; p, phosphorylated. |
AMPK activation by A-769662 restores
the protective effects of IPO in diabetic hearts
The present study investigated whether A-769662 is
able to restore IPO cardioprotection in diabetes, and whether these
effects may be affected by the autophagy inhibitor 3-MA. As
presented in Fig. 1,
administration of A-769662 alone failed to decrease myocardium
infarct size, and CK-MB and MDA level, and to increase SOD
activity. By contrast, A-769662 with IPO significantly decreased
the infarct size, decreased CK-MB and MDA expression, and elevated
SOD activity. All of these effects were reversed by treatment with
the autophagy inhibitor 3-MA, although 3-MA alone did not influence
the infarct size, CK-MB and MDA release, and SOD activity in
diabetic rats following IR insult. Hemodynamic parameters
reflecting left ventricular function were analyzed in the present
study. Diabetic rats exhibited markedly decreased HR, LVSP, +dP/dt
and -dP/dt compared with age-matched non-diabetic animals at
baseline (data not presented). As presented in Table I, all of the hemodynamic parameters
were decreased in the diabetic and non-diabetic groups following 2
h reperfusion. IPO significantly increased the level of HR, LVSP,
+dP/dt and -dP/dt in non-diabetic animals. Treatment with A-769662
alone did not alter the hemodynamic parameters, compared with the
untreated group. However, A-769662 treatment with IPO increased the
levels of HR, LVSP, +dP/dt and -dP/dt in diabetic rats. Notably,
all of the alterations in hemodynamic parameters were reversed by
treatment with 3-MA.
 | Table I.Hemodynamic parameters as markers of
left ventricular function following 2 h reperfusion. |
Table I.
Hemodynamic parameters as markers of
left ventricular function following 2 h reperfusion.
| Groups | HR, bpm | LVSP, mmHg | +dP/dt, mmHg/s | -dP/dt, mmHg/s |
|---|
| N+S |
361±14 |
122±5 |
6362±130 |
4872±130 |
| N+IR |
280±8a |
89±4a |
4416±151a |
3237±154a |
| N+IPO |
338±14b |
112±4b |
5368±176c |
4465±135c |
| D+S |
288±10a |
100±4a |
4641±166a |
3537±163a |
| D+IR |
169±9c |
53±4c |
2781±158c |
2225±112c |
| D+IR+A |
178±8 |
57±4 |
2841±184 |
2349±248 |
| D+IPO |
201±12 |
67±5 |
2628±168 |
2506±129 |
| D+IPO+A |
269±12d |
91±4d |
4358±149d |
3212±143d |
| D+IPO+A+3-MA |
205±9e |
62±6e |
2561±143e |
2429±176e |
Effects of AMPK activation on
myocardial autophagy and the AMPK-mTOR signaling pathway in
diabetes
In order to investigate the underlying mechanisms,
the present study analyzed the effects of A-769662 on myocardial
autophagy status and the AMPK/mTOR signaling pathway. As presented
in Fig. 2, A-769662 administration
or IPO alone did not affect myocardial autophagy status and the
AMPK/mTOR signaling pathway in diabetes. However, A-769662 combined
with IPO increased the autophagosome number, LC3B/A ratio and AMPK
phosphorylation, and decreased p62 expression and mTOR
phosphorylation. However, these alterations were reversed by the
autophagy inhibitor 3-MA.
Effects of HG on HPO cardioprotection,
autophagy and the AMPK/mTOR signaling pathway in H9c2 cell
In vitro, H9c2 cells were exposed to HG
conditions for 48 h to simulate the diabetic myocardium. As
presented in Fig. 3, HG insult led
to decreased cell viability and SOD activity, and increased LDH and
MDA release. These alterations were further increased by HR in the
LG and HG groups. HPO significantly increased cell viability and
SOD activity, and decreased LDH and MDA release in LG medium
cultured cells only. As presented in Fig. 4, a decreased LC3B/A ratio and
decreased phosphorylated AMPK expression, with increased p62
expression and phosphorylated mTOR expression, was detected in H9c2
cells exposed to HG, compared with the LG group. Following HR
insult, the autophagy level and activity of the AMPK/mTOR signaling
pathway were upregulated in the LG group, and were further
increased by HPO. In the HG groups, HR and HPO did not
significantly affect the autophagy level and the AMPK/mTOR
signaling pathway.
