Introduction
Osteoarthritis (OA) is one of the most common forms
of arthritis. The slow development of the disease affects joint
structures, which leads to pain and disability in the elderly.
Various risk factors are associated with the initiation and
progression of OA, including demographic characteristics and family
history, obesity and metabolic syndrome, nutritional factors, bone
density and bone mass, and smoking (1). The primary pathological features of
OA are cartilage degeneration, marginal osteophyte formation and
subchondral bone sclerosis (2).
Current treatment strategies are limited to relieving joint pain
and improving joint function, and eventually end in artificial
joint replacement as no therapeutic strategies that halt the
progression of the disease exist (2). Therefore, it is particularly
important to clarify the etiology and pathogenesis of OA, which
remain unclear. Articular cartilage degeneration is one of the
major factors behind the development of OA. Chondrocytes and
extracellular matrix are the major components of cartilage, and
chondrocyte hypertrophy and extracellular matrix damage may lead to
cartilage degeneration (3,4).
Parathyroid hormone (PTH) has roles in the anabolic
and catabolic metabolism of G-protein-coupled receptor signaling
proteins (5). PTH 1–34 (also
termed teriparatide) is the only drug approved by the US Food and
Drug Administration for the treatment of osteoporosis (6). Its action is mediated by the
parathyroid hormone 1 receptor (PTH1R), which is expressed in
chondrocytes (7). Previous studies
have demonstrated that PTH 1–34 may prevent OA (8–10).
PTH 1–34 inhibits cartilage degeneration and promotes cartilage
regeneration following meniscal/ligamentous injury-induced OA in
mice (9). PTH 1–34 also prevents
the degeneration of articular cartilage and retains the subchondral
bone microstructure in spontaneous OA of guinea pigs (8). Furthermore, PTH 1–34 inhibits the
terminal differentiation of human articular chondrocytes with
azacytidine-induced and papain-induced OA in rats (10). However, the specific functions of
PTH 1–34 in OA are yet to be established.
Wnt/β-catenin signaling pathways have a central role
in the maintenance of cartilage homeostasis (3). In cartilage, moderate activity of Wnt
is essential for chondrocyte proliferation and the maintenance of
typical cartilage characteristics (4,11,12).
However, excessive activity of the Wnt/β-catenin signaling pathway
may result in the hypertrophy of chondrocytes and the degradation
of the extracellular matrix of cartilage (11). In cartilage, ablation of β-catenin
increases aggrecan and collagen X (12). Therefore, excessive or inadequate
β-catenin may damage the homeostasis of articular cartilage. PTH
and Wnt/β-catenin exist in complex associations, which are
important for the pathogenesis of OA (3). Sclerostin and dickkopf Wnt signaling
pathway inhibitor 1 (DKK1) are upstream inhibitors of the
Wnt/β-catenin signaling pathway and are used in the research of OA
(13). Runt-related transcription
factor 2 (RUNX2), which is highly expressed in hypertrophic
chondrocytes, regulates the transcription of hypertrophic markers,
including collagen X and matrix metallopeptidase (MMP) 13 (3). The Wnt/β-catenin pathway promotes
RUNX2 expression (14). In
cartilage, the specific association between PTH and RUNX2 is not
yet established, and its role in the pathogenesis of OA remains
unclear. The present study was designed to investigated the effect
of PTH 1–34 on cartilage degeneration and the association between
PTH 1–34 and the Wnt/β-catenin signaling pathway following anterior
cruciate ligament transection (ACLT) and partial medial
meniscectomy (MMx) -induced OA in rats.
Materials and methods
Animal models and treatment
All experiments were approved by the Ningxia Medical
University Animal Care and Use Committee (Yinchuan, China). All
animal model procedures were carried out according to the
principles and guidelines of ethical animal studies. A total of 64
healthy male Sprague-Dawley rats, 10 weeks old and 300–325 g were
used in the following experiments. The animals were housed at
22±2°C with 55±5% humidity, free access to food and water and 12-h
light/dark cycle. Experiments were performed on the right knees of
rats in this study. The rats were anesthetized by an
intraperitoneal injection of 10% chloral hydrate (0.3 ml/100 g;
Tianjin Guangfu Fine Chemical Research Institute, Tianjin, China).
