Introduction
Atherosclerosis is currently recognized as the
outcome of the formation of multiple atheromatous plaques in the
arteries, which has been characterized as arterial chronic
degeneration and inflammation (1).
Recruitment of leukocytes from the blood to the arterial wall is an
early and critical step in the inflammatory cascade (1,2).
Collagen and proteoglycans then accumulate, which results in
arterial hardening and thickening, leading to a loss of flexibility
of the arteries (3). Once the
disease is established, it may manifest with angina pectoris,
myocardial infarction and other potentially fatal diseases, thus
has a high mortality rate (3–5). The
process and cause of arterial thickening is complex, including the
involvement of inflammatory cells (epithelial cells, monocytes,
macrophages, smooth muscle and platelets), molecular factors
(lipoprotein, growth hormones, cholesterol, fat or collagen) and
cytokines (interleukins, tumor necrosis factors and chemokines)
(3,6).
Gap junctions act a bridge for communication between
two adjacent cells via connexins, arranged in a hexamer membrane
structure (1,7–9). The
exchange of information and energy material are between adjacent
cells is mediated through gap junction intercellular space
connection communication (gap junction intercellular communication)
(7). These communication channels
allow for the transport of ions, small molecular metabolites and
secondary signaling molecules, allowing material exchange between
cells and electrical coupling (1,8).
Studies have demonstrated that the expression of the
junction proteins, including tight junction proteins and gap
junction proteins is closely associated with atherosclerosis. Tight
junction protein (TJP) is an adhesive structure between the cells
(10,11). Cell adhesion is involved in
maintaining stability of organizational homeostasis, and is a
factor for cellular movement, regulation of permeability, cell
differentiation and cell proliferation (2,12).
Tight junctions is composed of a group of proteins including the
claudin family, junction adhesion molecules (JAMs) and the closed
small ring protein ZO family (4,12,13);
previous studies have demonstrated that these proteins do not take
part in gap junction formation, however facilitate the release of
small functional molecules such as adenosine triphosphate (ATP)
(1,3,8).
Connexin 43 (Cx43) is predominantly expressed in myocardial tissue,
macrophages, connective tissue cells, endothelial cells and smooth
muscle cells (14,15). Cx43 forms a half channel and then
is transported to the plasma membrane by the Golgi apparatus.
Previous studies have demonstrated that is Cx43 is essential for
the differentiation and development of the heart; abnormal
expression of Cx43 can lead to a variety of cardiovascular diseases
including congenital malformation of the heart (6,8,9,14,15).
It has been reported that the upregulation of Cx43
expression enhanced monocyte-endothelial adhesion and vice versa
when downregulated. This mechanism was associated with Cx43-induced
vascular cell adhesion molecules and intercellular cell adhesion
molecules suggesting that local regulation of endothelial Cx43
expression within the vasculature regulates monocyte endothelial
adhesion and the development of atherosclerosis (16).
A previous study indicated that circulating
monocytes expressing low levels of Cx37 protect against
atherosclerosis by regulating monocyte adhesion, however the
expression of other connexins remains low (17). However upon stimulation with
cytokines including tumor necrosis factor (TNF) α and interferon γ,
human blood monocytes were demonstrated to express high levels of
Cx43 (18).
A previous study focused on the association between
tight junction and atherosclerosis progression, observing that
blocking myosin light chain kinase by inhibitor ML7 improved
vascular endothelial dysfunction and atherosclerosis (19). This mechanism is explained by the
regulation of TJP1 (ZO-1) and occludins in the atherosclerosis
model, which implies overexpression of tight junctions may serve
important roles in the pathogenesis.
It is currently recognized that monocytes originate
from progenitors in the bone marrow and they reach the circulation
as two major subgroups, the classical CD14(high)CD16−
monocytes and the CD14(low)CD16+ monocytes (20).
