Introduction
Cardiovascular disease is a common disease
worldwide. Essential hypertension (EH) and coronary heart disease
are the most common types of cardiovascular disease. Among them, EH
affects ~1 billion people worldwide and ~130 million individuals in
China (1). In addition, EH is
associated with an increased risk for stroke and renal dysfunction,
and it represents one of the greatest public health concerns
worldwide. At present, the molecular mechanism underlying EH
remains largely unknown. It is generally believed that EH is a
complex and multifactorial disorder, which may be caused by single
gene defects or environmental conditions. Among these genetic
factors, the maternal inheritance of EH has been observed in
numerous families, indicating that variation in mitochondrial DNA
(mtDNA) is involved in the pathogenesis of EH (2,3).
Previous studies have identified some mtDNA pathogenic mutations
including the 12S ribosomal (r)RNA A1555 G mutation (4), the transfer (t)RNAMet
A4435 G mutation (5), and the
tRNAMet/tRNAGln A4401G and tRNAIle
A4295G mutations (6,7). These mtDNA mutations, mainly located
at tRNA genes, may lead to failures in tRNA metabolism, and
subsequently result in defects in mitochondrial translation, thus
causing mitochondrial dysfunction which in implicated in EH
pathophysiology. Therefore, mtDNA mutations may have potential as
novel biomarkers for the early detection, prevention and management
of maternally inherited EH.
However, the frequency of these mt-tRNA mutations in
Han Chinese subjects with EH remains to be elucidated. To
understand the contribution of mitochondrial variants to EH, we
have initiated an extensive mutational screening program for mtDNA
in a large cohort of EH subjects at the Hanchuan People's Hospital
(Hanchuan, China). The present study described a Chinese pedigree
with EH. Analysis of the entire mitochondrial genome resulted in
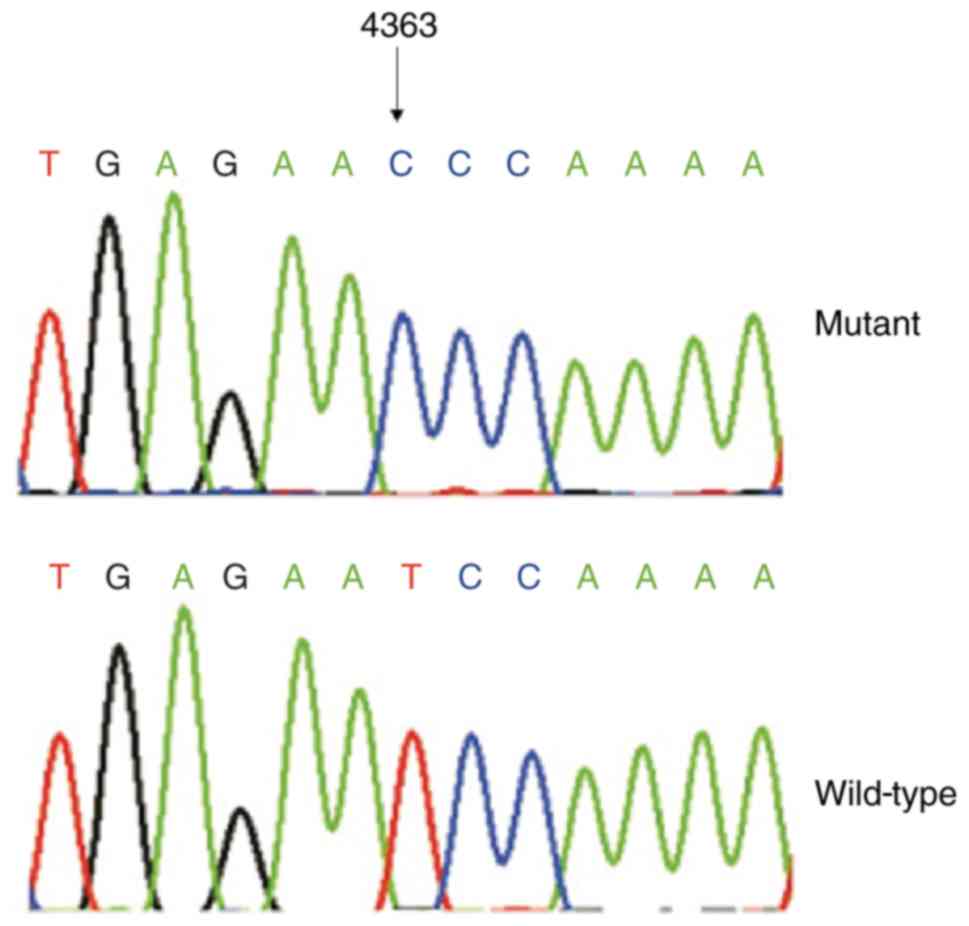
identification of a homoplasmic tRNAGln T4363C mutation.