In the OA animal models, the right knee joint cavity was exposed
using the medial parapatellar approach. The patella was dislocated
laterally when the knee was placed in full flexion. The ACLT in
combination with MMx (ACLT + MMx) was performed as previously
described (15) and confirmed with
the anterior drawer test. (16)
Subsequently, the joint surface was washed with sterile saline
solution and the knee was closed. In the sham operation, the same
procedures were performed with the exception of ACLT + MMx
(15). Rats were randomly assigned
to four groups: Sham-operated rats with normal saline
(NS)-treatment (n=16); ACLT + MMx rats with NS-treatment (n=16);
sham-operated rats treated with PTH 1–34 (n=16); and ACLT + MMx
rats treated with PTH 1–34 (n=16). PTH 1–34 (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was administered by subcutaneous
injection (15 µg/kg/day) five successive days per week
(Monday-Friday) from the first postoperative day until sacrifice at
2 or 6 weeks. Rats not receiving PTH 1–34 received NS at the same
dose and duration. Following the sacrifice of rats with carbon
dioxide, right knees were divided and the distal femurs were
removed for subsequent experiments.
Histological analysis
Femurs were fixed in 4% paraformaldehyde for 24 h,
decalcified in 10% EDTA for 6 weeks and embedded in paraffin. They
were cut into 4 µm-thick sections and mounted on common slides for
staining with hematoxylin and eosin and safranin O. The
histopathological features of cartilage were analyzed using the
scoring system modified by Mankin et al (17).
Immunohistochemical staining of PTH1R,
sclerostin, DKK1, β-catenin and RUNX2
To investigate the association between PTH 1–34 and
factors associated with the Wnt/β-catenin pathway, the present
study measured the expression of PTH1R, sclerostin, DKK1, β-catenin
and RUNX2 in cartilage by immunohistochemical analysis. Paraffin
sections (4 µm) of joint tissue were routinely deparaffinized,
rehydrated and treated with 0.1% trypsin for antigen retrieval for
15 min at 37°C, and subsequently incubated with 3%
H2O2 solution for 10 min. Sections were
blocked with normal goat serum (Wuhan Boster Biological Technology,
Ltd., Wuhan, China) for 8 min at 37°C and incubated overnight at
4°C with anti-rabbit sclerostin (1:50; ab63097), DKK1 (1:100;
ab93017), β-catenin (1:100; ab16051) and RUNX2 (1:100; ab23981)
polyclonal antibodies (Abcam, Cambridge, UK) and anti-rabbit PTH1R
polyclonal antibody (1:100; BA3170; Wuhan Boster Biological
Technology, Ltd.). Sections were then treated with the Two-Step IHC
Detection Reagent (PV-9001; Origene Technologies, Inc., Rockville,
MD, USA). Briefly, sections were incubated with reagent 1 (Reaction
enhancement solution) for 20 min at 37°C and then incubated with
reagent 2 (Enhanced enzyme-labeled goat anti-rabbit IgG polymer)
for 30 min at 37°C. The color brown was developed using
3,3′-diaminobenzidine (Origene Technologies, Inc.). For negative
controls, PBS without antibody was used for incubation.
Counterstaining was carried out with hematoxylin. The positive
cells were stained yellow or brown. All sections were analyzed
using Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA) at ×400 magnification to quantify protein expression; 5 random
fields were randomly selected in each slide and the numbers of
positive cells were counted and expressed as a percentage of the
total cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Femurs were thoroughly cleaned and the muscle,
ligament and joint capsule were simultaneously removed, while
damage to the femur was avoided. The femur was rapidly frozen with
liquid nitrogen and articular cartilage was obtained for further
investigation. Following treatment, total RNA was extracted with an
E.Z.N.A Total RNA kit (Omega Bio-Tek, Inc., Norcross, GA, USA) from
articular cartilage according to the manufacturer's protocol.