Endoglin [CD105, type III transforming growth factor
(TGF)-β receptor] and mothers against decapentaplegic homolog
(SMAD) pathways were identified to be involved in the negative
regulation pathway of inflammation resulting in the release of
TGF-β, interleukin 4 and other immune suppressive proteins
(21). In addition, the role of
endoglin in the monocyte circulation system, including
cell-mediated vascular repair, has been well-studied (22). Therefore, it is critical to
understand the immunological homeostasis of monocytes and possible
correlation between cellular junctions and the endoglin/SMAD
pathway during atherosclerosis.
CD14 marker-positive isolation is an optimal way to
study monocytic gap junctions and tight junctions from patients and
normal controls. In the present study, the focus was predominantly
on the expression level of key gap junction proteins (Cx37, Cx40,
Cx43, Cx45 and Cx46), TJPs (claudin-1, occludin-1 and ZO-1), and
the inflammation regulating genes, endoglin and SMAD1, in
atherosclerotic patients and normal controls. Further combination
analysis of these genes with public datasets were performed to draw
a more comprehensive correlation between atherosclerosis and
junction proteins, and how they act on immune regulatory
pathways.
Materials and methods
Patients and samples
The subjects were divided into two groups with the
results of the coronary angiogram. A total of 25 outpatients with
indications of stent implantation and 25 normal controls that were
under routine physical examination were admitted into Shaoxing
Second Hospital (Shaoxing, China). The protocols were reviewed and
approved by the Animal Ethical and Welfare Committee of Shaoxing
Second Hospital. Informed consent was obtained from each patient.
CD14+ fluorescent antibody at 5 µl/million cells (cat.
no. 367115; BioLegend, San Diego, CA, USA) and flow cytometry (BD
Biosciences, Franklin Lakes, NJ, USA) were applied on isolated
monocytes from peripheral blood mononuclear cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the monocytes of patients was
extracted using TRIzol (cat. no. 15596018, Thermo Fisher
Scientific, Inc., Waltham, MA, USA) reagent. The quantity and
quality of RNA were confirmed using a NanoDrop 1000 thermocycler
(NanoDrop; Thermo Fisher Scientific, Inc.) and 1.0% agarose (Hushi,
Shanghai, China; cat. no. CAS:9002-18-0) electrophoresis. The
primers were designed using Primer 6.0 software (Premier Biosoft
International, Palo Alto, CA, USA) and were synthesized by Shanghai
Generay Biotech Co., Ltd. (Shanghai, China) (Table I).
 | Table I.Primers designed using Primer 6.0
software and synthesized from Generay Biotech. |
Table I.
Primers designed using Primer 6.0
software and synthesized from Generay Biotech.
| Gene name | Primer sequence (5′
to 3′) | Amplicon
size/NCBI.NM |
|---|
| H-GAPDH-157 bp-F |
GAGTCCACTGGCGTCTTCAC | 157 bp |
| H-GAPDH-157 bp-R |
TGCTGATGATCTTGAGGCTGTT | NM_001256799.2 |
| H-CX37-F |
AGTTCCTCTTCGTCAGCACAC | 239 bp |
| H-CX37-R |
GAGCACACTGGCGACATAGG | NM_002060.2 |
| H-CX40-F |
TGGAAGAAGATCAGACAGCGATT | 218 bp |
| H-CX40-R |
CCTCGTACTTGCTCGGTGAC | NM_005266.6 |
| H-CX43-F |
CTGGTGGTGTCCTTGGTGTC | 182 bp |
| H-CX43-R |
GGTGAGGAGCAGCCATTGAA | NM_000165.4 |
| H-CX45-F |
ACCGAACTGTCCAATGCTAAGA | 142 bp |
| H-CX45-R |
AGCGTTCCTGAGCCATCCT | NM_001080383.1 |
| H-CX46-F |
TGTTCATCTTCCGCATCTTGGT | 119 bp |
| H-CX46-R |
CCTGTCGTAGCAGACGTTCTC | NM_021954.3 |
| H-Claudin-1-F |
AGGTCTTGCCGCCTTGGTA | 173 bp |
| H-Claudin-1-R |
GACAGGAACAGGAGAGCAGTG | NM_001307.5 |
| H-Endoglin-F |