In addition, to determine whether mitochondrial genetic background
may serve an active role in EH, the present study conducted
polymerase chain reaction (PCR)-Sanger sequencing for the fragments
spanning the mitochondrial genome, and used RNA Fold Webserver to
predict the potential pathogenicity of the tRNAGln
T4363C mutation.
Materials and methods
Subjects
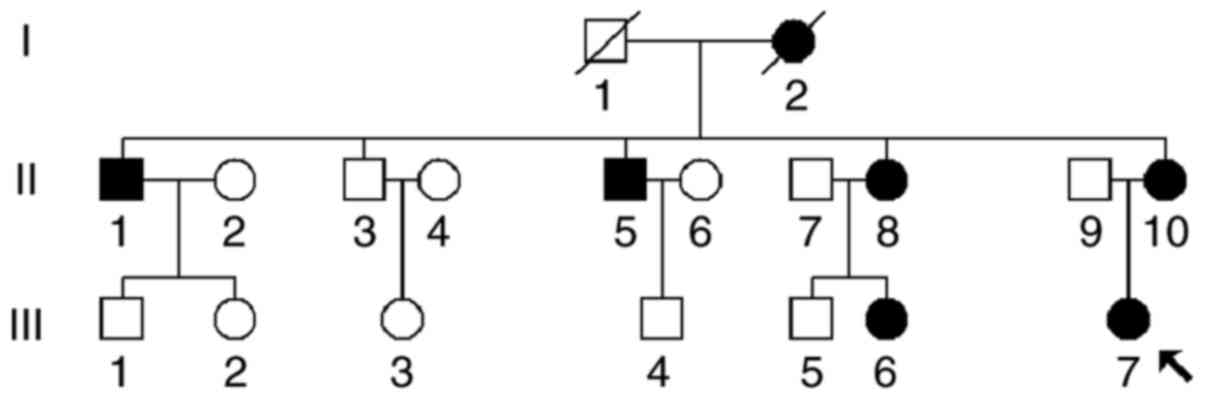
A Han Chinese family (Fig. 1) was recruited at the Department of
Cardiology, Hanchuan People's Hospital. The individuals were
interviewed, and detailed demographics, anthropometrics, vital
parameters and medical history were recorded. Furthermore, 300 DNA
samples were collected form age and gender-matched healthy
participants from the same area, which were used as controls. The
present study was approved by the Ethics Committee of Hanchuan
People's Hospital, and written informed consent was obtained from
all individuals or relatives prior to enrollment in the present
study.
Blood pressure (BP) measurement
Members of the Chinese family underwent a complete
examination, including physical examination, clinical laboratory
evaluation and routine electrocardiography. Using an electronic
measuring device, two doctors determined the systolic and diastolic
BP of each individual; BP measurements were repeated three times.
According to the World Health Organization International Society of
Hypertension (8), EH was defined
as a systolic BP >140 mmHg or a diastolic BP >90 mmHg.