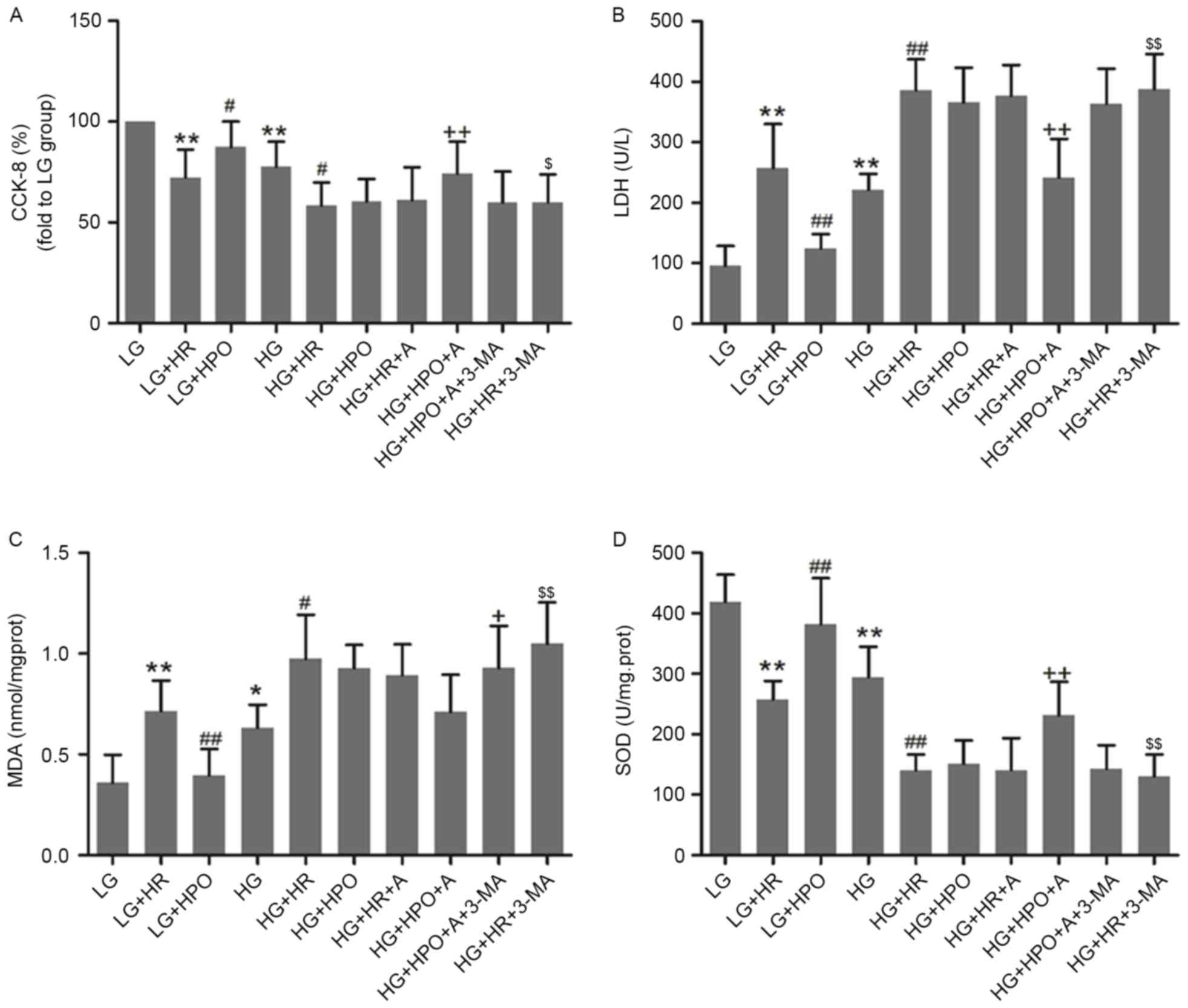 | Figure 3.Effects of HPO, with or without A, on
H9c2 cells cultured in HG or LG conditions. (A) Results of CCK-8
assay. The expression of (B) LDH, (C) MDA and (D) SOD was analyzed.
