Introduction
Hematopoietic progenitor kinase 1 (HPK1), also known
as mitogen-activated protein kinase kinase kinase kinase 1 (MAP4K1)
is a 97 kDa serine/threonine protein kinase, which belongs to the
germinal center kinase family (1,2). It
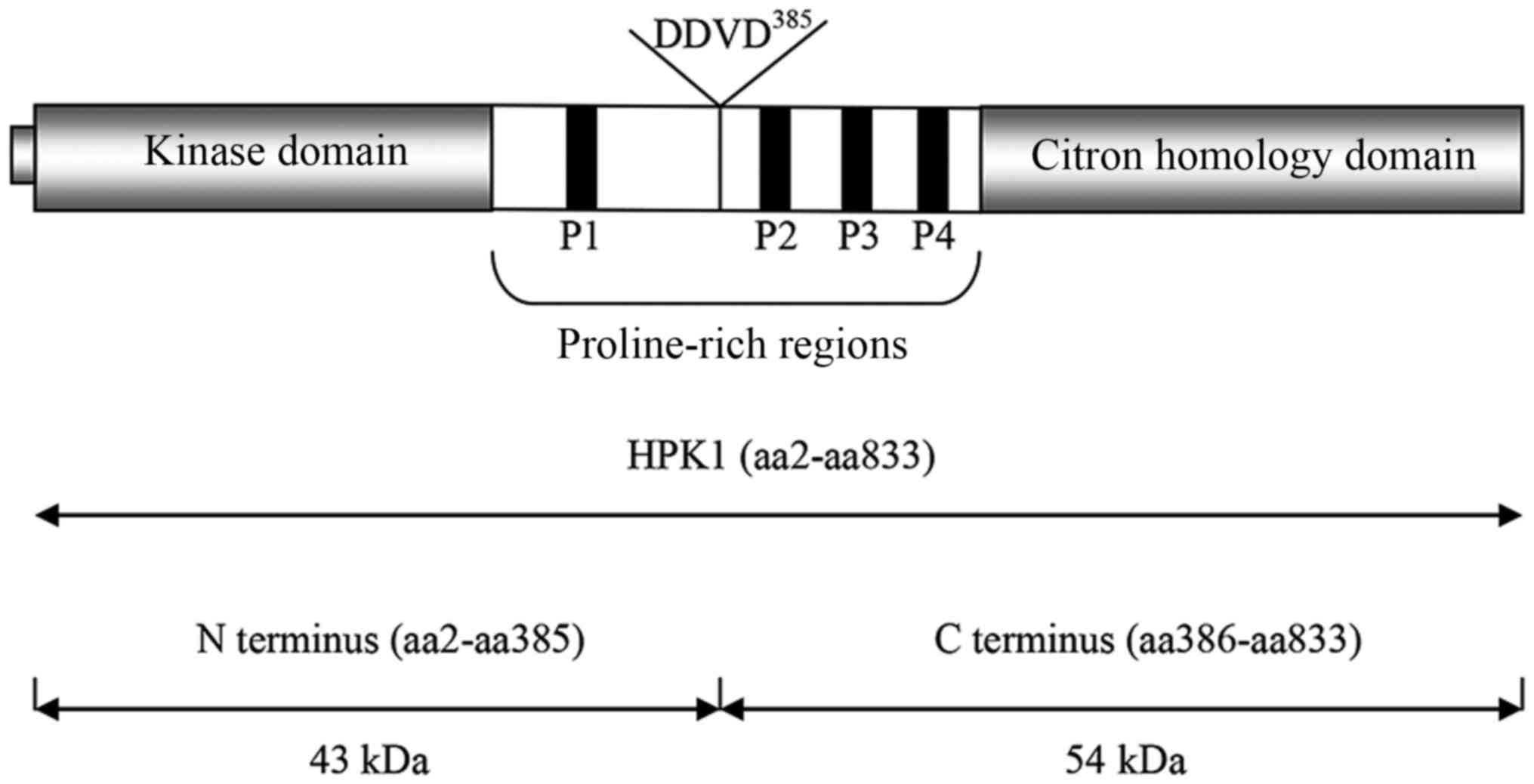
is comprised of an N-terminal Ste20-like kinase domain and four
proline-rich regions (P1, P2, P3 and P4, respectively). P1, P2 and
P4 contain the typical class II binding motif, which can bind
adaptor proteins containing Src homology 3 (SH3) domains (3–5).
HPK1 also has a citron homology (CNH) domain at its C-terminus,
which may act as a regulatory domain and is involved in molecular
interactions (6–8) (Fig.
1). It contains 13 potential tyrosine phosphorylation residues,
a number of which can be phosphorylated by ζ-chain associated
protein kinase 70 kDa (ZAP-70), providing potential docking sites
for Src homology 2 (SH2)-containing molecules (9).
The activation of HPK1 is involved in adaptor
protein binding, relocating to the plasma membrane,
autophosphorylation and transphosphorylation by protein kinase D1
(a serine/threonine kinase) and protein tyrosine kinases (PTKs)
(4,8,10,11).
It can be activated by a variety of stimuli (2,12),
including epidermal growth factor (EGF) (13,14),
prostaglandin E2 (PGE2) (15), transforming growth factor-β (TGF-β)
(16,17), erythropoietin (EPO) (18), T cell receptor (TCR) and B cell
receptor (BCR) stimulation (2,4,7,19).
During T cell activation, HPK1 functions upstream of protein kinase
C (PKC) and Ras (6).
Roles of HPK1 in immunity
HPK1 has a variety of roles in immunity. As a
serine/threonine protein kinase, HPK1 serves as an MAP4K and can
transmit signals through mitogen-activated protein kinase kinase
kinase (MAP3K), including mitogen-activated protein kinase kinase
(MEKK) 1, mixed-lineage protein kinase 3 and TGF-β activated
kinase-1 (TAK1) to mitogen-activated protein kinase kinase (MAP2K),
including MAPK kinase (MKK) 4 and MKK7, finally resulting in the
activation of c-Jun N-terminal kinase (JNK) (16,17,20–22).
HPK1 can activate the α and β subunits of the
inhibitor of nuclear factor-κB (IκB) kinase (IKK) complex,
resulting in IκB degradation and nuclear factor-κB (NF-κB)
activation (23).
HPK1 is cleaved into two fragments by caspase-3
during apoptosis at Asp385 within a conserved DDVD385
motif: An N-terminal region (43 kDa; aa2-aa385), which contains the
kinase domain and P1, and a C-terminal region (54 kDa;
aa386-aa833), which contains the CNH domain and the other three
proline-rich regions (8,19,24,25)
(Fig. 1). The N-terminal domain of
HPK1 retains the ability to activate JNK, whereas the C-terminal
domain of HPK1 becomes an inhibitor of NF-κB (7,24,25).
HPK1 also induces the expression of Fas ligand (FasL) in T cells
(2,26).
The JNK family is involved in various T cell
responses, which include Th1/Th2 differentiation, cellular
proliferation and apoptosis (26,27).
It has been shown that the activation of NF-κB can protect cells
from cellular apoptosis (23,28,29),
whereas FasL is a known inducer of apoptosis in T cells (26,28,29).
The ability of HPK1 to regulate JNK, NF-κB and FasL simultaneously
indicates its complex and conflicting role in controlling cellular
proliferation and apoptosis.
In addition to regulating cellular proliferation and
apoptosis, HPK1 is a negative regulator of TCR/BCR signaling and
T/B cell-mediated immune responses (1,11,30–32).
Shui et al found that HPK1−/− mouse T cells are
hyperproliferative in response to stimulation with anti-CD3 and
anti-CD28 antibodies, and these cells produce increased IL-2, IL-4
and interferon (IFN)-γ when immunized with T cell-dependent
antigens (6). In addition, T
cell-dependent humoral responses in HPK1−/− mice are
more marked, compared with those in controls and HPK1−/−
mice have been shown to exhibit more severe autoimmune phenotypes
in an experimental autoimmune encephalomyelitis model of multiple
sclerosis in mice (6). As with T
cells, HPK1 negatively regulates Ras-related protein 1
(RAP1)-mediated B cell adhesion; HPK1−/− mice B cells
appear hyperresponsive to a range of stimuli (12).
In humans, HPK1 is important roles in the
pathogenesis of systemic lupus erythematosus (SLE). It has been
shown that the mRNA and protein levels of HPK1 decreased
significantly in CD4+ T cells from patients with SLE,
compared with those in healthy controls, and its mRNA expression
was negatively correlated with disease activity in the patients
with SLE. When the expression of HPK1 was knocked down in healthy
CD4+ T cells using small interfering (si) RNA, increased
T cell proliferation, overproduction of IFNγ and excessive B cell
stimulation was observed. By contrast, plasmid-induced
overexpression of HPK1 in SLE CD4+ T cells resulted in
reduced T cell proliferative activity, and decreased IFNγ and IgG
synthesis. Therefore, HPK1 may serve as a novel target in effective
SLE therapy (31). In addition to
SLE, microarray analyses have suggested that the level of HPK1 is
downregulated in the peripheral blood cells of patients with
psoriatic arthritis (33,34).