Analysis of mitochondrial genome
mutations
To screen mutations in the mitochondrial genome,
genomic DNA was extracted from blood samples using the Puregene DNA
Isolation kit (Gentra Systems, Inc., Minneapolis, MN USA). The
complete mitochondrial genomes of matrilineal relatives (II-1,
II-3, II-5, II-8, II-10, III-5, III-6 and III-7) were amplified by
PCR, using a previously described method (9). Following PCR amplification and
electrophoresis, the 24 fragments spanning the mitochondrial genome
were purified and analyzed using an ABI 3700 automated DNA
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Furthermore, genetic variants were identified in
the mitochondrial genome by comparing the sequence data with the
Cambridge reference sequence (NC_012920) (10).
Phylogenetic conservation
analysis
The entire mitochondrial sequence variants in the
matrilineal relatives with EH in the Chinese pedigree were assigned
to the Asia mitochondrial haplogroups, as described by Kong et
al (11). Furthermore, 10
vertebrates' mtDNA sequences were selected to assess evolutionary
conservation. The conservation index (CI) was calculated by
comparing the human nucleotide variants with 9 other vertebrates. A
CI >75% was considered as having functional significance.
Prediction of the secondary structure
of tRNAGln with and without the T4363C mutation
To determine whether the T4363C mutation affected
tRNAGln structure, the RNA Fold Webserver program
(http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi)
was used to predict the minimum free energy (MFE) secondary
structure of the wild-type tRNAGln and the mutant
tRNAGln carrying the T4363C mutation (12). The wild-type sequence of
tRNAGln was:
5′-TAGGATGGGGTGTGATAGGTGGCACGGAGAATTTTGGATTCTCAGGGATGGGTTCGATTCTCATAGTCCTAG-3′,
whereas the sequence of tRNAGln carrying the T4363C
mutation was:
5′-TAGGATGGGGTGTGATAGGTGGCACGGAGAATTTTGGGTTCTCAGGGATGGGTTCGATTCTCATAGTCCTAG-3′.
The structure was predicted using the loop based energy model and
dynamic programming algorithm, as described by Zuker and Stiegler
(13).
Statistical analysis
Statistical analyses were performed using SPSS 17.0
(SPSS Inc., Chicago, IL, USA). Differences in categorical variables
were assessed with Fisher's exact test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Clinical characterization of the
Chinese pedigree carrying EH
The proband (III-7) was a 45-year-old woman born in
Wenzhou, who now lived in Hanchuang. The patient had suffered from
EH for ~5 years, and her BP was 150/95 mmHg. Recently, she visited
the Department of Cardiology, Hanchuan People's Hospital for
treatment of EH. A comprehensive examination, including physical
examination, clinical laboratory assessment of risk factors for EH
and electrocardiography, indicated that she did not carry other
abnormalities, such as diabetes mellitus, myopia, deafness, cancer,
and renal and neurological disorders. Therefore, she suffered from
only one syndrome: EH. According to the family history of the
patient, it was determined that 7 individuals from her family
suffered from a variable degree of hypertension. The grandmother
(I-2) of the proband had succumbed several years ago, due to high
BP (180/95 mmHg). As presented in Fig.
1, the pattern of transmission in this family was maternal
inheritance. As presented in Table
I, the age of onset of EH in the pedigree ranged between 39 and
63 years, with an average of 53 years.
 | Table I.Summary of clinical data for the
matrilineal relatives in a family with essential hypertension. |
Table I.