The results are representative of ≥3 independent experiments.
*P<0.05 and **P<0.01 vs. LG group; #P<0.05 and
##P<0.01 vs. LG+HR group; +P<0.05 and
++P<0.01 vs. HG+HR group; and $P<0.05
and $$P<0.01 vs. HG+HPO+A group. LG, low glucose
medium; HG, high glucose medium; HR, hypoxia reoxygenation; HPO,
hypoxia post-conditioning; A, A-769662; CCK-8, Cell Counting Kit-8;
LDH, lactate dehydrogenase; MDA, malondialdehyde; SOD, superoxide
dismutase. |
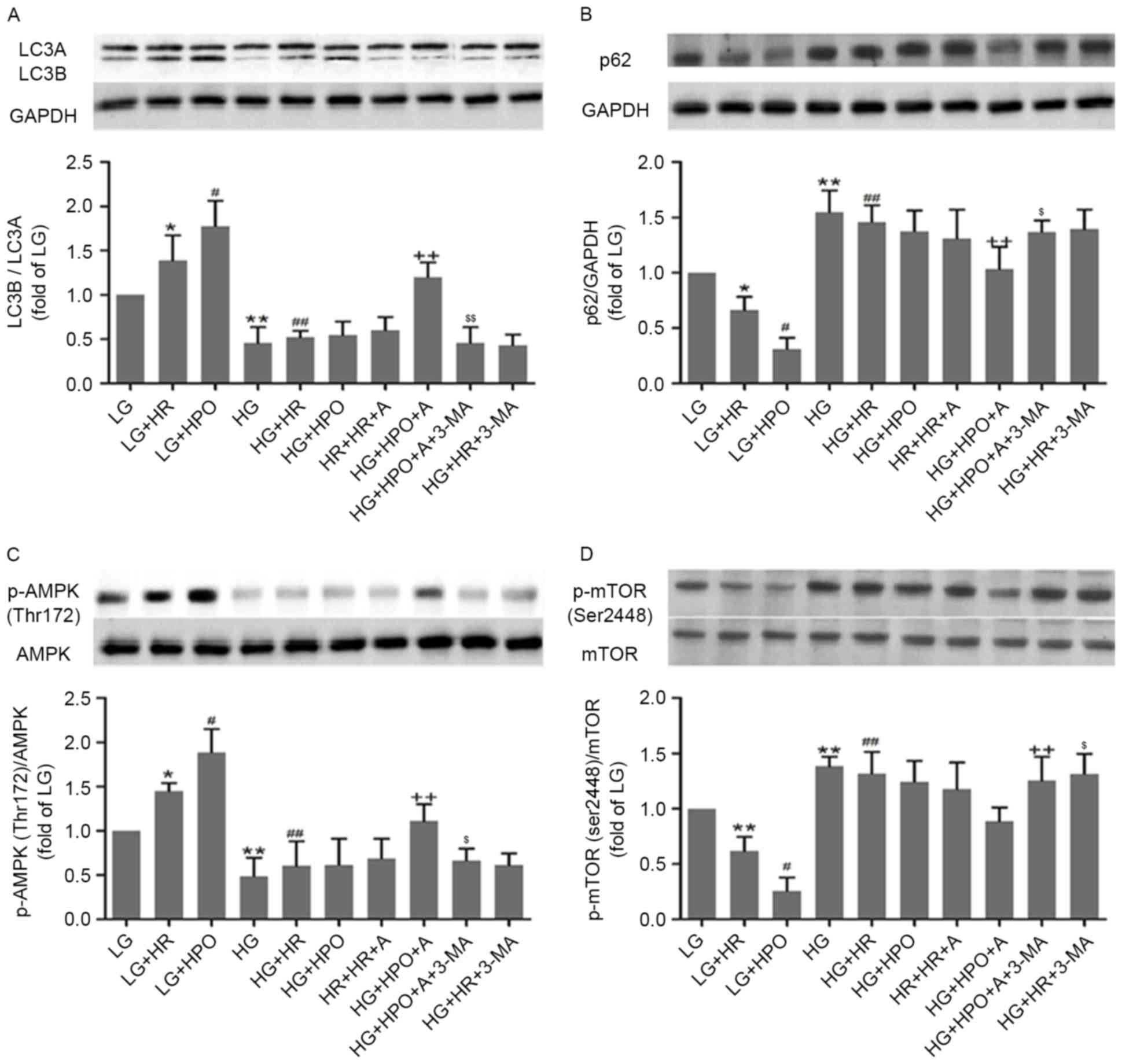 | Figure 4.Effects of HPO, with or without A, on
autophagy status, and AMPK and mTOR expression and phosphorylation,
in H9c2 cells treated with 4 h of hypoxia followed by 2 h of
reoxygenation. Analysis of (A) LC3B/A ratio, (B) expression of p62,
(C) AMPK phosphorylation and (D) mTOR phosphorylation. The results
are representative of ≥3 independent experiments. *P<0.05 and
**P<0.01 vs LG group; #P<0.05 and
##P<0.01 vs LG+HR group; and ++P<0.05
vs. LG+HPO group; $P<0.05 and $$P<0.01
vs HG+HPO+A group. LG, low glucose medium; HG, high glucose medium;
HR, hypoxia reoxygenation; HPO, hypoxia post-conditioning; A,
A-769662; LC3, microtubule associated protein 1 light chain 3;
AMPK, 5′-AMP-activated protein kinase catalytic subunit α-1; mTOR,
mammalian target of rapamycin; p62, nuclear pore glycoprotein p62;