Subsequently, total RNA was reverse transcribed to cDNA using
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
protocol. Specific transcripts were quantified by qPCR using Maxima
SYBR-Green qPCR Kit (Thermo Fisher Scientific, Inc.), and analyzed
with an ABI 7500 Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The following gene-specific primers were
used: PTH1R, TCT CCT TAC CCA GGC AGA TG (forward) and CAT TGC ATC
CTC TCC ACA GA (reverse); β-catenin, ACC ATC GAG AGG GCT TGT TG
(forward) and CGC ACT GCC ATT TTA GCT CC (reverse); and GAPDH, GAA
GGT GAA GGT CGG AGT C (forward) and GAA GAT GGT GAT GGG ATT TC
(reverse). PCR was performed at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec, and 60°C for 1 min. Expression of PTH1R
and β-catenin mRNA was calculated using the ΔΔCq method (18), levels were normalized to the
expression of GAPDH. Each experiment was repeated three times with
each rat.
Statistical analysis
SPSS version 20.0 (SPSS, Inc., Chicago, IL, USA) was
used for statistical analysis. Results were analyzed using two-way
factorial design analysis of variation followed by
Student-Newman-Keuls post-hoc test. Data are presented as the mean
± standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Histological assessments
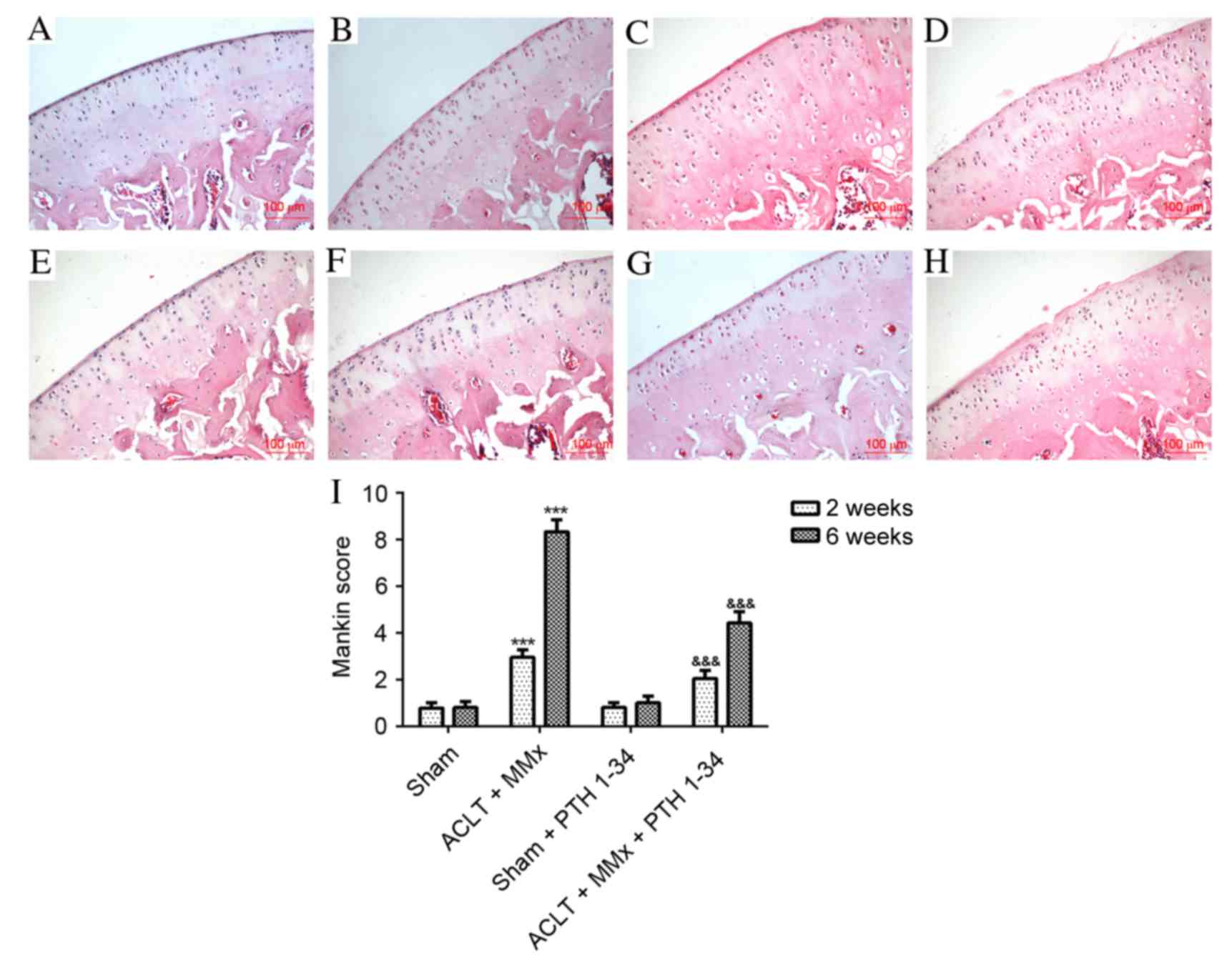
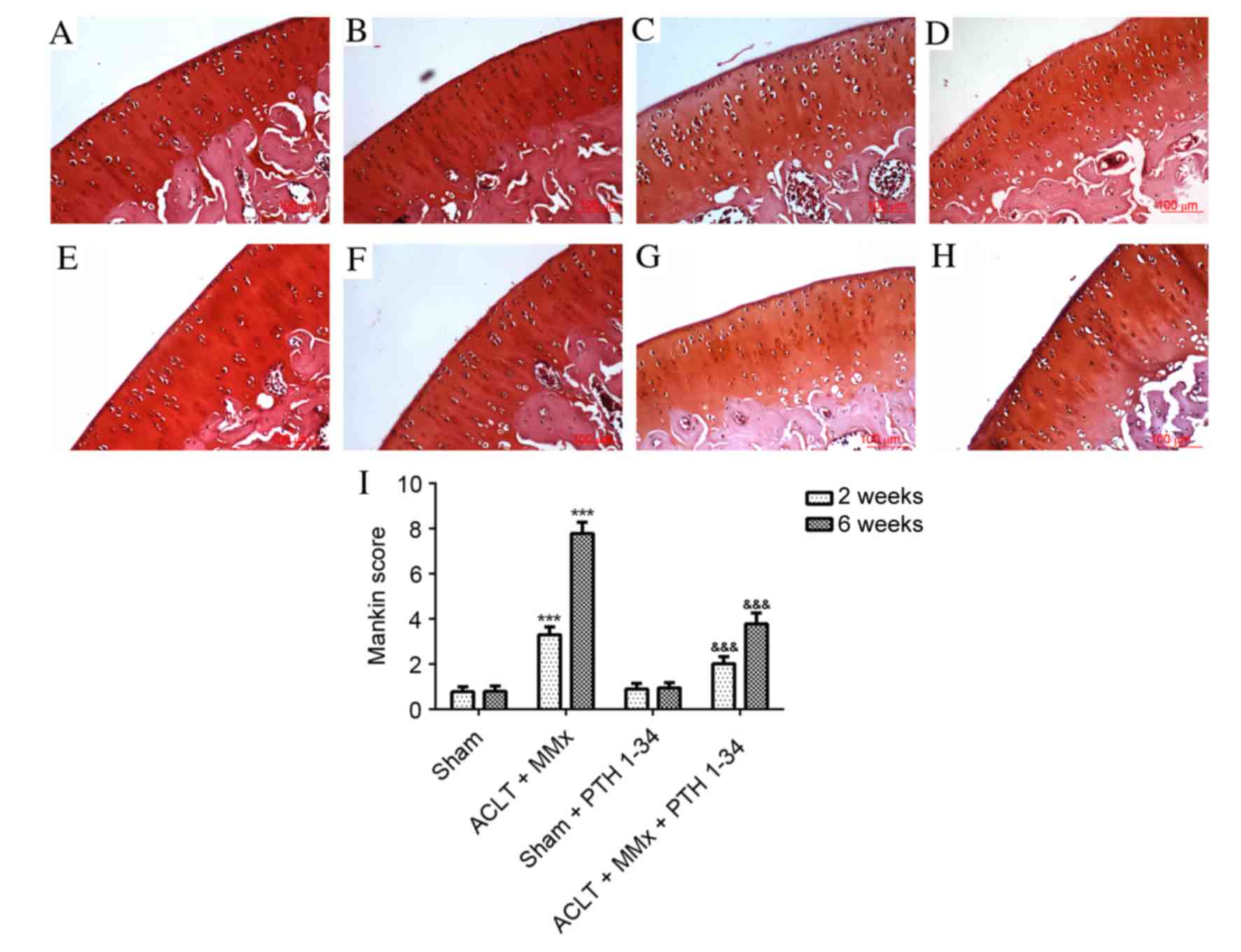
The present study applied hematoxylin and eosin
staining (Fig. 1), safranin O
staining (Fig. 2) and the Mankin
score (Figs. 1 and 2) to evaluate the effect of PTH 1–34 on
the knee joints of ACLT + MMx rats. The articular cartilage in
NS-treated sham rats and PTH 1–34-treated sham rats at 2 and 6
weeks had a normal, smooth surface and structure, and no