CGACGCCAACCACAACAT | 156 bp |
| H-Endoglin-R |
ACGAAGGATGCCACAATGC | NM_000118.3 |
| H-Occludin-1-F |
TCGCTGCCAATGCTCATCTG | 206 bp |
| H-Occludin-1-R |
GCCTCCAAGGAAGAGACTGAAG | NM_005985.3 |
| H-SMAD-F |
CATGCCACTCAACGCCACTT | 295 bp |
| H-SMAD-R |
AACCGCCTGAACATCTCCTCT | NM_005900.2 |
| H-ZO-1-F |
GCGGATGGTGCTACAAGTGAT | 138 bp |
| H-ZO-1-R |
GCCTTCTGTGTCTGTGTCTTCA | NM_001301025.1 |
For gene specific quantitation, reverse
transcription was performed based on the specification of the
ReverTra Ace qPCR RT kit (Toyobo Co., Ltd., Osaka, Japan) to
synthesize the first strand of cDNA and RT-qPCR were conducted on
FTC-3000 (Funglyn Biotech, Inc., Richmond Hill, ON, Canada) with
SYBR Green Fast qPCR kit (Kapa Biosystems, Inc., Wilmington, MA,
USA) (Table II). Thermal cycling
parameters were set as follows: 95°C for 3 min (enzyme activation),
the next stage was repeated 40 times; 95°C for 5 sec
(denaturation), and 60°C for 30 sec (annealing/extension/data
acquisition). Data were analyzed by the 2−ΔΔCq algorithm
(23).
| Table II.Quantitative polymerase chain
reaction components (20 µl). |
Table II.
Quantitative polymerase chain
reaction components (20 µl).
| PCR components | Volume, µl |
|---|
| RNase-free
H2O | 7.2 |
| 2X Realtime PCR
master mix | 10 |
| Forward primer, 10
µM | 0.4 |
| Reverse primer, 10
µM | 0.4 |
| cDNA template | 2 |
Combined analysis of microarray data
from coronary heart disease (CHD) study
The microarray dataset GSE71226 was downloaded from
NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). This dataset
included the microarray gene expression profiling of peripheral
blood of three Chinese patients with CHD and three Chinese healthy
controls. The raw data were normalized with R project limma
packages in Bioconductor (https://www.bioconductor.org/) with default settings.
Fold change of gene expression and corresponding t-test P-values
were calculated between patients with CHD and healthy individuals.
Differentially expressed genes (DEGs) were defined as genes that
met the criteria of fold change value >1.5 and t-test P<0.05.
DEGs were used with the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway database (http://www.genome.jp/kegg/) to determine the
biological function of these DEGs. Enriched pathways were
determined by those yielding a significant value from Fisher's
exact test (P<0.05).
Statistical analysis
Student's t-test was used to filter the DEGs between
control groups and patient groups (Fold change value >1.5 and
t-test P<0.05), and a Fisher's exact test was performed to
determine the enriched pathways. Statistical analyses were
performed using GraphPad Prism 5 software (GraphPad Software, Inc.,
La Jolla, CA, USA) and SPSS version 17.0 statistical software
program/package (SPSS, Inc., Chicago, IL, USA).
Results
Gene expression quantification of cell
junction genes in CHD patients
To further clarify the important role of the cell
junction in the pathogenesis of atherosclerosis, eight genes that
were involved in the gap junction and tight junction pathways in
CHD patients were investigated by RT-qPCR. As presented in Fig. 1, Cx43 and Cx46, the major
components of the gap junctions localized on the cell surface, were
significantly increased (P<0.05) in patients with CHD. In
addition, Cx45 was marginally enhanced in patients with CHD
(P>0.05). However, the levels of Cx37 and Cx40 were
significantly downregulated in patients with CHD compared with
normal controls (P<0.05). In terms of TJPs, claudin-1,
occludin-1 and ZO-1 were all significantly increased in in patients
with CHD (P<0.05).