In addition to autoimmune diseases, HPK1 is involved
in the pathogenesis of cancer. It has been found that the level of
HPK1 is altered in acute myeloid leukemia (35,36),
bladder urothelial carcinoma (37), extramammary Paget's disease
(38) and colon carcinoma
(39). However, unlike in
autoimmune diseases, the expression of HPK1 is increased in these
types of cancer. Of note, it has been reported that inhibiting HPK1
promotes resistance to cancer, and this results from the role of
HPK1 in immunity; HPK1 is a negative regulator of T cell and
dendritic cell (DC) activation. Mature HPK1−/− bone
marrow-derived DCs (BMDCs) are superior to their wild-type
counterpart in stimulating T cell proliferation in vivo and
in vitro. This is due to mature HPK1−/− BMDCs
expressing higher levels of costimulatory molecules CD80, CD86 and
I-Ab, and higher levels of proinflammatory cytokines
interleukin (IL)-1β, IL-6, IL-12 and tumor necrosis factor (TNF)-α.
HPK1−/− BMDCs are significantly resistant to
lipopolysaccharide-induced apoptosis. The vaccination of Lewis lung
carcinoma-bearing mice with HPK1−/− BMDCs can result in
complete tumor eradication (1,40).
Alzabin et al (11) demonstrated that HPK1−/−
mouse T cells are resistant to TCR- and PGE2-induced
apoptosis, and they can withstand PGE2-mediated
suppression of T cell proliferation, producing increased IL-2 and
IFNγ; following the transfer of a Lewis lung carcinoma tumor cell
line (3LL) intravenously into wild-type or HPK1−/− mice,
the number and size of tumors were smaller in the
HPK1−/− mice, compared with those in wild-type
counterparts, and this tumor inhibition was T cell-mediated. When
HPK1−/− mice splenocytes were co-cultured with the 3LL
cells, they were five times more effective in killing 3LL cells
than the wild-type splenocytes. Following intravenous injection of
wild-type or HPK1−/− T cells with 3LL cells into T
cell-deficient mice, the number and size of tumors were markedly
reduced in the lungs of the mice exposed to HPK1−/− T
cells. These results suggest that reducing the level of HPK1 leads
to an enhanced anti-tumor response by T cells (11,40).
HPK1 is also involved in the inflammatory response.
It is important for chemokine (C-X-C motif) ligand 1-induced
lymphocyte function associated antigen 1 (LFA-1)-mediated
polymorphonuclear neutrophil (PMN) adhesion to intercellular
adhesion molecule 1 (ICAM)-1 during the acute inflammation. In
addition, HPK1 is involved in CD11/CD18-mediated adhesion
strengthening, spread and the directed mechanotactic crawling of
PMNs under flow conditions. PMN adhesion and extravasation are
severely defective in HPK1-deficient mice (41).
Geisberger et al (42) reported that HPK1 is cleaved into N-
and C-terminal fragments during monocytic differentiation, and the
N-terminal region of HPK1 can induce the differentiation of
progenitor cells towards the monocytic lineage (19). HPK1 interacts with the cytoplasmic
tail of membrane IgE. This may represent a missing link to upstream
regulatory elements in HPK1 activation (42).
Adaptor proteins
Adaptor proteins are defined as proteins, which
exert no enzymatic activity (kinase or phosphatase) or
transcriptional activity, but contain noncovalent protein-protein
interaction domains, thus being able to selectively form
multi-protein complexes and transmit upstream signals to downstream
targets (14,43,44).
They usually localize close to the plasma membrane, and a number
are directly associated with surface receptors or the cytoskeleton
(18). Adaptor proteins are
essential in relaying extracellular signals from the plasma
membrane to the cell nucleus.
Those adaptor proteins, which interact with HPK1,
usually contain SH2/SH3 domains. The SH2 domains can bind
phosphorylated tyrosine residue, and the SH3 domains can bind the
consensus sequence (PXXP), which is the minimal sequence
requirement for SH3 domain ligands and is located within the
proline-rich regions of HPK1 (14,25,45).
With multiple binding sites, these adaptor proteins can integrate
and diversify cellular signals by linking different signaling
proteins to create various combinations of multi-protein complexes
(45,46).
Upon receiving upstream signals, these SH2/SH3
adaptor proteins can recruit HPK1 from the cytoplasm to activated
receptors, which have been tyrosine-phosphorylated, localized at
the plasma membrane or glycolipid-enriched microdomains (GEMs or
lipid rafts), which facilitates the subsequent activation of HPK1
and downstream signaling events (13,14,47).
Certain adaptor proteins can also link PTKs to HPK1 (9,14).
This review aims to describe those adaptor proteins,
which interact with HPK1, focusing particularly on their types of
interaction with HPK1 and the effects of these interactions.
CARD11
Caspase recruitment domain family, member 11
(CARD11), also known as CARMA1 and Bimp3, belongs to the CARD
family and to the membrane-associated guanylate kinase (MAGUK)
family. The MAGUK family members usually contain between one and
three PDZ domains (named after the domain-containing PSD-95, Dlg
and ZO-1 proteins). CARD11 contains one PDZ domain, followed by SH3
and GUK domains (48).
In addition to its C-terminal MAGUK-like features,
CARD11 contains a CARD within its N-terminus. A coiled-coil domain
is present after the CARD, and a linker region is present between
the coiled-coil domain and PDZ domain (48).
It has been shown there is a constitutive
association between HPK1 and CARD11 in T cells. The interaction
between HPK1 and CARD11 is mainly mediated by the coiled-coil
domain and CARD of CARD11; and HPK1 phosphorylates the linker
region of CARD11 upon TCR stimulation (49). The activation of NF-κB in response
to antigen receptor stimulation is mediated by the IKK complex, and
phosphorylation of the linker region of CARD11 is a precondition
for IKK activation. Following phosphorylation, CARD11 interacts
with B-cell lymphoma 10 and mucosa associated lymphoid tissue
lymphoma translocation gene 1, and induces activation of the IKK
complex. The loss of CARD11 causes disruption of IKK activation.
Therefore, CARD11 is important for the activation of NF-κB via HPK1
in lymphocytes (48,49). Ser551 in CARD11 is an important
residue for the TCR-induced activation of NF-κB though HPK1, with
Ser549 having an auxiliary role (49).
It has been reported that HPK1 and PKCθ alone can
increase the CARD11-dependent activity of NF-κB, and their combined
expression is able to further augment the activation of NF-κB. This
indicates that CARD11 can integrate HPK1-mediated and PKCθ-mediated
signaling events to enhance the activation of NF-κB (49).
HS1
Hematopoietic lineage cell-specific protein 1 (HS1)
is mainly expressed in hematopoietic cells (3,47,50).
It contains several characteristic structures, including an
N-terminal actin-binding domain, proline-rich sequences and a
C-terminal SH3 domain (3,47). As an actin-binding protein, HS1 can
bind to the actin cytoskeleton and orchestrate TCR or BCR signaling
(51). It is reported to be
involved in the activation of T/B cells and differentiation of
erythroid cells. Upon hematopoietic cell antigen receptor
engagement, it is tyrosine-phosphorylated by the Src family and
Syk/ZAP-70 family (3,18,50).
In hematopoietic cells, HS1 is constitutively
associated with HPK1 through its SH3 domain (3,18).
However, the sites responsible for their binding in HPK1 remain to
be elucidated. By synthesizing a series of peptide analogs derived
from the proline-rich regions of HPK1 and examining their ability
to bind the SH3 domain of HS1, it has been found that the typical
class II PXXPXK binding motif is not sufficient to induce the
interaction with the SH3 domain of HS1 (3,52).
An additive lysine, which locates two (in P2) or five (in P4)
residues following the basal PXXPXK motif (PXXPXKXKX in P2 or
PXXPXKXXXXK in P4) is required for the high-affinity binding of the
HS1 SH3 domain (3). The finding
that the cleaved HS1 SH3 domain and full-length HS1 exhibit similar
affinities with the proline-rich regions of HPK1 demonstrates that
the whole HS1 does not affect the affinity of its SH3 domain and
the proline-rich regions of HPK1, and that the SH3 domain contains
all binding recognition determinants (3).
The effects of the association between HS1 and HPK1
remain to be elucidated, however, it is suggested that this
interaction forms a possible link between the HPK1-JNK pathway and
HS1 signaling cascades (18).
HIP-55
HPK1-interacting protein of 55 kDa (HIP-55), also
termed DBNL, SH3P7 and mAbp1, is a novel member of the drebrin/Abp1
family of actin-binding proteins. The drebrin/Abp1 family is
conserved from yeast to mammals, and is characterized by the
presence of a homologous N-terminal actin-depolymerizing factor
homology domain (50,53). This family includes SH3P7 in mice
(47,53) and Abp1 in yeast (53). The drebrin/Abp1 family, which binds
filamentous actin but not actin monomers, is involved in multiple
signaling events (53).