p, phosphorylated. |
AMPK activation with A-769662 restores
the protective effects of HPO in HG-exposed H9c2 cells
In order to confirm whether AMPK activation restores
the protective effects of HPO in H9c2 cell lines exposed to HG,
cells were pretreated with the AMPK agonist A-769662. As presented
in Fig. 3, A-769662 or HPO alone
did not confer protective effects to combat HR injury in cells
exposed to HG conditions. By contrast, A-769662 with HPO protected
H9c2 cells exposed to HG conditions from HR injury, as evidenced by
increased CCK-8 and SOD activity, and reduced LDH and MDA release.
All of the observed protective effects were reversed by treatment
with 3-MA.
Effects of HPO with AMPK activation on
autophagy in HG-exposed H9c2 cells
As presented in Fig.
4, pretreatment with HPO or A-769662 alone did not affect
autophagy status or the AMPK/mTOR signaling pathway. However, HPO
with A-769662 significantly activated AMPK/mTOR-regulated
autophagy, as evidenced by an increased LC3B/A ratio and increased
AMPK phosphorylation, in combination with decreased p62 expression
and m-TOR phosphorylation. All of these alterations were reversed
by treatment with the autophagy inhibitor 3-MA, which demonstrated
that A-769663 and HPO confer their combined protective effects by
activating AMPK-regulated autophagy.