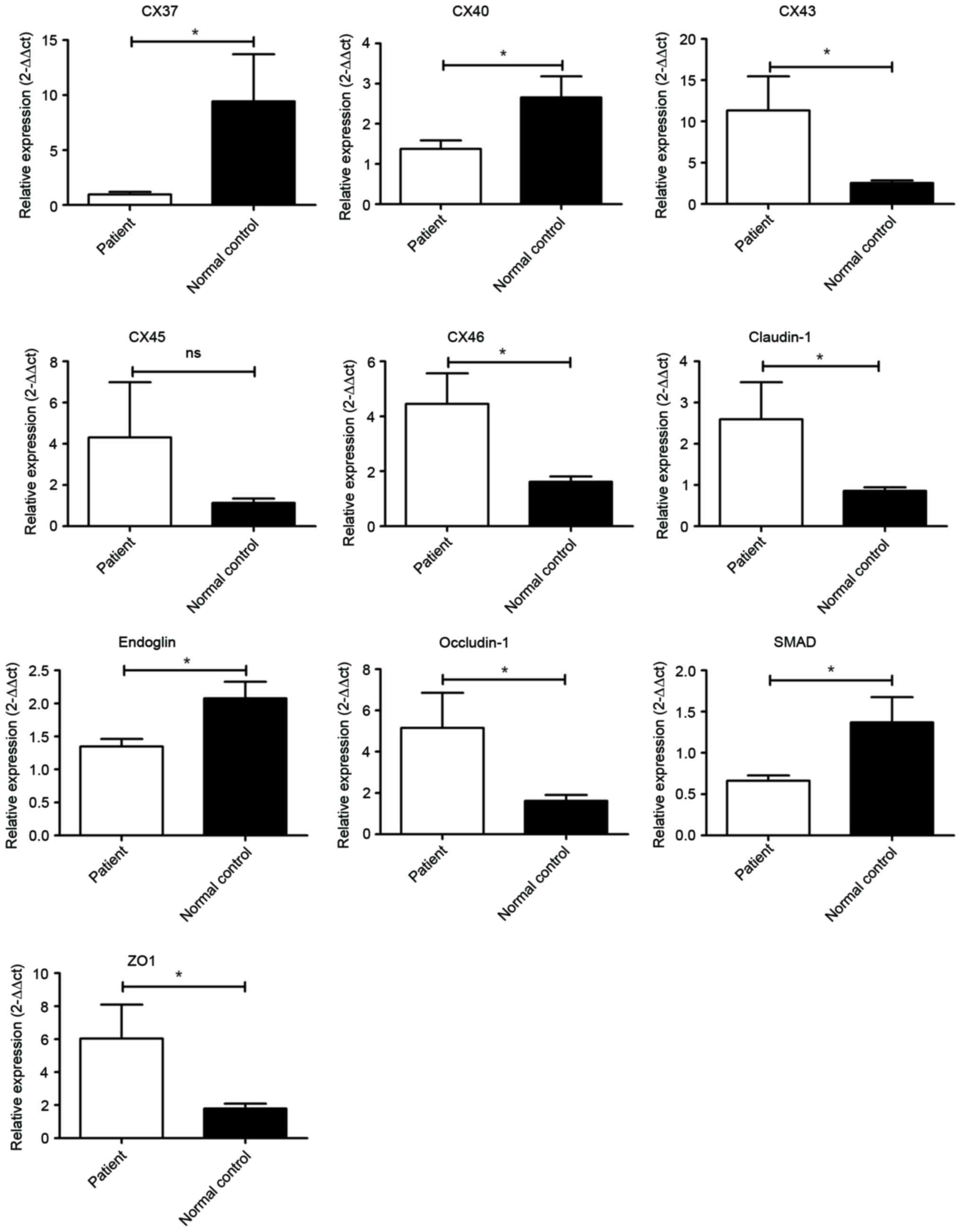 | Figure 1.Transcriptional analysis of genes
promoting atherosclerosis. Cx43, Cx45, Cx46, claudin-1, occludin-1
and ZO-1 were significantly elevated in patients. However, Cx37,
Cx40, endoglin and SMAD1 were downregulated in patients.