HIP-55 is a ubiquitously expressed protein, which
consists of an actin-binding domain at its N-terminus, two
consensus tyrosine phosphorylation sites, and an SH3 domain at its
C-terminus (50,53,54).
Its C-terminal SH3 domain is homologous with HS1 (50).
HIP-55 has no actin polymerization activity
(53). It binds to filamentous
actin and colocalizes with actin filaments (54,55).
As an actin-binding protein, HIP-55 may affect immune synapse
formation and TCR signaling by regulating cytoskeletal
reorganization (54). HIP-55 is
able to inducibly interact with and is phosphorylated by upstream
ZAP-70 and Fyn following TCR stimulation (53–55).
It is possible that HIP-55 may be involved in regulating the
activity of ZAP-70 by modulating the recruitment of ZAP-70 to TCR
via cytoskeletal reorganization in TCR signaling (54). It is also known that HIP-55 is
involved in BCR signaling. As Syk shares similar regulatory
mechanisms with ZAP-70, it is possible that HIP-55 may interact
with Syk, and affect B cells or other antigen-presenting cells
(APCs) via Syk (54).
In Jurkat T cells, HIP-55 constitutively binds HPK1,
and can increase the kinase activity of HPK1 (53,56).
This interaction is mediated by the SH3 domain of HIP-55 and the P2
domain of HPK1 (51,56).
Using 293T cells, the coexpression of HIP-55 with
HPK1 has been shown to significantly increase HPK1 and JNK kinase
activity (55,56). The activation of JNK by HIP-55 is
mediated by HPK1, as transfection with the kinase-dead HPK1 mutant
(HPK1-K46M) completely inhibits the activation of JNK by HIP-55
(56). The knockdown of HIP-55 by
RNA interference (RNAi) in Jurkat T cells or the knockout of HIP-55
in mouse T cells results in a marked decrease in the activity of
HPK1 and JNK, but not that of extracellular signal-regulated kinase
(ERK) kinase upon TCR stimulation (54,55).
Therefore, HIP-55 serves as an upstream activator of HPK1 and the
JNK signaling pathway (56).
HIP-55 also cooperates with HPK1 in inhibiting the
activity of nuclear factor of activated T cell (NFAT) in T cells.
Overexpressing HIP-55 or HPK1 alone leads to inhibition of the
transcriptional activity of NFAT following TCR crosslinking, and
this inhibitory effect is further augmented by the coexpression of
two proteins. Transfection with the kinase-dead HPK1 mutant
(HPK1-K46E), or knockdown of either HIP-55 or HPK1 by RNAi, leads
to the activity of NFAT increasing in these systems (10,53).
Upon TCR/BCR stimulation, HIP-55 and HPK1
translocate to the T/B cell-APC junction and GEMs (53–55).
The SH3 domain and actin-binding domain of HIP-55 are essential for
its locating in the T/B cell-APC interface (53). It has been suggested that HIP-55
may bind HPK1 and mediate the recruitment of HPK1 to the T/B
cell-APC interface, where HPK1 interacts with its substrates and
downregulates the TCR/BCR signaling pathway (10,47,53).
GRB2 family
The growth factor receptor-bound protein 2 (GRB2)
family consists of growth factor GRB2, GRB2-related adaptor protein
(GRAP) and GRB2-related adaptor downstream of Shc (GADS), also
termed GRAP2, GRAPL, Grf40, Grid and Mona (5,43,45).
The GRB2 family contains one N- and one C-terminal SH3 domain
flanking a central SH2 domain; and GADS possesses a 120-amino acid
unique region, which is rich in glutamine and proline residues and
is between the SH2 domain and C-terminal SH3 domain (5,45). A
database search indicated that this unique region has no apparent
homology with other proteins (45). GRAP has a similar SH2 and SH3
binding profile to GRB2 (5,57).
GADS shares 40–50% sequence homology in the SH3 domains and 57%
homology in the SH2 domain with GRB2 and GRAP (45), however, the SH3 domains of GADS
cannot interact with certain proline-rich proteins (5,43).
GRB2 is expressed ubiquitously, whereas the expression of GRAP and
GADS are restricted to hemopoietic cells (5,45).
GRB2 and GRAP constitutively associate with HPK1
(9,13,57).
The N-terminal SH3 domain of GRB2 binds to the consensus motif
(PXXPXR/K) (14,45,58).
It has been shown that P1, P2 and P4 of HPK1 bind to the N-terminal
SH3 domain of GRB2, and P2 is the primary site for GRB2 binding to
HPK1, whereas P4 is the last of the three (13,14,58).
However, the exact binding domain of HPK1 to GRAP remains to be
elucidated.
GADS and HPK1 can form a complex in cells, and the
TCR-induced phosphorylation of HPK1 is able to increase its ability
to bind GADS (5,25,45).
The C-terminal SH3 domain of GADS mediates the binding with P2
(45) or P4 (5) of HPK1, and the SH2 domain is
associated with the tyrosine phosphorylation of HPK1 (5).
Previous X-ray crystallography studies have found
that the typical β-barrel SH3 domain in GADS presents the binding
pocket to HPK1; there are 16 structured residues in HPK1 composing
a peptide, which mediates the HPK1-GADS interaction. The specific
structural characteristics of the peptide comprise a 310
helix between residues Arg9 and Lys12, and a polyproline type II
(PPII) helix between residues Gln2 and Pro7. The PXVPXRXXK residues
of this peptide are essential for mouse HPK1 binding to the GADS
C-terminal SH3 domain, and the residues RXXK, which form the
310 helix, are the core motif. However, in the human
HPK1 sequence, the arginine can be substituted with a lysine,
leaving only the last lysine residue as a strictly conserved
position. A PXXP motif, which belongs to the PPII helix, is also
essential for the interaction of GADS and HPK1 (59).
In T cells, the SH2 and SH3 domains of GRB2 and GRAP
bind to similar proteins (5); in B
cells, the absence of GRB2 or GRAP only mildly reduces the kinase
activity of HPK1 upon BCR stimulation, whereas the absence of both
in B cells severely impairs HPK1 kinase activity upon BCR
stimulation (9,57). GRAP null mice exhibit no apparent
phenotypes (5). These findings
suggest that GRB2 and GRAP may share overlapping functions.
GRAP is able to inhibit the ERK pathway without
affecting the activation of JNK (60), which may provide an explanation for
how HPK1 negatively affects the ERK pathway in T cells.
In T cells, it has been suggested that GRB2 and GRAP
couple PTKs to the Ras pathway through the recruitment of GRB2-son
of sevenless homolog (SOS) or the GRAP-SOS complex to
tyrosine-phosphorylated linker for activation of T cells (LAT)
(5,57). By contrast, GRB2 has been shown to
couple upstream PTKs, including Src, to HPK1 (13,18).
As GRB2 can couple PTKs to the Ras pathway and to HPK1, it may
serve as a molecular bridge between these two biochemical pathways
and lead to interpathway cross-talk between these signaling
pathways (13).
GRB2 couples HPK1 not only to the TCR signaling
pathway, but also to the EGF receptor signaling pathway. Upon EGF
stimulation, GRB2 mediates the recruitment of HPK1 to the
autophosphorylated EGF receptor tail and to the Shc docking
protein, and HPK1 is phosphorylated on tyrosine (9,13).
The cotransfection of GADS and HPK1 increases the
kinase activity of HPK1, suggesting that GADS may recruit HPK1 to
the proximity of other HPK1 activators; the coexpression of GADS
and HPK1 also increases HPK-mediated activation of the JNK pathway,
and the additive effect is mediated by the specific GADS-HPK1
interaction; in Jurkat T cells, the cotransfection of HPK1 and GADS
also has an additive effect by increasing the transcriptional
activation of IL-2 following TCR stimulation (45). It may be that, upon TCR
stimulation, GADS couples TCR and CD28 with PTKs to HPK1,
facilitating the phosphorylation and activation of HPK1 by PTKs,
and transmitting the signals downstream to result in activation of
the JNK pathway and the transcription of IL-2 (45).
GADS facilitates the phosphorylation of HPK1,
however, HPK1 also phosphorylates Thr254 (mouse) or Thr262 (human)
of GADS, which mediates the binding of 14-3-3ζ to GADS (61). GADS can also inducibly associate
with LAT in T cells, but it does not form a complex with SOS.
Instead, GADS binds to the SH2 domain-containing leukocyte protein
of 76 kDa (SLP-76)/B-cell linker (BLNK), which is important in
transmitting the signaling of TCR/BCR. Therefore, through its
association with SLP-76/BLNK and LAT, GADS is central in
transmitting the signaling from the activated TCR/BCR (5,62).