Discussion
The present study demonstrated that
hyperglycemia-induced AMPK downregulation contributed to the
ineffectiveness of IPO cardioprotection, and that the underlying
mechanism may involve myocardial autophagy inhibition. AMPK
activation by A-769662 restored the sensitivity of the diabetic
myocardium to IPO, possibly by improving autophagy status. To the
best of our knowledge, the present study was the first to
investigate the effects of AMPK inhibition on IPO cardioprotection
in hyperglycemic cardiomyocytes, and the roles of
AMPK/mTOR-regulated autophagy in this pathophysiological
process.
IPO is an approach whereby brief cycles of
ischemia-reperfusion are applied directly following the continued
occlusion of a coronary artery, which was first proposed by Zhao
et al (2) in 2003. IPO has
been demonstrated to be an effective way to relieve myocardial IR
injury in animal (4,5) and clinical trials (25,26),
and the underlying mechanisms are associated with activation of the
reperfusion injury salvage kinase pathway and the Janus
kinase/signal transducer and activator of transcription pathway, by
inhibiting mitochondrial permeability transition pore opening and
antioxidation (27,28). However, according to a number of
studies (4,5,7), IPO
appears to be unable to induce cardioprotection in diabetes, due in
part to severe oxidative stress. Therefore, the present study aimed
to investigate the in-depth mechanisms associated with the
inefficiency of IPO in diabetes.
Autophagy is an important mechanism in cellular
metabolism and survival; it is a dynamic process, which is
comprised of autophagosome formation and autolysosomal clearance.
Under physiological conditions, a baseline level of autophagy is
required to maintain cardiac homeostasis, and autophagy may be
activated in response to stress (13). However, excessive autophagy results
in programmed cell death (29).
The conversion of LC3A to LC3B is a marker of autophagosome
formation, and an increased ratio of LC3B/A demonstrates an
increase in autophagy and a decrease in autolysosome degradation.
p62/sequestome 1, a protein adaptor which is able to bind
ubiquitinated cargo designated for autophagic breakdown, was
observed to reflect myocardial autophagy status; it is an improved
marker of autophagic flux for measuring LC3B/LC3A ratio and levels
of p62 (30). Previous studies
have demonstrated that autophagy was involved in the pathological
process of IR injury. Autophagy was reported to be elevated during
ischemia, although whether this is beneficial or detrimental to
target organs remains controversial. Huang et al (31) demonstrated that myocardial
autophagy inhibition mediated by berberine leads to a decrease in
IR-induced myocardium infarct size and cardiac dysfunction, and
similar conclusions were drawn in a brain research study by Gao
et al (32). By contrast,
upregulation of autophagy has been demonstrated to be a potential
method of protection against IR injury. Buss et al (10) and Wei et al (22) demonstrated that autophagy
activation mitigated IR injury. Zhao et al (14) reported that autophagy mediated by
acetylcholine attenuated HR injury in H9c2 cells, evidenced by
increased cell viability and decreased apoptosis. The results of
the present study demonstrated that myocardial autophagy was
significantly increased following IR insult in the non-diabetic
heart, which was increased further following treatment with IPO.
However, autophagy inactivation was observed in the diabetic heart,
which was consistent with a previous study (20). In addition, as an endogenous
protection strategy, autophagic responses failed to be activated by
IR or IPO in diabetic hearts; therefore, it was hypothesized that
the ineffectiveness of IPO is associated with the inactivation of
autophagy in diabetes.