Summary of clinical data for the
matrilineal relatives in a family with essential hypertension.
| Subject | Sex | Age at test | Age of onset | Diastolic blood
pressure (mmHg) | Systolic blood
pressure (mmHg) | Occurrence of the
T4363C mutation |
|---|
| II-1 | Male | 66 | 61 | 95 | 145 | Yes |
| II-3 | Male | 63 | / | 80 | 120 | Yes |
| II-5 | Male | 68 | 63 | 80 | 150 | Yes |
| II-8 | Female | 65 | 60 | 100 | 160 | Yes |
| II-10 | Female | 61 | 59 | 95 | 175 | Yes |
| III-6 | Female | 41 | 39 | 90 | 145 | Yes |
| III-7 | Female | 45 | 40 | 95 | 150 | Yes |
| III-5 | Male | 46 | / | 75 | 130 | Yes |
| III-3 | Female | 40 | / | 80 | 135 | No |
| III-1 | Male | 36 | / | 75 | 135 | No |
Mutational analysis of the
mitochondrial genome
As shown in Fig. 1,
the pattern of transmission of EH in this family was consistent
with maternal inheritance, indicating that mitochondrial genome
mutations may be the molecular basis for this disease. To determine
the contribution of mtDNA mutations to EH, PCR amplification of the
mitochondrial genome was conducted on samples from matrilineal
relatives (II-1, II-3, II-5, II-8, II-10, III-5, III-6 and III-7)
and the PCR fragments were subsequently sequenced from each
affected individual. As presented in Table II, after comparing with the
Cambridge reference sequence by phylogenetic analysis, 25 genetic
polymorphisms were identified, belonging to human mitochondrial
haplogroup B4 (11). Of these,
there were 7 variants in the D-loop gene, 2 known variants in the
12S rRNA gene and 1 variant in the 16S rRNA gene, as well as a 9-bp
common deletion in the conjunction between the tRNALys
and cytochrome c oxidase subunit 2 genes. The missense
mutations included NADH dehydrogenase subunit 2 C5263T mutation
(A265V), ATPase subunit 6 A8701G (T59A) and A8860G (T112A)