The three members of the GRB2 family are known to be
involved in forming distinct signaling complexes during TCR
stimulation (5,14), indicating that these adaptors are
involved in the TCR signaling pathway. Although the majority of
signaling molecules involved in TCR signaling have their
counterparts in B cells, no functional analog of LAT has been
identified in B cells (62),
therefore, the mechanisms mediating the recruitment of the GRB2
family to BCR remain to be elucidated.
LAT
LAT is a 36–38 kDa palmitoylated
transmembrane-associated adaptor protein. It is localized in GEMs
(9,54,55).
Following TCR stimulation, LAT can be heavily tyrosine
phosphorylated by the Syk/ZAP-70 family (9,47).
It subsequently serves as a docking protein for several SH2
domain-containing signaling molecules, including SLP-76, GRB2,
GADS, phospholipase C (PLC)-γ1, and the p85 subunit of
phosphatidylinositol 3-kinase (PI3K) (9,54,55).
The LAT-associated proteins recruit various signaling molecules to
GEMs, where these signaling molecules are activated and promote
multiple downstream signaling events, including the activation of
MAPK, release of intracellular calcium and increased production of
IL-2 (54,55).
It has been shown that the presence of LAT is
required for TCR-induced HPK1 activation (4). In Jurkat T cells, LAT can form an
inducible complex with HPK1 as early as 1 min post-TCR stimulation,
and LAT recruits HPK1 into GEMs, leading to the activation of HPK1
by PTKs (6,9). The proline-rich regions of HPK1 are
critical to the HPK1-LAT interaction. As LAT does not contain an
SH3 domain for its direct binding to the HPK1 proline-rich regions,
the inducible HPK1-LAT complex formation is likely via SH2/SH3
adaptors. It is possible that GRB2 or GADS mediate the HPK1-LAT
interaction, which bind LAT through the SH2 domain and interact
with HPK1 by the SH3 domain. GRB2 or GADS may bring HPK1 to the LAT
signaling complex, where HPK1 interacts with its effector molecules
and substrates upon TCR stimulation (9,47).
SLP-76 family
The SLP-76 family includes SLP-76, its B-cell
homolog BLNK (also termed SLP-65 or BASH), and cytokine-dependent
hematopoietic cell linker (CLNK; also termed MIST) (57,62,63).
They all contain a similar structure with an N-terminal acidic
region, which contains a tyrosine-based SH2 domain-binding motif, a
central proline-rich region, which interacts with SH3 domains, and
a C-terminal SH2 domain, which interacts with phosphotyrosine
(46,63–65).
SLP-76 is widely expressed in T cells, natural
killer (NK) cells, myeloid cells, platelets and mast cells; BLNK
accumulates predominantly in B cells, whereas, CLNK is contained
exclusively in cytokine-stimulated hemopoietic cells, including
IL-2-induced T cells and NK cells, and IL-3-induced myeloid cells
and mast cells (64).
SLP-76 and BLNK are adaptor proteins specific to T
cells and B cells, respectively, and they are crucial in the signal
transmission of TCR and BCR (59,63,66).
Upon TCR or BCR stimulation, SLP-76 and BLNK are tyrosine
phosphorylated by the Syk/ZAP-70 family members, and translocate
into and recruit several signaling molecules into GEMs, where the
activated TCR and PTKs can activate downstream effectors (6,62,63).
They can interact with several components of TCR or BCR signaling
pathways, including Vav, non-catalytic region of tyrosine kinase
adaptor protein (NCK), GADS, GRB2, Btk, Itk, Cb1, PLC-γ1 and PLC-γ2
(47,62,63).
They also bind HPK1 following TCR or BCR stimulation (6,32,47).
It has been reported that GRB2, GADS and LAT interact with
SLP-76/BLNK, and may mediate the interactions between SLP-76/BLNK
and HPK1 (62,63,67).
The majority of HPK1-adaptor proteins interactions
are mediated by the proline-rich regions of HPK1 and the SH3
domains of adaptor proteins; however, the HPK1-SLP-76 and HPK-BLNK
interactions are mediated by Tyr379 (mouse) or Tyr381 (human),
located in the N-terminal of HPK1 P2, and phosphorylated by the
Syk/ZAP-70 family following TCR or BCR stimulation and the SH2
domains of SLP-76 and BLNK (10,62,63,65).
When T cells lack SLP-76 or B cells lack BLNK, the
kinase activity of HPK1 induced by TCR or BCR stimulation is
reduced (57,62,63).
It is noted that BLNK contributes to the activation, but not to the
tyrosine-phosphorylation, of HPK1 by BCR stimulation (63).
SLP-76 is involved in activating HPK1, however,
HPK1 also phosphorylates the serine and threonine residues of
SLP-76 (6,51,67).
It has been shown that HPK1 is able to directly phosphorylate the
serine residues of SLP-76, consequently creating 14-3-3-binding
sites, the critical one being phosphorylated Ser376, on SLP-76 and
inducing the binding of 14-3-3ζ (67) and τ (6) isoforms to serine-phosphorylated
SLP-76. The protein 14-3-3ζ and τ are negative regulators of the
TCR signaling pathway (6,67). They can bind and inhibit PI3K and
PKCθ. PI3K can trigger TCR-induced calcium signaling, and PKCθ can
promote TCR-induced ERK phosphorylation (6). PKCθ is also able to increase the
activity of RAP1 (51). RAP1 can
mediate LFA-1 integrin activation through its binding to RAP
ligand, a regulator for cell adhesion and polarization enriched in
lymphoid tissues, which assists T cells in adhering to ICAM-1 and
spreading (51). Upon TCR
stimulation, adhesion and degranulation promoting adaptor protein
(ADAP) binds SLP-76, and their binding causes membrane recruitment
of active RAP1. HPK1, which also binds SLP-76, competes with ADAP
for SLP-76 and reduces the recruitment of RAP1 (51). The phosphorylation of Ser376 also
mediates SLP-76 proteasomal degradation and Lys30 ubiquitination.
The Lys-30 ubiquitination of SLP-76 can attenuate the activation of
JNK and ERK (66). Lasserre et
al (61) reported that,
following activation, HPK1 is incorporated into SLP-76-containing
microclusters, subsequently phosphorylating SLP-76 on Ser376 and
GADS on Thr254 (mouse) or Thr262 (human), respectively. These
modifications recruit the 14-3-3ζ protein dimer, which in turn
dissociates the SLP-76-GADS-14-3-3ζ complex from phosphorylated LAT
and prevents its incorporation into a new one, consequently
reducing the persistence of SLP-76-containing microclusters and
downregulating TCR signaling (61). These results explain, at least in
part, the mechanisms through which HPK1 causes downregulation of
the TCR signaling pathway and inhibition of T cell activity.
The TCR-induced phosphorylation of SLP-76 tyrosine
is higher in HPK1−/− T cells and thymocytes than in
wild-type cells, and it has been found that several serine and
threonine residues are located in the proximity of three primary
phosphotyrosine sites (Tyr113, Tyr128 and Tyr145) on SLP-76.
Therefore, it is possible that the phosphorylation of SLP-76 serine
and threonine residues mediated by HPK1 changes the conformation of
SLP-76, and results in altered protein-protein interactions and
decreased tyrosine phosphorylation, creating further effects
(6). Sauer et al (62) reported that HPK1 can inhibit the
activation of activator protein-1 (AP-1), and this depends on it
antagonizing the recruitment of positive effectors into the SLP-76
SH2 domain.
Following BCR stimulation, HPK1 is also able to
phosphorylate BLNK Thr152, which mediates BLNK/14-3-3 binding.
Thr152 phosphorylation also induces BLNK proteasomal degradation
and ubiquitination at Lys37, Lys38 and Lys42. It has been found
that an E3 ligase is recruited to BLNK via 14-3-3, mediating and
inducing BLNK ubiquitination. These ubiquitination sites can
inhibit the activation of ERK, JNK and IKK. All of these, in turn,
attenuate BCR signaling and B cell activation (32).
Unlike SLP-76, HPK1 can induce BLNK to interact
with all seven isoforms of 14-3-3, with the relative affinity order
γ > τ > η > ε > β > δ > ζ. In contrast to
HPK1-deficient T cells, in which the SLP-76/14-3-3 interaction is
only partially lost, BLNK/14-3-3 binding is undetectable in
HPK1-deficient B cells, suggesting HPK1 is involved exclusively in
B cells (32).
As mentioned previously, HPK1 can activate JNK
(16,17,20–22)
and IKK (23). However, through
SLP-76/BLNK, HPK1 also has an inhibitory effect on the activation
of JNK and IKK. In an HPK1-overexpression system, the expression of
SLP-76/BLNK is limited for HPK1-mediated feedback inhibition.
Consequently, the positive effect of HPK1 on the activation of JNK
and IKK can overcome the inhibitory effect. However, in
HPK1-deficient cells, the inhibitory role of HPK1 is predominant
(32,66).
CLNK is the third member of the SLP-76 family. It
is a cytokine inducible (IL-2 and IL-3) adaptor protein (47). Unlike SLP-76 and BLNK, CLNK does
not interact with Vav, NCK, GRB2, GADS or PLC-γ (64), and it is not required for immune
system functions (68).