AMPK is a heterotrimeric complex which consists of a
catalytic α subunit and two regulatory subunits, β and γ. The
serine/threonine kinase activity of AMPK is mediated by the α
subunit, and is characterized by the presence of a threonine
residue (Thr172) in a loop that must be phosphorylated for
activation to occur (33). AMPK
protein is expressed in the majority of mammalian tissues,
including those of the cardiovascular system; it is a
highly-conserved sensor of the cellular energy status and serves a
role in regulating cellular biological activity. mTOR, an
additional highly-conserved serine/threonine protein kinase, is
important for cell growth, proliferation and differentiation. AMPK
has been demonstrated to be an upstream protein, which is able to
negatively regulate mTOR in a directly or indirect manner (12). Previous studies have indicated that
the AMPK/mTOR signaling pathway is associated with autophagy
regulation, which serves a role in the occurrence and development
of a number of diseases. Guo et al (19) and Zhao et al (14) observed that an IR insult
upregulated AMPK phosphorylation and downregulated mTOR
phosphorylation, with an increased level of autophagy in
cardiomyocytes in vivo and in vitro. Consistent with
the previous studies mentioned above, the results of the present
study demonstrated that AMPK/mTOR pathway activity was promoted by
IR insult in the non-diabetic myocardium, and that IPO further
activated the AMPK/mTOR pathway, in combination with an increased
level of autophagy. However, in the diabetic myocardium,
phosphorylation of AMPK was inhibited, which was consistent with
studies by Guo et al (20)
and Viollet et al (34). In
addition, the present study demonstrated that IR and IPO were
unable to activate the AMPK/mTOR signaling pathway efficiently in
diabetes.
In order to confirm whether hyperglycemia-induced
AMPK inhibition contributes to the ineffectiveness of IPO
cardioprotection by decreasing myocardial autophagy, the AMPK
agonist 769662 and the autophagy inhibitor 3-MA were applied in
vivo and in vitro. A-769662 was observed to activate
AMPK efficiently by allosteric inhibition of AMPK dephosphorylation
at the Thr172 site, in a previous study (35). An additional previous study
demonstrated that A-769662 did not affect the total expression of
AMPK, although it significantly increased the phosphorylation of
AMPK at Thr172. AMPK activation by A-769662 was reported to exert
cardioprotection by increasing the expression level of a downstream
signaling pathway involving endothelial NO synthase, thereby
stimulating NO release (21). Kim
et al (36) demonstrated
that pretreatment with A-769662 in vivo decreased infarct
size in C57Bl/6 mice undergoing left coronary artery occlusion and
reperfusion. Similarly, Paiva et al (37) demonstrated that directly enhancing
AMPK activation with A-769662 at reperfusion protects the IR rat
myocardium against infarction. Notably, all of the above previous
studies were performed in non-diabetic conditions. In the present
study, A-769662 nor IPO alone did not attenuate IR injury in
diabetic hearts. By contrast, A-769662 administration in
combination with IPO treatment significantly protected diabetic
hearts from IR injury, with a simultaneous increase in autophagy
being observed. In addition, it was observed that the protection
mediated by A-762669 with IPO was reversed by the autophagy
inhibitor 3-MA, with a decrease in the myocardial autophagy level,
which further demonstrated that autophagy is associated with the
protective mechanism of IPO. The results obtained from cultured
H9c2 cells in the present study were consistent with the in
vivo experiments.
In conclusion, the present study confirmed the
ineffectiveness of IPO cardioprotection in diabetes, and
demonstrated that hyperglycemia-induced AMPK inhibition underlies
this ineffectiveness, in part by decreased myocardial autophagy.
AMPK activation mediated by A-769662 restored the sensitivity of
diabetic hearts to IPO cardioprotection, through autophagy
activation. Therefore, the present study demonstrated that
targeting AMPK may elicit IPO cardioprotection in human
diabetes.
References
|
1
|
Beckman JA, Paneni F, Cosentino F and
Creager MA: Diabetes and vascular disease: Pathophysiology,
clinical consequences, and medical therapy: Part II. Eur Heart J.
34:2444–2452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F,
Wang NP, Guyton RA and Vinten-Johansen J: Inhibition of myocardial
injury by ischemic postconditioning during reperfusion: Comparison
with ischemic preconditioning. Am J Physiol Heart Circ Physiol.