mutations, NADH dehydrogenase subunit 3 A10398G (T114A) mutation
and cytochrome B C14766T (I7T) mutation. All of these genetic
variants can be found by searching Google and specific databases,
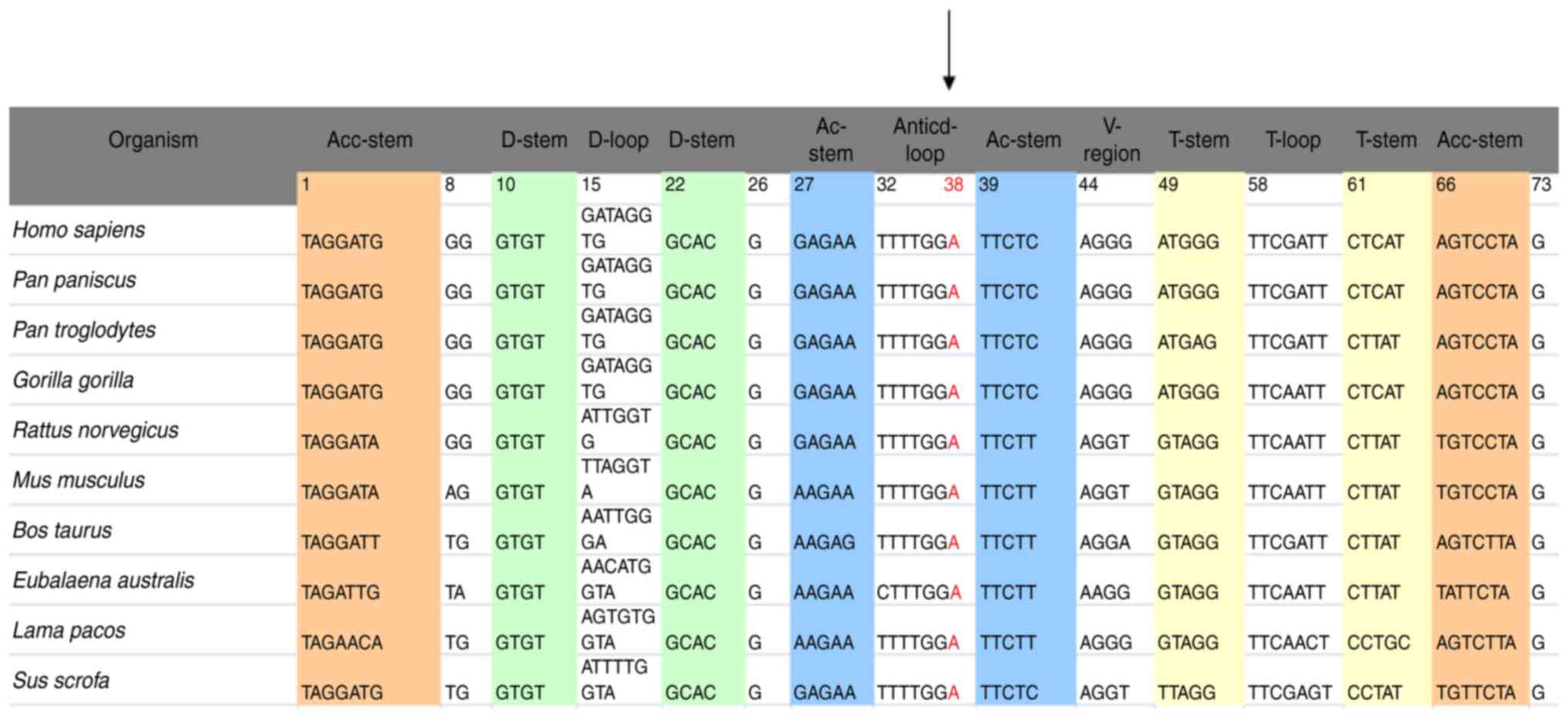
and therefore should not be regarded as novel (14). Furthermore, evolutionary
conservation was assessed for these identified variants in 9
organisms, including mice (15),
cattle (16) and Xenopus
laevis (17). We found that
other variants were not conserved, with the exception of the T4363C
mutation (Figs. 2 and 3). Notably, some matrilineal relatives
(II-3 and III-5) carried the T4363C mutation, but did not have high
BP. Fisher's exact frequency difference test demonstrated that the
T4363C mutation was significant when compared with the frequency in
control samples (P<0.05).
| Table II.Mitochondrial DNA sequence variants in
a family with essential hypertension. |
Table II.
Mitochondrial DNA sequence variants in
a family with essential hypertension.
| Gene | Position | Replacement | Conservation
(H/B/M/X) | Members carrying
these mutations |
|---|
| D-loop | 73 | A to G |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 152 | T to C |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 263 | A to G |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 310 | InsC |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 16,136 | T to C |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 16,189 | T to C |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 16,519 | T to C |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| 12S rRNA | 750 | A to G | A/A/A/- | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 827 | A to G | A/A/A/A | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| 16S rRNA | 3,107 | delC |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| ND1 | 3,970 | C to T |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| tRNA