*P<0.05, patient compared with normal control. ZO-1, tight
junction protein 1; SMAD1, mothers against decapentaplegic homolog
1. |
In addition, expression of endoglin and SMAD1 were
significantly reduced in patients with CHD.
Microarray data analysis of CHD
To obtain genes associated with CHD, DEGs were
identified by calculating the gene expression fold change and
t-test P-value from peripheral blood monocytic cells between
patients and healthy individuals. In total, 1,774 genes were
identified as DEGs. All DEGs identified were used with the KEGG
database for pathway enrichment analysis. The tight junction
pathway was significantly enriched and 18 DEGs were involved in
this pathway, which includes the claudin-1 and occludin-1 genes.
The results indicated that there was an alteration of the tight
junction pathway in patients with CHD (Fig. 2).
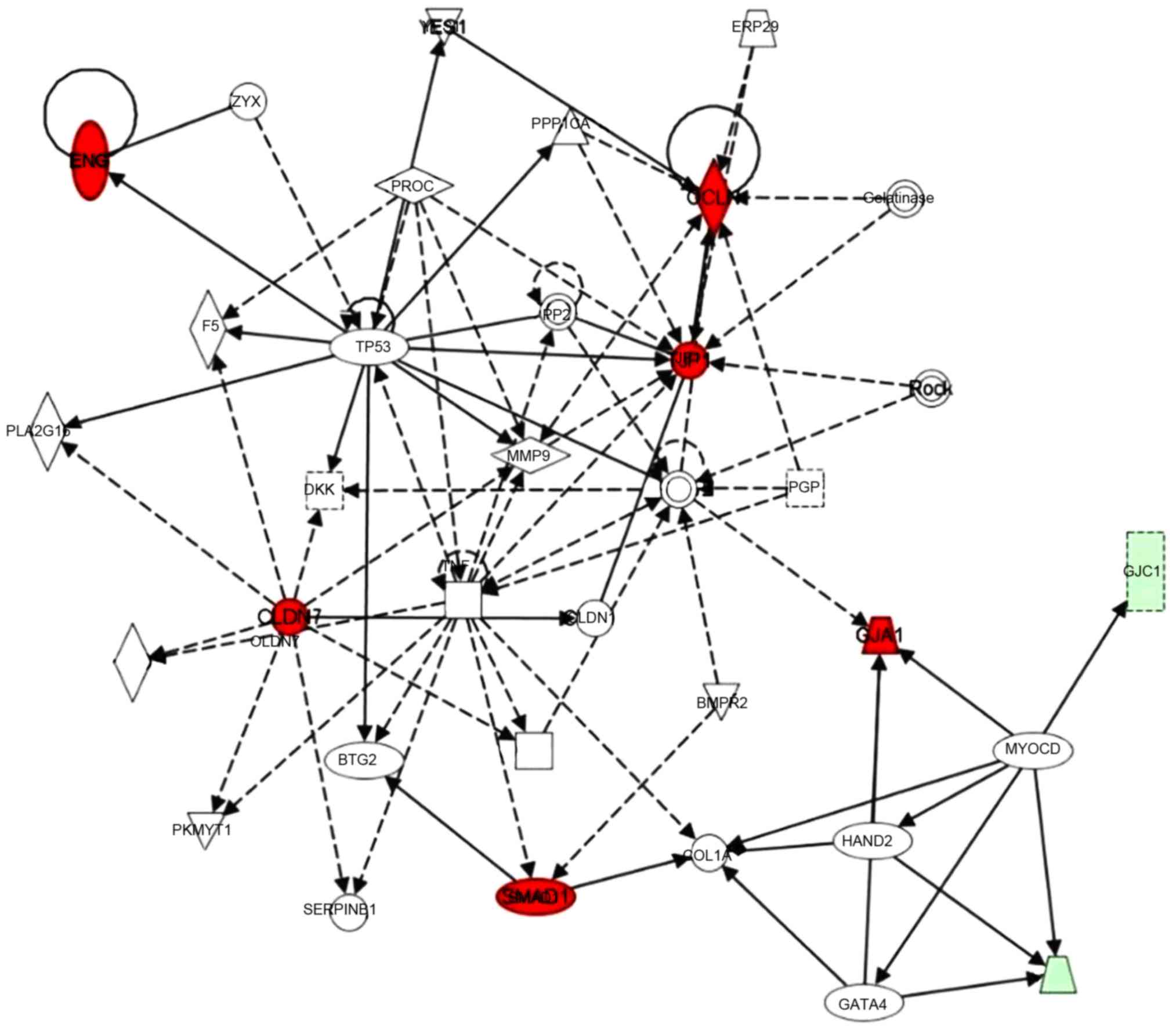
A gene interaction network was constructed base on
these significantly regulated genes using Ingenuity Pathway
Analysis (IPA) tools. Claudin-7, occludin-1, ZO-1 and SMAD1 were
identified as hub genes in this network. TNF was also involved in
this network, and the myocardin gene connected three connexin
family members (Cx43 as a core gene, and Cx33 and Cx45; in red
circles). In addition, chemokine (C-X3-C motif) ligand 1, matrix
metalloproteinase 9, tumor protein 53 and several other genes were
also involved in this network (Fig.
3).
Discussion
The present study provided evidence that the key
factor of cell junctions were significantly altered in patients
with CHD with atherosclerosis, which indicated an association
between cell junctions and atherosclerosis. The claudin-1,
occludin-1 and ZO-1 were significantly enhanced in atherosclerosis,
indicating that tight junction pathway was activated during the
pathogenesis of atherosclerosis. In addition, the gene expression
of 5 connexin members involved in the gap junction pathway were
quantified, Cx43 and Cx46 were significantly upregulated in
atherosclerosis. Combined in silico lab analysis also agreed
with the evidence that connexins and tight junctions were altered
in association with monocytic inflammation regulations. Further
inflammatory factors including endoglin and SMAD were evaluated to
be downregulated in the pathway. The presence of different cell