Upon TCR engagement, CLNK binds HPK1 with its SH2
domain. The conserved residue, Arg335, within the CLNK SH2 domain,
and phosphorylated Tyr379 (mouse) or Tyr381 (human) within HPK1 is
essential for their interaction (10,64).
However, deletion of the CLNK SH2 domain only partially (~75%)
reduces the ability of CLNK to bind with HPK1 in Cos-1 cells.
Therefore, there may be additional motifs involved in the CLNK-HPK1
binding (64). The association
between CLNK-HPK1 is significantly higher, compared with that of
SLP-76-HPK1 (10,64). The expression of CLNK is able to
augment the extent of tyrosine phosphorylation and kinase activity
of HPK1, and CLNK is also phosphorylated by HPK1 (10,64).
The coexpression of CLNK and Lck can result in HPK1
membrane recruitment, which in lymphocytes results in the
recruitment of HPK1 to the contact site of T-cell-APC conjugates,
where HPK1 is tyrosine phosphorylated and activated. However, CLNK
alone is incapable of localizing HPK1 to the plasma membrane; HPK1
appears to divert CLNK away from the plasma membrane (10).
It has been reported that the expression of HPK1
alone is able to inhibit the activity of the IL-2 promoter, and the
introduction of CLNK alone can increase the activation of the IL-2
promoter, although the coexpression of CLNK and HPK1 causes more
pronounced activation of the IL-2 promoter, compared with CLNK
alone. Their cooperation requires the enzymatic activity of HPK1,
as the kinase-inactive HPK1 (K46E) inhibits the ability of CLNK to
upregulate IL-2 promoter activity (64).
It has been suggested that CLNK may permit the
involvement of HPK1 in a signaling pathway, which differs from that
used by HPK1 alone. In this signaling pathway, HPK1 turns from an
inhibitor to a positive regulator of IL-2 promoter activation. This
change may be due to a CLNK-mediated change in HPK1 cellular
localization or a modification in its substrate specificity
(64).
CLNK and SLP-76 may transmit immunoreceptor
signaling in a similar manner but using different sets of partners.
CLNK can restore antigen receptor-induced transcriptional events in
a manner analogous to that of SLP-76 in SLP-76-deficient cells,
indicating that CLNK is capable as a functional substitute for
SLP-76 in immunoreceptor signaling (64).
Through CLNK, certain requirements for
immunoreceptor signaling, including the formation of a proper
SLP-76-GADS-LAT complex in T cells, may be alleviated, resulting in
the facilitation of cell activation. In T cells, this property may
reduce the requirements for engagement of coreceptors (CD4 and CD8)
and costimulatory molecules (CD28), which has been described in
memory T cells (64). Therefore,
the interaction between CLNK and HPK1 indicates the possibility
that HPK1 can be involved in T cells which have been activated by
cytokines, for example memory T cells (47).
CRK family
V-crk avian sarcoma virus CT10 oncogene homolog
(CRK) adaptor proteins consist of CRK I-, CRK II- and CRK-like
protein (CRKL) (14,69). All CRK proteins contain SH2 and SH3
domains (58,69). CRKI and CRKII result from
alternative RNA splicing. CRK I consists of one N-terminal SH2
domain followed by one SH3 domain, and CRK II consists of one
N-terminal SH2 domain followed by two tandem SH3 domains; CRKL,
which is cloned from chronic myelogenous leukemia cells, shares 60%
overall homology of CRK II and contains two SH3 domains (14,43).
CRK I is expressed in embryonic cells, whereas CRK II and CRKL are
widely expressed (43).
In Jurkat T cells, CRK inducibly binds HPK1
following TCR stimulation for 10 min, with CRKL forming a
constitutive complex with HPK1 (9). The P2 and P4 of HPK1, which contain
the consensus motif (PXLPXK), may be involved in recognition of the
CRK family (14,70,71).
This consensus motif is unique and incompatible with the majority
of other SH3 domain-binding motifs (58). HPK1 P2, the sequence of which is
fully conserved between mouse and humans, is considered to be the
primary site for the CRK family binding. The Lys400 of P2 is
important in the recognition of CRK and CRKL by HPK1 (14). In terms of HPK1 P4, its sequence is
not conserved between humans and mouse; therefore, it is not likely
to be essential for the interaction with CRK/CRKL (14,58).
The members of the CRK family preferentially use their first SH3
domain to bind HPK1 (9,14,58),
and the binding is independent of the catalytically activity of
HPK1, as the inactive HPK1 can form more stable binding with the
CRK proteins than active HPK1 (58).
HPK1 can phosphorylate CRK mainly on threonine,
with weaker phosphorylation on serine, whereas HPK1 phosphorylates
CRKL mainly on serine with weaker phosphorylation on threonine
(14,47).
Upon TCR stimulation, CRK and CRKL are shown to
form inducible complexes with tyrosine kinases ZAP-70 and Fyn,
respectively (9). Therefore, CRK
and CRKL may couple HPK1 to ZAP-70 and Fyn, respectively, to become
involved in the TCR-mediated signaling events following TCR
engagement. In addition to TCR stimulation, HPK1 is
tyrosine-phosphorylated by EGF stimulation and is recruited to the
EGF receptor tail via the CRK adaptors, suggesting that the CRK
adaptors are also involved in the recruitment of HPK1 to the EGF
receptor (9,14).
In 293T cells, the CRK family can activate HPK1,
and shares common downstream effectors, including TAK1, MEKK1 and
MKK4, for the activation of JNK with HPK1; the CRK family can
cooperate with HPK1 to activate JNK synergistically when they are
coexpressed. These data indicate that the CRK proteins function as
upstream regulators of the HPK1-mediated JNK signaling cascade
(14). In contrast to the results
in 293T cells, the overexpression of CRKL and HPK1 does not further
activate JNK in Cos-7 cells (58).
These results indicate the role of CRKL in modulating the
activation of HPK1 and activity of JNK depends on the cell types or
the accessory proteins of every cell type (14). Upon stimulation, the CRKL-HPK1
interaction may also translocate HPK1 from the cytoplasm to areas
of signal transduction, including GEMs (47).
CRK interacts with two guanine nucleotide exchange
factors for Ras family members, SOS and RAP guanine nucleotide
exchange factor 1 (RAPGEF1), also known as C3G. SOS is a known
activator of Ras, and RAPGEF1 is an exchange factor of RAP1A and
RAP1B. CRK may be involved in the activation of JNK through RAPGEF1
(14,43). CRKL has also been shown to activate
the Ras and JNK signaling pathways in cells (14). HPK1 has the potential to inhibit
RAP1 by sequestration of RAPGEF1 though the interaction of HPK1 and
CRKL, leading to the inhibition of T cell activity (51).
BAM32
B-cell adaptor molecule of 32 kDa (BAM32), also
known as DAPP1 and PHISH, is a recently identified adaptor protein,
which is restricted primarily to hematopoietic cells with
particularly high expression in B cells (72). BAM32 consists of an N-terminal SH2
domain and a C-terminal pleckstrin homology (PH) domain. It is
recruited to the plasma membrane in a PI3K-dependent manner
following BCR crosslinking, and appears to function as a dual
adaptor for phosphotyrosine and 3-phosphoinosides (46). BAM32−/− B cells are
defective in T cell-independent, but normal in T cell-dependent
responses to antigens in vivo (46).
BAM32 associates with HPK1, and this is not
dependent on BCR stimulation (46); it can directly localize fHPK1 to
the plasma membrane (46). BAM32
is important for the activation of HPK1, ERK and JNK following BCR
stimulation. BAM32−/− B cells show defects in the
activities of HPK1, ERK and JNK in response to BCR crosslinking,
however, p38 is not affected in the absence of BAM32 (47,50).
The exact association between BAM32 and HPK1 requires further
investigation.
NCK
NCK includes NCKα and NCKβ, NCKα is encoded by the
nck gene, whereas NCKβ, also known GRB4, is encoded by a novel
gene. NCKβ shares 68% amino acid identity with NCKα (43). The NCK proteins contain one SH2
domain and three SH3 domains, and can be phosphorylated on serine,
threonine and tyrosine residues (47).
NCKα can interact directly with HPK1 (9,13),
although the interaction of NCKβ with HPK1 remains to be
elucidated. Using the yeast two-hybrid assay, the three NCK SH3
domains are able to interact with any of the four proline-rich
motifs of HPK1, indicating that NCK may recognize multiple HPK1
proline-rich regions (13). It has
been shown that the binding between NCK and HPK1 occurs primarily
through the second SH3 domain of NCK, which is also known to bind
the SOS and Abl proteins (58).
In Jurkat T cells, the interaction between HPK1 and
NCK occurs following TCR stimulation for 1 min, and the HPK1-NCK
complex is sustained until 10 min of TCR stimulation. The kinetics
of the interaction between HPK1 and NCK correlates with that of the
activation of HPK1 during TCR stimulation, suggesting that NCK may
be involved in the regulation of HPK1 during TCR stimulation
(9).