Gln | 4,363 | T to C | Y/Y/Y/Y | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| ND2 | 4,715 | A to G | G/G/G/G | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 5,263 | C to T (Ala to
Val) | A/A/I/F | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| CO1 | 7,028 | C to T | A/A/A/A | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| NC_7 | 8,281–8,289 | 9-bp del |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| A6 | 8,701 | A to G (Thr to
Ala) | T/S/L/Q | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 8,860 | A to G (Thr to
Ala) | T/A/A/T | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| CO3 | 9,540 | T to C |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| ND3 | 10,398 | A to G (Thr to
Ala) | T/T/T/A | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 10,400 | C to T | T/T/T/A | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| ND5 | 12,705 | C to T | I/L/L/T | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
| Cytb | 14,766 | C to T (Thr to
Ile) | T/S/T/S | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 14,783 | T to C | I/I/I/I | II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
|
| 15,301 | G to A |
| II-1, II-3, II-5,
II-8, II-10, III-5, III-6, III-7 |
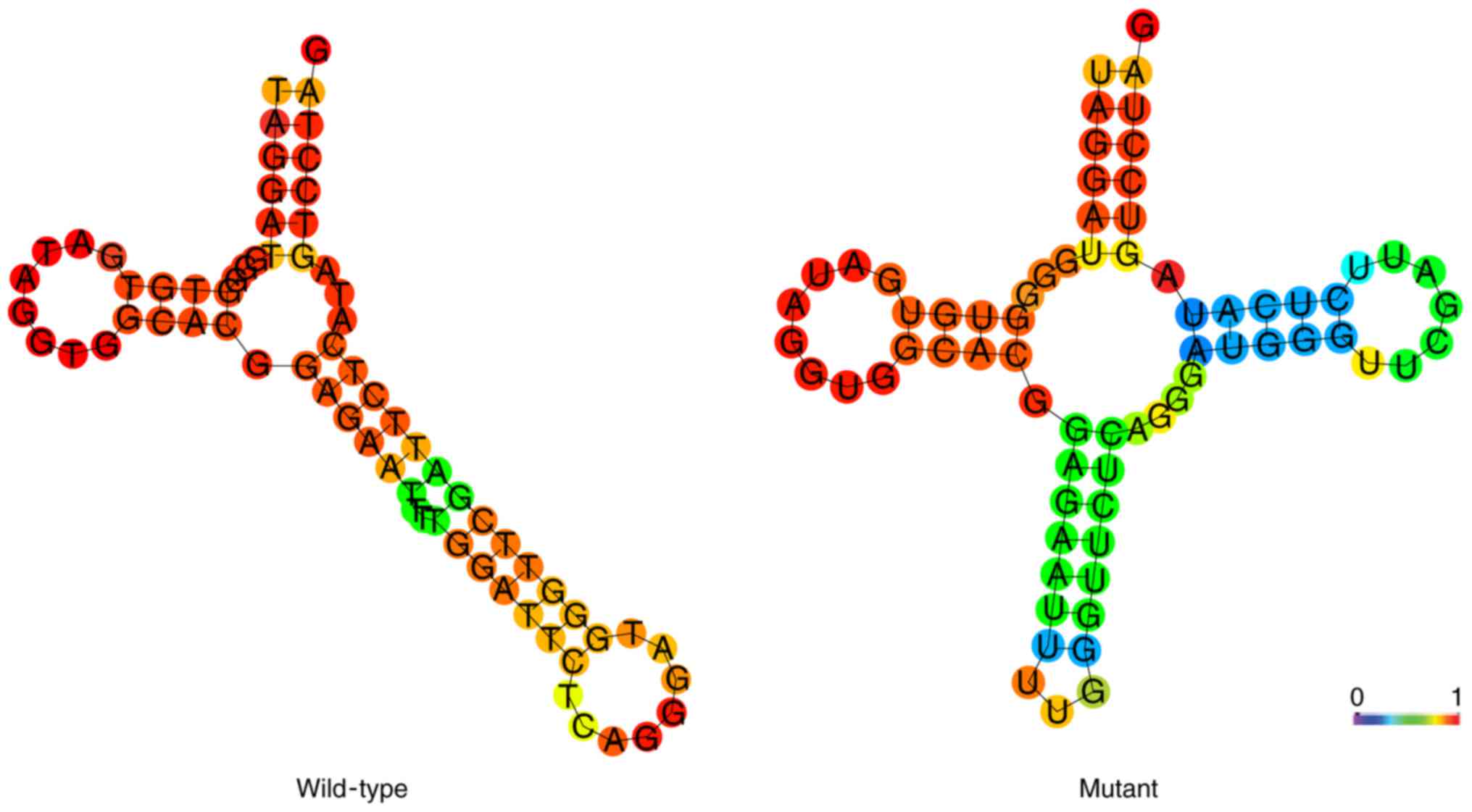
T4363C mutation induces structural
alterations to tRNAGln
To determine whether the T4363C mutation induced
secondary structure alterations to tRNAGln, the RNA Fold
program was used to predict the MFE structure of tRNAGln
with and without the T4363C mutation (12). As presented in Fig. 4, this mutation appeared to alter
the secondary structure of tRNAGln, thus suggesting that
the T4363C mutation may serve an important role in the development
of EH.