junction contacts is an essential characteristic of different cell
systems, with a high degree of cell-to-cell integration and
communication (24,25). It can be hypothesized that the
disturbances in functioning of gap junctions may be the cause of
the disintegration of the cellular network in atherosclerotic
disease.
The importance of gap junction-mediated
intercellular communication in the development of the pathogenesis
of atherosclerosis has been discussed previously (15,26).
A previous study indicated that high levels of Cx43 expression were
associated with reduced contractile ability and an increase in the
synthetic phenotype in smooth muscle cells (27). It was previously reported that the
upregulation of Cx43 enhanced monocyte-endothelial adhesion and
therefore the suggested mechanism was that Cx43 induced an increase
in monocyte adherance onto vascular endothelial cells during the
development of atherosclerosis (16). The results of the current study
from both the wet lab and dry lab supported this hypothesis, that
Cx43 was upregulated in atherosclerotic patients. In addition, Cx45
and Cx46 were identified to present with the same pattern as Cx43,
exhibiting synergistic function or association. Cx37 and Cx40 were
additionally observed to exhibit reduced expression in monocytes in
the current study, resulting in altered ATP release during the
monocyte-endothelial adhesion bioprocess.
Intercellular tight junctions serve important roles
in the stability of endothelial barriers as they regulate
paracellular permeability. They are composed of families including
claudins, occludins and JAMs (2,12).
ZO-1, as a junctional adaptor protein, in addition to the
transmembrane proteins of the claudin and JAM families, interacts
with multiple junction components and transduces molecules in the
signaling pathways (24,25).
As reported by Collins et al (28), increased levels of occludin and
ZO-1 and increased localization to cell-cell junctions resulted in
a parallel reduction in transendothelial protein permeability. The
changes of TJP expression were also accompanied by alterations in
their phosphorylation state. Cheng et al (19) reported that the myosin light chain
kinase (MLCK) inhibitor ML7 was used to improve atherosclerosis by
regulating the expression of ZO-1 and occludin through MLCK and MLC
phosphorylation in a high-fat diet fed rabbit model (29).