285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bochaton T, Crola-Da-Silva C, Pillot B,
Villedieu C, Ferreras L, Alam MR, Thibault H, Strina M, Gharib A,
Ovize M and Baetz D: Inhibition of myocardial reperfusion injury by
ischemic postconditioning requires sirtuin 3-mediated deacetylation
of cyclophilin D. J Mol Cell Cardiol. 84:61–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xue R, Lei S, Xia ZY, Wu Y, Meng Q, Zhan
L, Su W, Liu H, Xu J, Liu Z, et al: Selective inhibition of PTEN
preserves ischaemic post-conditioning cardioprotection in
STZ-induced Type 1 diabetic rats: Role of the PI3K/Akt and
JAK2/STAT3 pathways. Clin Sci (Lond). 130:377–392. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu M, Zhou B, Xia ZY, Zhao B, Lei SQ,
Yang QJ, Xue R, Leng Y, Xu JJ and Xia Z: Hyperglycemia-induced
inhibition of DJ-1 expression compromised the effectiveness of
ischemic postconditioning cardioprotection in rats. Oxid Med Cell
Longev. 2013:5649022013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drenger B, Ostrovsky IA, Barak M,
Nechemia-Arbely Y, Ziv E and Axelrod JH: Diabetes blockade of
sevoflurane postconditioning is not restored by insulin in the rat
heart: Phosphorylated signal transducer and activator of
transcription 3- and phosphatidylinositol 3-kinase-mediated
inhibition. Anesthesiology. 114:1364–1372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Przyklenk K, Maynard M, Greiner DL and
Whittaker P: Cardioprotection with postconditioning: Loss of
efficacy in murine models of type-2 and type-1 diabetes. Antioxid
Redox Signal. 14:781–790. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buss SJ, Riffel JH, Katus HA and Hardt SE:
Augmentation of autophagy by mTOR-inhibition in myocardial
infarction: When size matters. Autophagy. 6:304–306. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
She C, Zhu LQ, Zhen YF, Wang XD and Dong
QR: Activation of AMPK protects against hydrogen peroxide-induced
osteoblast apoptosis through autophagy induction and NADPH
maintenance: New implications for osteonecrosis treatment? Cell
Signal. 26:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang
H, Ren J and Zang WJ: Acetylcholine mediates AMPK-dependent
autophagic cytoprotection in H9c2 cells during
hypoxia/reoxygenation injury. Cell Physiol Biochem. 32:601–613.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS,
Cao L, Wang HF, Shi JQ, Gao L, Qin H, et al: Ischemic
preconditioning provides neuroprotection by induction of
AMP-activated protein kinase-dependent autophagy in a rat model of
ischemic stroke. Mol Neurobiol. 51:220–229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nepal S and Park PH: Activation of
autophagy by globular adiponectin attenuates ethanol-induced
apoptosis in HepG2 cells: Involvement of AMPK/FoxO3A axis. Biochim
Biophys Acta. 1833:2111–2125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang LT, Chen BL, Wu CT, Huang KH, Chiang
CK and Liu S Hwa: Protective role of AMP-activated protein
kinase-evoked autophagy on an in vitro model of
ischemia/reperfusion-induced renal tubular cell injury. PLoS One.
8:e798142013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pauly M, Daussin F, Burelle Y, Li T, Godin
R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A,
Coisy-Quivy M, et al: AMPK activation stimulates autophagy and
ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J
Pathol. 181:583–592. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo L, Xu JM and Mo XY: Ischemic
postconditioning regulates cardiomyocyte autophagic activity
following ischemia/reperfusion injury. Mol Med Rep. 12:1169–1176.