Discussion
The present study investigated the contribution of
mitochondrial mutations in the clinical manifestation of EH in a
Han Chinese family. Notably, members of this pedigree presented
with hypertension as the sole phenotype. Clinical and genetic
assessment revealed a variable degree of EH, with differing
severities and age of onset. Notably, the age of onset of EH in
matrilineal relatives (II-1, II-3, II-5, II-8, II-10, III-5, III-6
and III-7) ranged between 39 to 63 years, with an average age of 53
years. Furthermore, it was observed that compared with the first
and second generation, the members in the third generation in this
family had an earlier age of onset of EH; indicating that screening
for the presence of pathogenic mtDNA mutations may be useful for
the early diagnosis and prevention of EH.
Analysis of the mutations in the mitochondrial
genome identified 25 genetic polymorphisms belonging to human
mitochondrial haplogroup B4d. Of them, the tRNAGlnT4363C
mutation is of particular interest. This mutation was present in 6
matrilineal relatives with EH, but was also present in 2
matrilineal relatives without EH. Notably, the T4363C mutation was
localized at the immediate 3′ end of the anticodon, corresponding
to position 38 of tRNAGln (18). Notably, the nucleotide at this
position is highly conserved among9 other vertebrates, and is often
modified during tRNAGln processing and function. Thus,
the T4363C mutation may reduce the steady-state level of
tRNAGln (19). Previous
studies have reported that the T4363C mutation is associated with
deafness, developmental delay and pseudoexfoliation glaucoma
(20,21). Furthermore, the results of an RNA
Fold analysis indicated that the T4363C mutation altered the
structure of tRNAGln, strongly suggesting that this
mutation will result in the failure of tRNAGln
metabolism, consequently impairing mitochondrial translation and
finally leading to mitochondrial dysfunction associated with
EH.
In conclusion, the identification of a homoplasmic
tRNAGln T4363C mutation in members of this Chinese
pedigree suggested that this mutation may serve an active role in
the pathogenesis of EH. However, the family members (II-3 and
III-5) that carried the T4363C mutation but did not suffer from EH
suggested that environmental factors, nuclear gene and epigenetic
modifications may also serve important roles in the pathogenesis of
EH. It is recommended that the T4363C mutation in
tRNAGln may be considered a risk factor for the early
diagnosis of EH. Therefore, the present study provided a novel
insight into the molecular mechanism, prevention and potential
treatment of EH, particularly for those with a family history of
EH.
References
|
1
|
Gu D, Reynolds K, Wu X, Chen J, Duan X,
Muntner P, Huang G, Reynolds RF, Su S, Whelton PK, et al:
Prevalence, awareness, treatment, and control of hypertension in
China. Hypertension. 40:920–927. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wallace DC: Mitochondrial defects in
cardiomyopathy and neuromuscular disease. Am Heart J. 139:S70–S85.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwartz F, Duka A, Sun F, Cui J, Manolis
A and Gavras H: Mitochondrial genome mutations in hypertensive
individuals. Am J Hypertens. 17:629–635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Zheng J, Xue L, Meng Y, Wang Y,
Zheng B, Fang F, Shi S, Qiu Q, Jiang P, et al: The 12S rRNA A1555G
mutation in the mitochondrial haplogroup D5a is responsible for
maternally inherited hypertension and hearing loss in two Chinese
pedigrees. Eur J Hum Genet. 20:607–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu Z, Chen H, Meng Y, Wang Y, Xue L, Zhi
S, Qiu Q, Yang L, Mo JQ and Guan MX: The tRNAMet 4435A>G
mutation in the mitochondrial haplogroup G2a1 is responsible for
maternally inherited hypertension in a Chinese pedigree. Eur J Hum
Genet. 19:1181–1166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li R, Liu Y, Li Z, Yang L, Wang S and Guan
MX: Failures in mitochondrial tRNAMet and tRNAGln metabolism caused
by the novel 4401A>G mutation are involved in essential
hypertension in a Han Chinese Family. Hypertension. 54:329–337.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Z, Liu Y, Yang L, Wang S and Guan MX:
Maternally inherited hypertension is associated with the
mitochondrial tRNA(Ile) A4295G mutation in a Chinese family.