In the current study, a higher expression level of
TJPs was observed, which is in agreement with the previous findings
in animal model studies (19,28,29).
Increased TJP expression may contribute to more stable contact
after monocytes adhere to endothelia during the initial steps of
atherosclerotic pathogenesis.
In the investigation of the mechanisms involved, it
was identified that the TGF-β/endoglin and SMAD pathway were
suppressed in patients with atherosclerosis. The pathway was
reported to have a negative correlation between atherosclerosis and
its activation. Therefore it was hypothesized that reduced
expression of this immune regulatory pathway is correlated with the
generation of gap junctions and tight junctions. However, further
validation of a causal association between the endoglin pathway and
TJPs or gap junction proteins is required.
In conclusion, the present study demonstrated that
cell junction pathways including tight junctions and gap junctions
are actively involved in the progression of atherosclerosis.
Increased levels of TJPs, claudin-1, occludin-1 and
ZO-1, and gap junction proteins Cx43 and Cx46 were detected with
significance in atherosclerosis. The endoglin and SMAD inflammatory
regulatory pathways were suppressed in patients with
atherosclerosis, and silicon-based data analysis additionally
confirmed core roles of gap junctions and tight junctions in
monocytic inflammatory regulation. Accordingly, the genes
identified in the current study may represent a possible target for
prevention of atherosclerosis by blocking monocytic cell
junctions.
References
|
1
|
Scheckenbach KE, Crespin S, Kwak BR and
Chanson M: Connexin channel-dependent signaling pathways in
inflammation. J Vascular Res. 48:91–103. 2011. View Article : Google Scholar
|
|
2
|
Berardi DE and Tarbell JM: Stretch and
shear interactions affect intercellular junction protein expression
and turnover in endothelial cells. Cell Mol Bioeng. 2:320–331.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morel S, Burnier L and Kwak BR: Connexins
participate in the initiation and progression of atherosclerosis.
Semin Immunopathol. 31:49–61. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou T, He Q, Tong Y, Zhan R, Xu F, Fan D,
Guo X, Han H, Qin S and Chui D: Phospholipid transfer protein
(PLTP) deficiency impaired blood-brain barrier integrity by
increasing cerebrovascular oxidative stress. Biochem Biophys Res
Commun. 445:352–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wong CW, Burger F, Pelli G, Mach F and
Kwak BR: Dual benefit of reduced Cx43 on atherosclerosis in LDL
receptor-deficient mice. Cell Commun Adhes. 10:395–400. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei JM, Wang X, Gong H, Shi YJ and Zou Y:
Ginkgo suppresses atherosclerosis through downregulating the
expression of connexin 43 in rabbits. Arch Med Sci. 9:340–346.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ren Q, Riquelme MA, Xu J, Yan X, Nicholson
BJ, Gu S and Jiang JX: Cataract-causing mutation of human connexin
46 impairs gap junction, but increases hemichannel function and
cell death. PLoS One. 8:e747322013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Retamal MA: Connexin and Pannexin
hemichannels are regulated by redox potential. Front Physiol.
5:802014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan D, Wang Q, Wu D, Yu M, Zhang S, Li L,
Tao L and Harris AL: Monocyte-endothelial adhesion is modulated by
Cx43-stimulated ATP release from monocytes. Biochem Biophys Res
Commun. 420:536–541. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ba J, Peng H, Chen Y and Gao Y: Effects
and mechanism analysis of vascular endothelial growth factor and
salvianolic acid B on 125I-low density lipoprotein permeability of
the rabbit aortary endothelial cells. Cell Biochem Biophys.
70:1533–1538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Telo P, Lostaglio S and Dejana E:
Structure of intercellular junctions in the endothelium. Therapie.