2015.PubMed/NCBI
|
|
20
|
Guo Y, Yu W, Sun D, Wang J, Li C, Zhang R,
Babcock SA, Li Y, Liu M, Ma M, et al: A novel protective mechanism
for mitochondrial aldehyde dehydrogenase (ALDH2) in type i
diabetes-induced cardiac dysfunction: Role of AMPK-regulated
autophagy. Biochim Biophys Acta. 1852:319–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song T, Lv LY, Xu J, Tian ZY, Cui WY, Wang
QS, Qu G and Shi XM: Diet-induced obesity suppresses sevoflurane
preconditioning against myocardial ischemia-reperfusion injury:
Role of AMP-activated protein kinase pathway. Exp Biol Med
(Maywood). 236:1427–1436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei C, Li H, Han L, Zhang L and Yang X:
Activation of autophagy in ischemic postconditioning contributes to
cardioprotective effects against ischemia/reperfusion injury in rat
hearts. J Cardiovasc Pharmacol. 61:416–422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Swanlund JM, Kregel KC and Oberley TD:
Investigating autophagy: Quantitative morphometric analysis using
electron microscopy. Autophagy. 6:270–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang W, Yan J, Wang H, Shi M, Zhang M,
Yang W, Peng C and Li H: Rapamycin ameliorates inflammation and
fibrosis in the early phase of cirrhotic portal hypertension in
rats through inhibition of mTORC1 but not mTORC2. PLoS One.
9:e839082014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo W, Zhu M, Huang R and Zhang Y: A
comparison of cardiac post-conditioning and remote pre-conditioning
in paediatric cardiac surgery. Cardiol Young. 21:266–270. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Durdu S, Sirlak M, Cetintas D, Inan MB,
Eryılmaz S, Ozcinar E, Yazicioglu L, Elhan AH, Akar AR and Uysalel
A: The efficacies of modified mechanical post conditioning on
myocardial protection for patients undergoing coronary artery
bypass grafting. J Cardiothorac Surg. 7:732012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hausenloy DJ: Signalling pathways in
ischaemic postconditioning. Thromb Haemost. 101:626–634.
2009.PubMed/NCBI
|
|
28
|
Ovize M, Baxter GF, Di Lisa F, Ferdinandy
P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J,
Yellon DM, Schulz R, et al: Working Group of Cellular Biology of
Heart of European Society of Cardiology: Postconditioning and
protection from reperfusion injury: Where do we stand? Position
paper from the working group of cellular biology of the heart of
the european society of cardiology. Cardiovasc Res. 87:406–423.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Z, Han Z, Ye B, Dai Z, Shan P, Lu Z,
Dai K, Wang C and Huang W: Berberine alleviates cardiac
ischemia/reperfusion injury by inhibiting excessive autophagy in
cardiomyocytes. Eur J Pharmacol. 762:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L,
Su L and Zhang Y: Inhibition of autophagy contributes to ischemic
postconditioning-induced neuroprotection against focal cerebral
ischemia in rats. PLoS One. 7:e460922012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hawley SA, Davison M, Woods A, Davies SP,
Beri RK, Carling D and Hardie DG: Characterization of the
AMP-activated protein kinase kinase from rat liver and
identification of threonine 172 as the major site at which it
phosphorylates AMP-activated protein kinase. J Biol Chem.
271:27879–27887. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Viollet B, Lantier L, Devin-Leclerc J,
Hebrard S, Amouyal C, Mounier R, Foretz M and Andreelli F:
Targeting the AMPK pathway for the treatment of Type 2 diabetes.
Front Biosci (Landmark Ed). 14:3380–3400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sanders MJ, Ali ZS, Hegarty BD, Heath R,
Snowden MA and Carling D: Defining the mechanism of activation of
AMP-activated protein kinase by the small molecule A-769662, a
member of the thienopyridone family. J Biol Chem. 282:32539–32548.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim AS, Miller EJ, Wright TM, Li J, Qi D,
Atsina K, Zaha V, Sakamoto K and Young LH: A small molecule AMPK
activator protects the heart against ischemia-reperfusion injury. J
Mol Cell Cardiol. 51:24–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Paiva MA, Goncalves LM, Providência LA,
Davidson SM, Yellon DM and Mocanu MM: Transitory activation of AMPK
at reperfusion protects the ischaemic-reperfused rat myocardium
against infarction. Cardiovasc Drugs Ther. 24:25–32. 2010.
View Article : Google Scholar : PubMed/NCBI
|