Upon TCR stimulation, NCK and HPK1 translocate to
GEMs (9,53), where NCK can interact with the CD3ε
chain and modulate downstream signals (44). It has been suggested that NCK may
be involved in recruiting HPK1 to the activated TCR complex and
GEMs, and transmitting upstream signals (47).
The interaction between HPK1 and NCK is induced not
only by TCR stimulation, but also by EGF stimulation (13). The overexpression of GRB2 can
significantly decrease the association between HPK1 and NCK,
suggesting that these two adaptors can compete with each other for
binding HPK1 (13). NCK also
interacts with the SH2 domain binding motifs in the N-terminal
region of SLP-76, which encompass phosphorylated Tyr113, Tyr128 and
Tyr145 (61,67).
Conclusions and future directions
The features of the adaptor proteins described
above are summarized in Tables I
and II. As mentioned above, these
adaptor proteins do not interact with HPK1 separately, they also
associate with each other. Among these adaptors, LAT and
SLP-76/BLNK have a specific status; they serve as docking proteins
to form a platform for signal complexes. Following TCR/BCR
stimulation, LAT and SLP-76/BLNK are tyrosine phosphorylated by the
Syk/ZAP-70 family. SLP-76/BLNK is recruited onto LAT via their
association with the GRB2 family (61,62,67)
to form a central scaffold, and their phosphotyrosines provide
docking sites for SH2 and phosphotyrosine-binding domains of
enzymes and other adaptor proteins (10,62).
This promotes the activation of effector molecules, including
HPK1.
 | Table I.Adaptor proteins binding HPK1. |
Table I.
Adaptor proteins binding HPK1.
| Adaptor
protein | Structure | Mode of
binding | Binding domain
within HPK1 | Binding domain
within adaptor |
|---|
| CARD11 |
CARD-(coiled-coil)-linker-PDZ-SH3-GUK | Constitutive | Undetermined | CARD,
coiled-coil |
| HS1 | ABa-PRb-SH3 | Constitutive | Undetermined | SH3 |
| HIP-55 |
(ADF-H)-pTyr-SH3 | Constitutive | P2 | SH3 |
| GRB2 | SH3-SH2-SH3 | Constitutive | P1, p2, p4 | SH3(Nc) |
| GRAP | SH3-SH2-SH3 | Constitutive | Undetermined | Undetermined |
| GADS |
SH3-SH2-120aad-SH3 | Inducible | P2, p4 | SH3(Ce) |
| LAT | EXf-TMg-pTyr | Inducible | Indirect | Indirect |
| SLP-76 | pTyr-PR-SH2 | Inducible |
Ptyr379(mh), Ptyr381(hi) | SH2 |
| BLNK | pTyr-PR-SH2 | Inducible | Ptyr379(m),
Ptyr381(h) | SH2 |
| CLNK | pTyr-PR-SH2 | Inducible | Ptyr379(m),
Ptyr381(h) | SH2 |
| CRK I | SH2-SH3 | Inducible | P2, p4 | SH3 |
| CRK II | SH2-SH3-SH3 | Inducible | P2, p4 | SH3(N) |
| CRKL | SH2-SH3-SH3 | Constitutive | P2, p4 | SH3(N) |
| BAM32 | SH2-PH | Constitutive | Undetermined | Undetermined |
| NCK |
SH3-SH3-SH3-SH2 | Inducible | All SH3 | All PR |
| Table II.Functions of adaptor proteins. |
Table II.
Functions of adaptor proteins.
| Adaptor | Other name | Family | Function |
|---|
| CARD11 | CARMA1, Bimp3 | CARD/MAGUK | Activates IKK and
NF-κB |
| HS1 | – | – | Affects HPK1-JNK
pathway |
| HIP-55 | DBNL, SH3P7,
mAbp1 | drebrin/Abp1 | Activates HPK1 and
JNK; inhibits NFAT and TCR/BCR signaling |
| GRB2 | – | GRB2 | Activates HPK1;
affects Ras/EGFR/TCR/BCR signaling |
| GRAP | – | GRB2 | Activates HPK1;
inhibits ERK; affects Ras/TCR/BCR signaling |
| GADS | GRAP2, GRAPL,
Grf40, GRID, Mona | GRB2 | Activates HPK1, JNK
and IL-2; affects TCR/BCR signaling |
| LAT | – | – | Activates HPK1;
composes platform for signal complexes |
| SLP-76 | – | SLP-76 | Activates HPK1;
inhibits JNK, ERK; AP-1 and TCR signaling; composes platform for
signal complexes |
| BLNK | SLP-65, BASH | SLP-76 | Activates HPK1;
inhibits JNK, ERK, IKK and BCR signaling; composes platform for
signal complexes |
| CLNK | MIST | SLP-76 | Activates HPK1 and
IL-2; functionally substitutes for SLP-76 |
| CRK I | – | CRK | Activates HPK1 and
JNK |
| CRK II | – | CRK | Activates HPK1 and
JNK |
| CRKL | – | CRK | Activates HPK1 and
JNK |
| BAM32 | DAPP1, PHISH | – | Activates HPK1, JNK
and ERK |
| NCK | – | – | Activates HPK1;
affects TCR signaling |
There are several controversies surrounding the
data on HPK1. For example, it has been shown that the kinase-dead
mutant of HPK1 cannot inhibit the activation of JNK by anti-CD3 and
anti-CD28 co-stimulation (9,57);
another study reported that the kinase-dead mutant of HPK1 inhibits
the Vav-mediated activation of JNK following TCR stimulation
(73). Kiefer et al
(70) reported that HPK1 does not
affect the ERK family following transfection into Cos-1 cells,
whereas others have reported that HPK1 negatively affects the ERK
signaling pathway in an overexpression system using T cells
(57,62), and that HPK1−/− T cells
and thymocytes enhance the TCR-induced activation of ERK (6). Reports have indicated that HPK1
negatively affects the transcriptional activity of AP-1 (6,57,62,64),
whereas others have shown that HPK1 enhances the transcriptional
activity of AP-1 (14,16,39,45,71).
Previous studies have indicated that HPK1 is a negative regulator
of TCR-induced IL-2 gene transcription (4,6,11,64,67),
whereas others have reported that HPK1 can enhance the
transcriptional activity of IL-2 (14,24,45,49,71).
In terms of cellular proliferation, there is data
showing that HPK1 can assist PGE2 in suppressing T cell
proliferation (11), and that
HPK1−/− T cells are hyperproliferative in response to
TCR stimulation (6). Nagata et
al (18) confirmed that
antisense S-oligonuleotides to HPK1 mRNA can significantly inhibit
EPO-dependent FD-EPO cell growth in a dose-dependent manner.
It has been reported that the overexpression of
HPK1 promotes spontaneous apoptosis and anti-CD3-mediated apoptosis
of T cells (26);
HPK1−/− T cells have been shown to withstand
PGE2-mediated T cell apoptosis (11), however, Shui et al (6) reported that HPK1−/− T
cells undergo apoptosis in a similar manner to wild-type T cells
following stimulation with anti-CD3 or anti-Fas. Others have shown
that the siRNA-mediated knockdown of HPK1 increases T cell
apoptosis induced by TCR stimulation (7).
The conflicting findings reported may result from
using different cell types and stimuli. Which factors have
definitive roles in these events remain to be elucidated. It may be
that these adaptor proteins couple HPK1 with various effector
molecules, which leads HPK1 to different signaling pathways, and/or
alters the cellular localization of HPK1, or they may modify
substrate specificity to determine the functions of HPK1. This
requires further investigation.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81301359).
References
|
1
|
Alzabin S, Bhardwaj N, Kiefer F,
Sawasdikosol S and Burakoff S: Hematopoietic progenitor kinase 1 is
a negative regulator of dendritic cell activation. J Immunol.
182:6187–6194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou G, Boomer JS and Tan TH: Protein
phosphatase 4 is a positive regulator of hematopoietic progenitor
kinase 1. J Biol Chem. 279:49551–49561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siligardi G, Ruzza P, Hussain R, Cesaro L,
Brunati AM, Pinna LA and Donella-Deana A: The SH3 domain of HS1
protein recognizes lysine-rich polyproline motifs. Amino Acids.
42:1361–1370. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sawasdikosol S, Pyarajan S, Alzabin S,
Matejovic G and Burakoff SJ: Prostaglandin E2 activates HPK1 kinase
activity via a PKA-dependent pathway. J Biol Chem. 282:34693–34699.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu SK, Smith CA, Arnold R, Kiefer F and
McGlade CJ: The adaptor protein Gads (Grb2-related adaptor
downstream of Shc) is implicated in coupling hemopoietic progenitor
kinase-1 to the activated TCR. J Immunol. 165:1417–1426. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shui JW, Boomer JS, Han J, Xu J, Dement
GA, Zhou G and Tan TH: Hematopoietic progenitor kinase 1 negatively
regulates T cell receptor signaling and T cell-mediated immune
responses. Nat Immunol. 8:84–91. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brenner D, Golks A, Kiefer F, Krammer PH
and Arnold R: Activation or suppression of NFkappaB by HPK1
determines sensitivity to activation-induced cell death. EMBO J.