Biochem Biophys Res Commun. 367:906–911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
1999 World Health
Organization-International Society of Hypertension Guidelines for
the Management of Hypertension, . Guidelines Subcommittee. J
Hypertens. 17:151–183. 1999.PubMed/NCBI
|
|
9
|
Rieder MJ, Taylor SL, Tobe VO and
Nickerson DA: Automating the identification of DNA variations using
quality-based fluorescence re-sequencing: Analysis of the human
mitochondrial genome. Nucleic Acids Res. 26:967–973. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23:1471999. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kong QP, Bandelt HJ, Sun C, Yao YG, Salas
A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, et al: Updating the
East Asian mtDNA phylogeny: A prerequisite for the identification
of pathogenic mutations. Hum Mol Genet. 15:2076–2086. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gruber AR, Lorenz R, Bernhart SH, Neuböck
R and Hofacker IL: The Vienna RNA websuite. Nucleic Acids Res.
36:W70–W74. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zuker M and Stiegler P: Optimal computer
folding of large RNA sequences using thermodynamics and auxiliary
information. Nucleic Acid Res. 9:133–148. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bandelt HJ, Salas A, Taylor RW and Yao YG:
Exaggerated status of ‘novel’ and ‘pathogenic’ mtDNA sequence
variants due to inadequate database searches. Hum Mutat.
30:191–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bibb MJ, Van Etten RA, Wright CT, Walberg
MW and Clayton DA: Sequence and gene organization of mouse
mitochondrial DNA. Cell. 26:167–180. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gadaleta G, Pepe G, De Candia G,
Quagliariello C, Sbisà E and Saccone C: The complete nucleotide
sequence of the Rattus norvegicus mitochondrial genome: Cryptic
signals revealed by comparative analysis between vertebrates. J Mol
Evol. 28:497–516. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roe BA, Ma DP, Wilson RK and Wong JF: The
complete nucleotide sequence of the Xenopus laevis mitochondrial
genome. J Biol Chem. 260:9759–9774. 1985.PubMed/NCBI
|
|
18
|
Florentz C, Sohm B, Tryoen-Tóth P, Pütz J
and Sissler M: Human mitochondrial tRNAs in health and disease.
Cell Mol Life Sci. 60:1356–1375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bjork GR: Stable RNA modificationNeidhardt
FC, Curtiss R III, Ingraham JL, Lin ECC, Low BK, Magasanik B,
Reznikoff WS, Riley M, Schaechter M and Umbarger HE: Escherichia
coli and Salmonella: Cellular and Molecular Biology. American
Society for Microbiology; Washington, DC: pp. 861–886. 1996
|
|
20
|
Wong LJ, Liang MH, Kwon H, Park J, Bai RK
and Tan DJ: Comprehensive scanning of the entire mitochondrial
genome for mutations. Clin Chem. 48:1901–1912. 2002.PubMed/NCBI
|
|
21
|
Abu-Amero KK, Bosley TM and Morales J:
Analysis of nuclear and mitochondrial genes in patients with
pseudoexfoliation glaucoma. Mol Vis. 14:29–36. 2008.PubMed/NCBI
|