52:395–398. 1997.PubMed/NCBI
|
|
12
|
Lehman DM, Leach RJ, Johnson-Pais T,
Hamlington J, Fowler S, Almasy L, Duggirala R, Stern MP and Abboud
HE: Evaluation of tight junction protein 1 encoding zona occludens
1 as a candidate gene for albuminuria in a Mexican American
population. Exp Clin Endocrinol Diabetes. 114:432–437. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gan H, Wang G, Hao Q, Wang QJ and Tang H:
Protein kinase D promotes airway epithelial barrier dysfunction and
permeability through down-regulation of claudin-1. J Biol Chem.
289:204892014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Polacek D, Bech F, McKinsey JF and Davies
PF: Connexin43 gene expression in the rabbit arterial wall: Effects
of hypercholesterolemia, balloon injury and their combination. J
Vasc Res. 34:19–30. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chadjichristos CE, Matter CM, Roth I,
Sutter E, Pelli G, Lüscher TF, Chanson M and Kwak BR: Reduced
connexin43 expression limits neointima formation after balloon
distension injury in hypercholesterolemic mice. Circulation.
113:2835–2843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan D, Sun G, Zhang R, Luo C, Ge M, Luo G
and Hei Z: Connexin 43 expressed in endothelial cells modulates
monocyteendothelial adhesion by regulating cell adhesion proteins.
Mol Med Rep. 12:7146–7152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kameritsch P, Pogoda K and Pohl U:
Channel-independent influence of connexin 43 on cell migration.
Biochim Biophys Acta. 1818:1993–2001. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morel S and Kwak BR: Roles of connexins in
atherosclerosis and ischemia-reperfusion injury. Curr Pharm
Biotechnol. 13:17–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng X, Wang X, Wan Y, Zhou Q, Zhu H and
Wang Y: Myosin light chain kinase inhibitor ML7 improves vascular
endothelial dysfunction via tight junction regulation in a rabbit
model of atherosclerosis. Mol Med Rep. 12:4109–4116. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Decano JL, Mattson PC and Aikawa M:
Macrophages in Vascular Inflammation: Origins and Functions. Curr
Atheroscler Rep. 18:342016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rathouska J, Jezkova K, Nemeckova I and
Nachtigal P: Soluble endoglin, hypercholesterolemia and endothelial
dysfunction. Atherosclerosis. 243:383–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Laake LW, van den Driesche S, Post S,
Feijen A, Jansen MA, Driessens MH, Mager JJ, Snijder RJ, Westermann
CJ, Doevendans PA, et al: Endoglin has a crucial role in blood
cell-mediated vascular repair. Circulation. 114:2288–2297. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haseloff RF, Dithmer S, Winkler L, Wolburg
H and Blasig IE: Transmembrane proteins of the tight junctions at
the blood-brain barrier: Structural and functional aspects. Semin
Cell Dev Biol. 38:16–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tornavaca O, Chia M, Dufton N, Almagro LO,
Conway DE, Randi AM, Schwartz MA, Matter K and Balda MS: ZO-1
controls endothelial adherens junctions, cell-cell tension,
angiogenesis, and barrier formation. J Cell Biol. 208:821–838.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chadjichristos CE and Kwak BR: Connexins:
New genes in atherosclerosis. Ann Med. 39:402–411. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rennick RE, Connat JL, Burnstock G,
Rothery S, Severs NJ and Green CR: Expression of connexin43 gap
junctions between cultured vascular smooth muscle cells is
dependent upon phenotype. Cell Tissue Res. 271:323–332. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Collins NT, Cummins PM, Colgan OC,
Ferguson G, Birney YA, Murphy RP, Meade G and Cahill PA: Cyclic
strain-mediated regulation of vascular endothelial occludin and
ZO-1: Influence on intercellular tight junction assembly and
function. Arterioscler Thromb Vasc Biol. 26:62–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu HQ, Wang XB, Han JX, Hu ZP and Wang Y,
Zhou Q, Gui SY and Wang Y: Myosin light chain kinase inhibitor
attenuates atherosclerosis and permeability via reduced endothelial
tight junction in rabbits. Int J Cardiol. 168:5042–5043. 2013.
View Article : Google Scholar : PubMed/NCBI
|