24:4279–4290. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brenner D, Golks A, Becker M, Müller W,
Frey CR, Novak R, Melamed D, Kiefer F, Krammer PH and Arnold R:
Caspase-cleaved HPK1 induces CD95L-independent activation-induced
cell death in T and B lymphocytes. Blood. 110:3968–3977. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ling P, Meyer CF, Redmond LP, Shui JW,
Davis B, Rich RR, Hu MC, Wange RL and Tan TH: Involvement of
hematopoietic progenitor kinase 1 in T cell receptor signaling. J
Biol Chem. 276:18908–18914. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arnold R, Patzak IM, Neuhaus B,
Vancauwenbergh S, Veillette A, Van Lint J and Kiefer F: Activation
of hematopoietic progenitor kinase 1 involves relocation,
autophosphorylation, and transphosphorylation by protein kinase D1.
Mol Cell Biol. 25:2364–2383. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alzabin S, Pyarajan S, Yee H, Kiefer F,
Suzuki A, Burakoff S and Sawasdikosol S: Hematopoietic progenitor
kinase 1 is a critical component of prostaglandin E2-mediated
suppression of the anti-tumor immune response. Cancer Immunol
Immunother. 59:419–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Königsberger S, Peckl-Schmid D, Zaborsky
N, Patzak I, Kiefer F and Achatz G: HPK1 associates with SKAP-HOM
to negatively regulate Rap1-mediated B-lymphocyte adhesion. PLoS
One. 5(pii): e124682010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anafi M, Kiefer F, Gish GD, Mbamalu G,
Iscove NN and Pawson T: SH2/SH3 adaptor proteins can link tyrosine
kinases to a Ste20-related protein kinase, HPK1. J Biol Chem.
272:27804–27811. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ling P, Yao Z, Meyer CF, Wang XS, Oehrl W,
Feller SM and Tan TH: Interaction of hematopoietic progenitor
kinase 1 with adapter proteins Crk and CrkL leads to synergistic
activation of c-Jun N-terminal kinase. Mol Cell Biol. 19:1359–1368.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sawasdikosol S, Russo KM and Burakoff SJ:
Hematopoietic progenitor kinase 1 (HPK1) negatively regulates
prostaglandin E2-induced fos gene transcription. Blood.
101:3687–3689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou G, Lee SC, Yao Z and Tan TH:
Hematopoietic progenitor kinase 1 is a component of transforming
growth factor beta-induced c-Jun N-terminal kinase signaling
cascade. J Biol Chem. 274:13133–13138. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang W, Zhou G, Hu MC, Yao Z and Tan TH:
Activation of the hematopoietic progenitor kinase-1
(HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK)
pathway by transforming growth factor beta (TGF-beta)-activated
kinase (TAK1), a kinase mediator of TGF beta signal transduction. J
Biol Chem. 272:22771–22775. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagata Y, Kiefer F, Watanabe T and
Todokoro K: Activation of hematopoietic progenitor kinase-1 by
erythropoietin. Blood. 93:3347–3354. 1999.PubMed/NCBI
|
|
19
|
Arnold R, Frey CR, Müller W, Brenner D,
Krammer PH and Kiefer F: Sustained JNK signaling by proteolytically
processed HPK1 mediates IL-3 independent survival during monocytic
differentiation. Cell Death Differ. 14:568–575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen YR and Tan TH: Inhibition of the
c-Jun N-terminal kinase (JNK) signaling pathway by curcumin.
Oncogene. 17:173–178. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Balakireva M, Rossé C, Langevin J, Chien
YC, Gho M, Gonzy-Treboul G, Voegeling-Lemaire S, Aresta S, Lepesant
JA, Bellaiche Y, et al: The Ral/exocyst effector complex counters
c-Jun N-terminal kinase-dependent apoptosis in Drosophila
melanogaster. Mol Cell Biol. 26:8953–8963. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito Y, Pandey P, Sathyanarayana P, Ling P,
Rana A, Weichselbaum R, Tan TH, Kufe D and Kharbanda S: Interaction
of hematopoietic progenitor kinase 1 and c-Abl tyrosine kinase in
response to genotoxic stress. J Biol Chem. 276:18130–18138. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu MC, Wang Yp, Qiu WR, Mikhail A, Meyer
CF and Tan TH: Hematopoietic progenitor kinase-1 (HPK1) stress
response signaling pathway activates IkappaB kinases
(IKK-alpha/beta) and IKK-beta is a developmentally regulated
protein kinase. Oncogene. 18:5514–5524. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen YR, Meyer CF, Ahmed B, Yao Z and Tan
TH: Caspase-mediated cleavage and functional changes of
hematopoietic progenitor kinase 1 (HPK1). Oncogene. 18:7370–7377.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arnold R, Liou J, Drexler HC, Weiss A and
Kiefer F: Caspase-mediated cleavage of hematopoietic progenitor
kinase 1 (HPK1) converts an activator of NFkappaB into an inhibitor
of NFkappaB. J Biol Chem. 276:14675–14684. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schulze-Luehrmann J, Santner-Nanan B, Jha
MK, Schimpl A, Avots A and Serfling E: Hematopoietic progenitor
kinase 1 supports apoptosis of T lymphocytes. Blood. 100:954–960.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen YR and Tan TH: The c-Jun N-terminal
kinase pathway and apoptotic signaling (Review). Int J Oncol.
16:651–662. 2000.PubMed/NCBI
|
|
28
|
Brenner D, Krammer PH and Arnold R:
Concepts of activated T cell death. Crit Rev Oncol Hematol.
66:52–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Krammer PH, Arnold R and Lavrik IN: Life
and death in peripheral T cells. Nat Rev Immunol. 7:532–542. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang H, Song X, Logsdon C, Zhou G, Evans
DB, Abbruzzese JL, Hamilton SR, Tan TH and Wang H:
Proteasome-mediated degradation and functions of hematopoietic
progenitor kinase 1 in pancreatic cancer. Cancer Res. 69:1063–1070.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Q, Long H, Liao J, Zhao M, Liang G,
Wu X, Zhang P, Ding S, Luo S and Lu Q: Inhibited expression of
hematopoietic progenitor kinase 1 associated with loss of jumonji
domain containing 3 promoter binding contributes to autoimmunity in
systemic lupus erythematosus. J Autoimmun. 37:180–189. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Li JP, Kuo HK, Chiu LL, Dement GA,
Lan JL, Chen DY, Yang CY, Hu H and Tan TH: Down-regulation of B
cell receptor signaling by hematopoietic progenitor kinase 1
(HPK1)-mediated phosphorylation and ubiquitination of activated B
cell linker protein (BLNK). J Biol Chem. 287:11037–11048. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stoeckman AK, Baechler EC, Ortmann WA,
Behrens TW, Michet CJ and Peterson EJ: A distinct inflammatory gene
expression profile in patients with psoriatic arthritis. Genes
Immun. 7:583–591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Batliwalla FM, Li W, Ritchlin CT, Xiao X,
Brenner M, Laragione T, Shao T, Durham R, Kemshetti S, Schwarz E,
et al: Microarray analyses of peripheral blood cells identifies
unique gene expression signature in psoriatic arthritis. Mol Med.
11:21–29. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen-Deutsch X, Kutner A, Harrison JS and
Studzinski GP: The pan-caspase inhibitor Q-VD-OPh has anti-leukemia
effects and can interact with vitamin D analogs to increase HPK1
signaling in AML cells. Leuk Res. 36:884–888. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen-Deutsch X and Studzinski GP: Dual
role of hematopoietic progenitor kinase 1 (HPK1) as a positive
regulator of 1α,25-dihydroxyvitamin D-induced differentiation and
cell cycle arrest of AML cells and as a mediator of vitamin D
resistance. Cell Cycle. 11:1364–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Luo H, Li Y, Chen T, Wu S and Yang
L: hsa-miR-96 up-regulates MAP4K1 and IRS1 and may function as a
promising diagnostic marker in human bladder urothelial carcinomas.
Mol Med Rep. 5:260–265. 2012.PubMed/NCBI
|
|
38
|
Qian Y, Takeuchi S, Dugu L, Tsuji G, Xie
L, Nakahara T, Takahara M, Moroi Y, Tu YT and Furue M:
Hematopoietic progenitor kinase 1, mitogen-activated
protein/extracellular signal-related protein kinase kinase kinase 1
and phosphomitogen-activated protein kinase kinase 4 are
overexpressed in extramammary Paget disease. Am J Dermatopathol.
33:681–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang HS, Matthews CP, Clair T, Wang Q,
Baker AR, Li CC, Tan TH and Colburn NH: Tumorigenesis suppressor
Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase
kinase 1 expression to suppress colon carcinoma cell invasion. Mol
Cell Biol. 26:1297–1306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sawasdikosol S, Zha R, Yang B and Burakoff
S: HPK1 as a novel target for cancer immunotherapy. Immunol Res.
54:262–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jakob SM, Pick R, Brechtefeld D, Nussbaum
C, Kiefer F, Sperandio M and Walzog B: Hematopoietic progenitor
kinase 1 (HPK1) is required for LFA-1-mediated neutrophil
recruitment during the acute inflammatory response. Blood.
121:4184–4194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Geisberger R, Prlic M,
Achatz-Straussberger G, Oberndorfer I, Luger E, Lamers M, Crameri
R, Appenzeller U, Wienands J, Breitenbach M, et al: Phage display
based cloning of proteins interacting with the cytoplasmic tail of
membrane immunoglobulins. Dev Immunol. 9:127–134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Buday L: Membrane-targeting of signalling
molecules by SH2/SH3 domain-containing adaptor proteins. Biochim
Biophys Acta. 1422:187–204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilkinson B, Wang H and Rudd CE: Positive
and negative adaptors in T-cell signalling. Immunology.
111:368–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ma W, Xia C, Ling P, Qiu M, Luo Y, Tan TH
and Liu M: Leukocyte-specific adaptor protein Grap2 interacts with
hematopoietic progenitor kinase 1 (HPK1) to activate JNK signaling
pathway in T lymphocytes. Oncogene. 20:1703–1714. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Han A, Saijo K, Mecklenbräuker I,
Tarakhovsky A and Nussenzweig MC: Bam32 links the B cell receptor
to ERK and JNK and mediates B cell proliferation but not survival.
Immunity. 19:621–632. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boomer JS and Tan TH: Functional
interactions of HPK1 with adaptor proteins. J Cell Biochem.
95:34–44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thome M, Charton JE, Pelzer C and
Hailfinger S: Antigen receptor signaling to NF-kappaB via CARMA1,
BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2:a0030042010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brenner D, Brechmann M and Röhling S,
Tapernoux M, Mock T, Winter D, Lehmann WD, Kiefer F, Thome M,
Krammer PH and Röhling S: Phosphorylation of CARMA1 by HPK1 is
critical for NF-kappaB activation in T cells. Proc Natl Acad Sci
USA. 106:14508–14513. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen YR, Kori R, John B and Tan TH:
Caspase-mediated cleavage of actin-binding and
SH3-domain-containing proteins cortactin, HS1, and HIP-55 during
apoptosis. Biochem Biophys Res Commun. 288:981–989. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patzak IM, Königsberger S, Suzuki A, Mak
TW and Kiefer F: HPK1 competes with ADAP for SLP-76 binding and via
Rap1 negatively affects T-cell adhesion. Eur J Immunol.
40:3220–3225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ruzza P, Siligardi G, Donella-Deana A,
Calderan A, Hussain R, Rubini C, Cesaro L, Osler A, Guiotto A,
Pinna LA and Borin G: 4-Fluoroproline derivative peptides: Effect
on PPII conformation and SH3 affinity. J Pept Sci. 12:462–471.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Le Bras S, Foucault I, Foussat A, Brignone
C, Acuto O and Deckert M: Recruitment of the actin-binding protein
HIP-55 to the immunological synapse regulates T cell receptor
signaling and endocytosis. J Biol Chem. 279:15550–15560. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Han J, Shui JW, Zhang X, Zheng B, Han S
and Tan TH: HIP-55 is important for T-cell proliferation, cytokine
production, and immune responses. Mol Cell Biol. 25:6869–6878.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han J, Kori R, Shui JW, Chen YR, Yao Z and
Tan TH: The SH3 domain-containing adaptor HIP-55 mediates c-Jun
N-terminal kinase activation in T cell receptor signaling. J Biol
Chem. 278:52195–52202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ensenat D, Yao Z, Wang XS, Kori R, Zhou G,
Lee SC and Tan TH: A novel src homology 3 domain-containing adaptor
protein, HIP-55, that interacts with hematopoietic progenitor
kinase 1. J Biol Chem. 274:33945–33950. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liou J, Kiefer F, Dang A, Hashimoto A,
Cobb MH, Kurosaki T and Weiss A: HPK1 is activated by lymphocyte
antigen receptors and negatively regulates AP-1. Immunity.
12:399–408. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Oehrl W, Kardinal C, Ruf S, Adermann K,
Groffen J, Feng GS, Blenis J, Tan TH and Feller SM: The germinal
center kinase (GCK)-related protein kinases HPK1 and KHS are
candidates for highly selective signal transducers of Crk family
adapter proteins. Oncogene. 17:1893–1901. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lewitzky M, Harkiolaki M, Domart MC, Jones
EY and Feller SM: Mona/Gads SH3C binding to hematopoietic
progenitor kinase 1 (HPK1) combines an atypical SH3 binding motif,
R/KXXK, with a classical PXXP motif embedded in a polyproline type
II (PPII) helix. J Biol Chem. 279:28724–28732. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shen R, Ouyang YB, Qu CK, Alonso A,
Sperzel L, Mustelin T, Kaplan MH and Feng GS: Grap negatively
regulates T-cell receptor-elicited lymphocyte proliferation and
interleukin-2 induction. Mol Cell Biol. 22:3230–3236. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lasserre R, Cuche C, Blecher-Gonen R,
Libman E, Biquand E, Danckaert A, Yablonski D, Alcover A and Di
Bartolo V: Release of serine/threonine-phosphorylated adaptors from
signaling microclusters down-regulates T cell activation. J Cell
Biol. 195:839–853. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sauer K, Liou J, Singh SB, Yablonski D,
Weiss A and Perlmutter RM: Hematopoietic progenitor kinase 1
associates physically and functionally with the adaptor proteins B
cell linker protein and SLP-76 in lymphocytes. J Biol Chem.
276:45207–45216. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tsuji S, Okamoto M, Yamada K, Okamoto N,
Goitsuka R, Arnold R, Kiefer F and Kitamura D: B cell adaptor
containing src homology 2 domain (BASH) links B cell receptor
signaling to the activation of hematopoietic progenitor kinase 1. J
Exp Med. 194:529–539. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu J, Riou C, Davidson D, Minhas R, Robson
JD, Julius M, Arnold R, Kiefer F and Veillette A: Synergistic
regulation of immunoreceptor signaling by SLP-76-related adaptor
Clnk and serine/threonine protein kinase HPK-1. Mol Cell Biol.
21:6102–6112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Burns JC, Corbo E, Degen J, Gohil M,
Anterasian C, Schraven B, Koretzky GA, Kliche S and Jordan MS: The
SLP-76 Src homology 2 domain is required for T cell development and
activation. J Immunol. 187:4459–4466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang X, Li JP, Chiu LL, Lan JL, Chen DY,
Boomer J and Tan TH: Attenuation of T cell receptor signaling by
serine phosphorylation-mediated lysine 30 ubiquitination of SLP-76
protein. J Biol Chem. 287:34091–34100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Di Bartolo V, Montagne B, Salek M,
Jungwirth B, Carrette F, Fourtane J, Sol-Foulon N, Michel F,
Schwartz O, Lehmann WD and Acuto O: A novel pathway down-modulating
T cell activation involves HPK-1-dependent recruitment of 14-3-3
proteins on SLP-76. J Exp Med. 204:681–691. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Utting O, Sedgmen BJ, Watts TH, Shi X,
Rottapel R, Iulianella A, Lohnes D and Veillette A: Immune
functions in mice lacking Clnk, an SLP-76-related adaptor expressed
in a subset of immune cells. Mol Cell Biol. 24:6067–6075. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Girardin SE and Yaniv M: A direct
interaction between JNK1 and CrkII is critical for Rac1-induced JNK
activation. EMBO J. 20:3437–3446. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kiefer F, Tibbles LA, Anafi M, Janssen A,
Zanke BW, Lassam N, Pawson T, Woodgett JR and Iscove NN: HPK1, a
hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO
J. 15:7013–7025. 1996.PubMed/NCBI
|
|
71
|
Hu MC, Qiu WR, Wang X, Meyer CF and Tan
TH: Human HPK1, a novel human hematopoietic progenitor kinase that
activates the JNK/SAPK kinase cascade. Genes Dev. 10:2251–2264.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Marshall AJ, Niiro H, Lerner CG, Yun TJ,
Thomas S, Disteche CM and Clark EA: A novel B lymphocyte-associated
adaptor protein, Bam32, regulates antigen receptor signaling
downstream of phosphatidylinositol 3-kinase. J Exp Med.
191:1319–1332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hehner SP, Hofmann TG, Dienz O, Droge W
and Schmitz ML: Tyrosine-phosphorylated Vav1 as a point of
integration for T-cell receptor- and CD28-mediated activation of
JNK, p38, and interleukin-2 transcription. J Biol Chem.
275:18160–18171. 2000. View Article : Google Scholar : PubMed/NCBI
|