abnormalities were observed. Early histopathological changes were
observed after 2 weeks in NS-treated ACLT + MMx rats, including
chondrocytes with diffuse hyper-cellularity, a marginal reduction
in matrix staining and destruction of tidemark integrity. After 6
weeks in NS-treated ACLT + MMx rats, more serious histological
changes were observed, including structure clefts to transitional
zone, chondrocyte clusters, a severe reduction in matrix staining
and destruction of tidemark integrity. Histological scoring was
performed using the modified Mankin histological scores to evaluate
pathological changes of articular cartilage. After 2 and 6 weeks,
ACLT + MMx rats receiving PTH 1–34 treatment had reduced articular
cartilage degeneration, compared with the NS-treated ACLT + MMx
rats. The present study also observed that there were no marked
differences between the PTH 1–34-treated sham rats and NS-treated
sham rats. For the ACLT + MMx rats, the Mankin score increased with
time compared with the sham rats. The Mankin score of the
hematoxylin and eosin (Fig. 1) and
safranin O (Fig. 2) staining in
PTH 1–34-treated ACLT + MMx rats, decreased by 31 and 46%, and 36
and 51%, at 2 and 6 weeks, respectively, compared with NS-treated
ACLT + MMx rats.
Immunohistochemical analysis
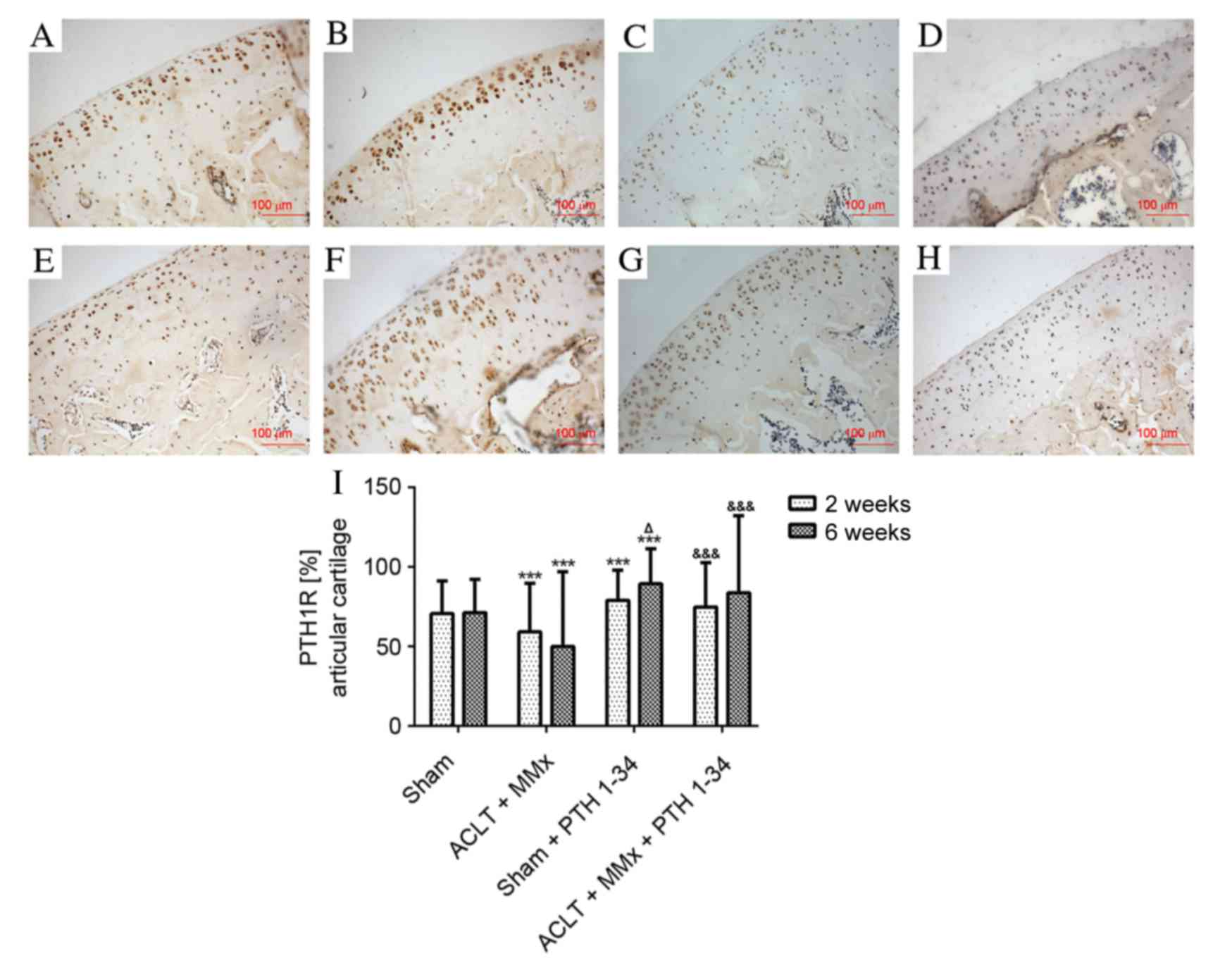
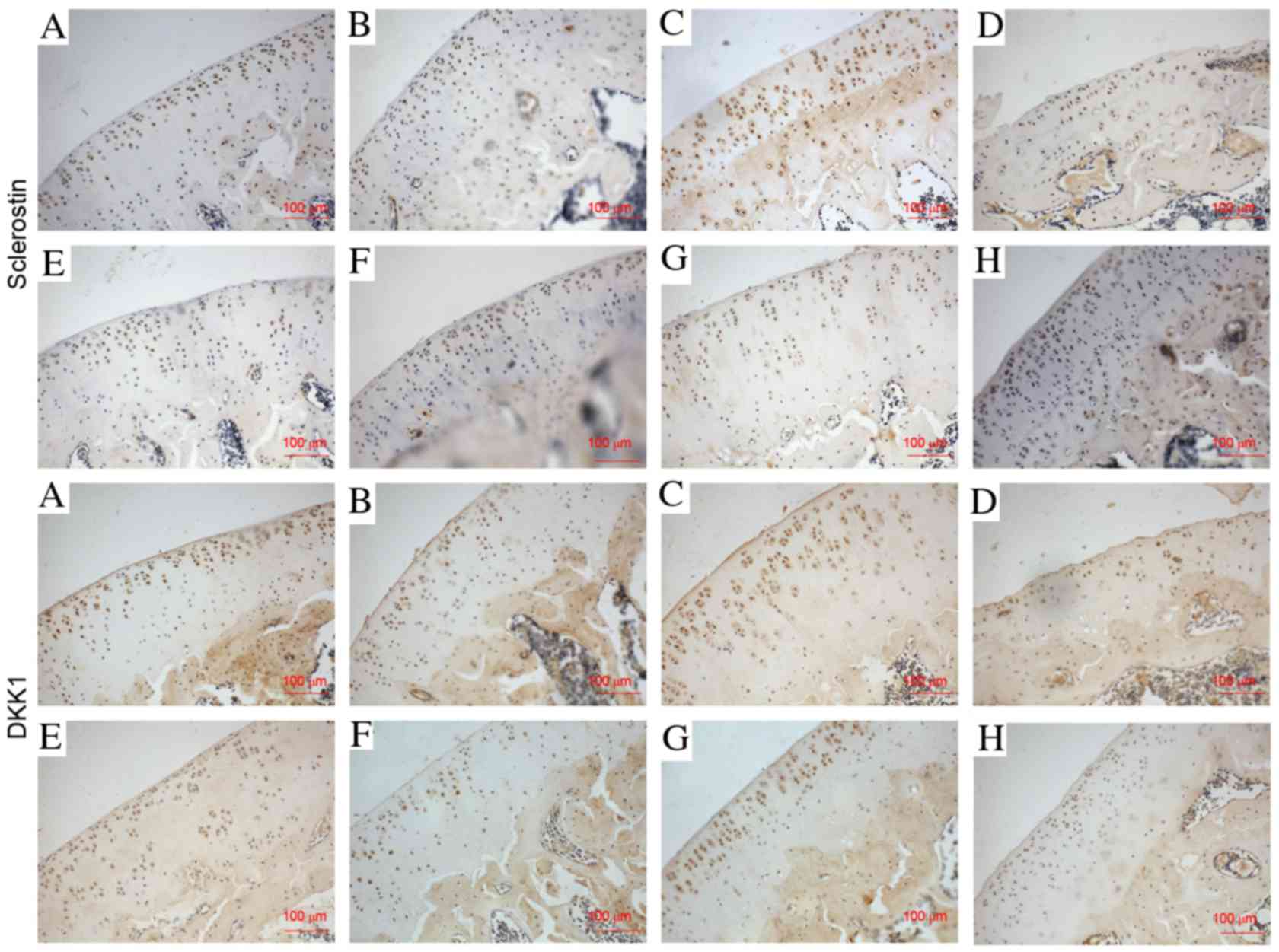
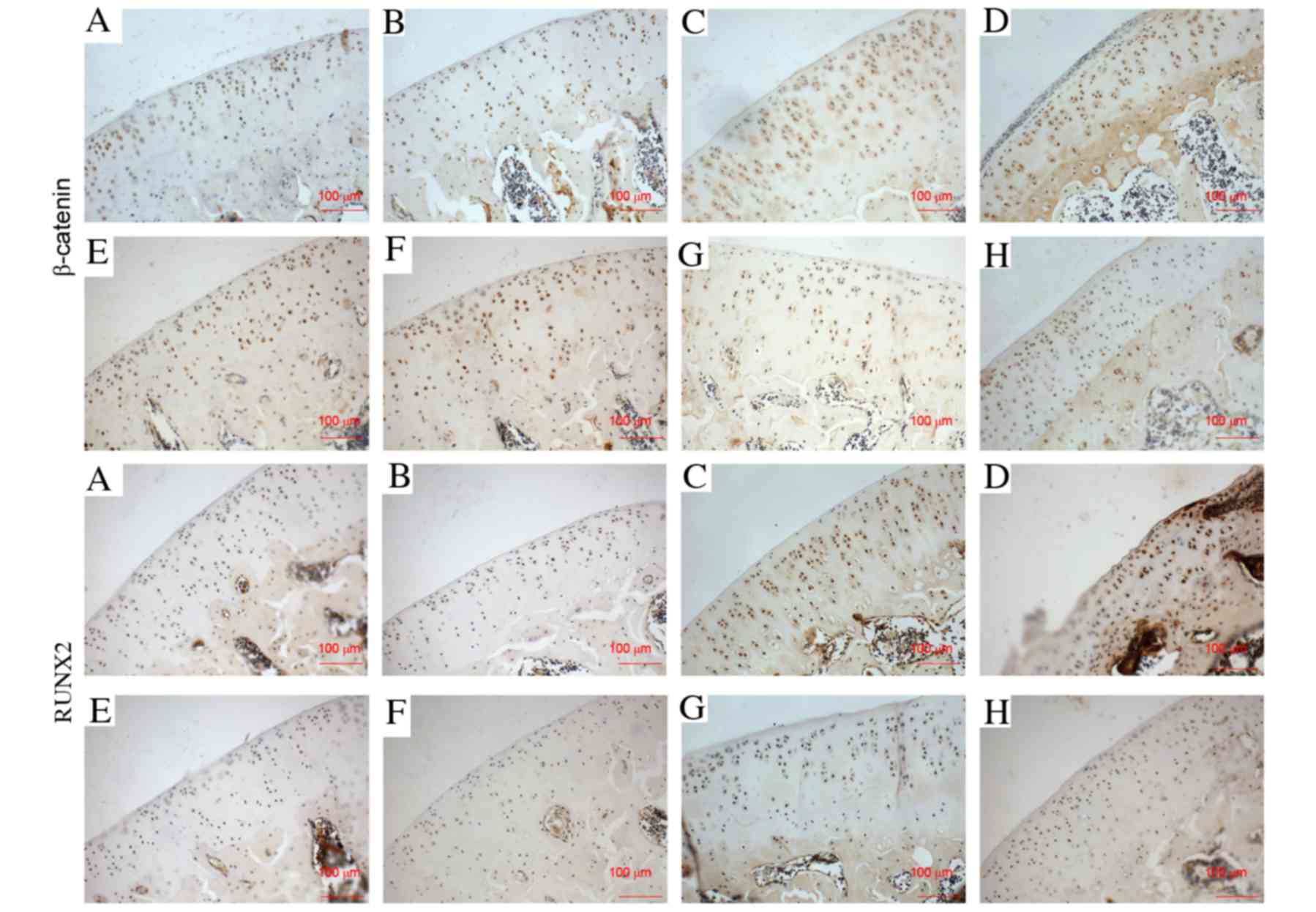
The expression of PTH1R, sclerostin, DKK1, β-catenin
and RUNX2 in cartilage was evaluated by immunohistochemistry
assays. The expression of PTH1R was significantly decreased
(P<0.001; Fig. 3), while
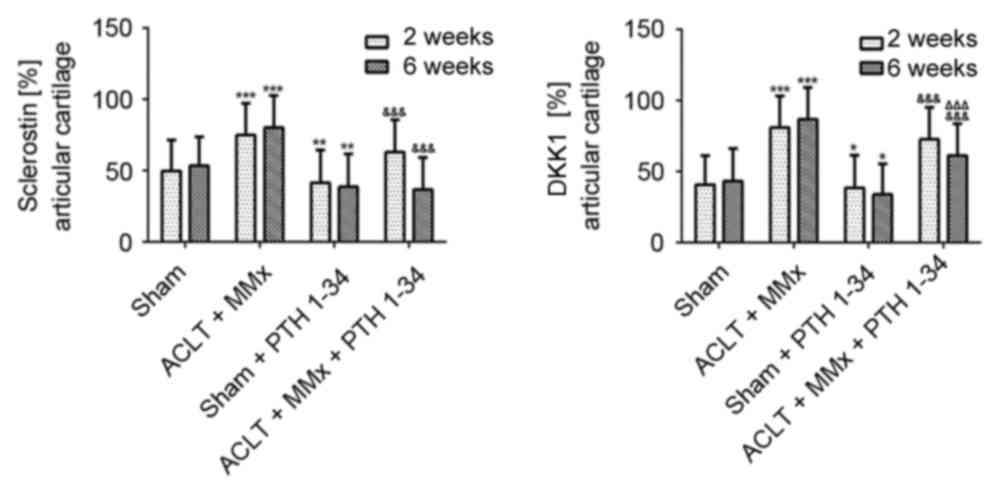
sclerostin and DKK1 (Figs. 4 and
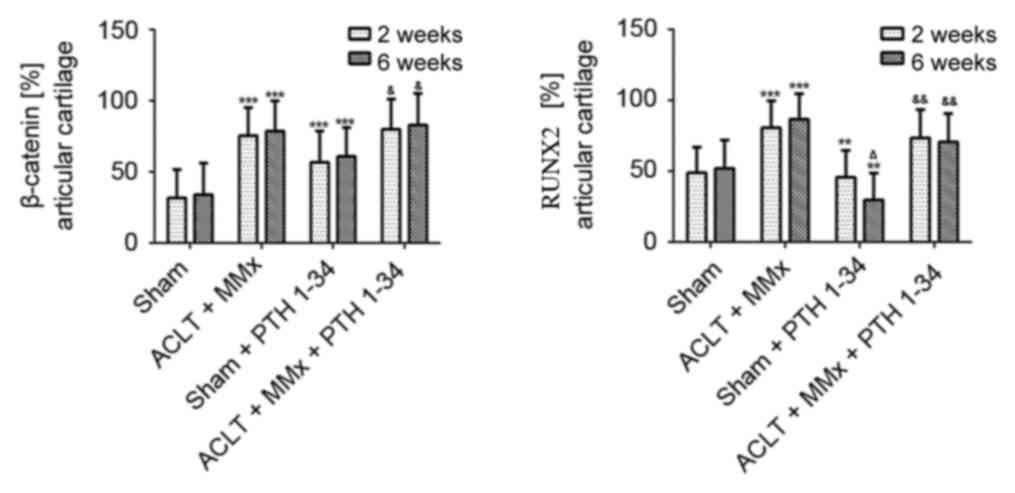
5), and β-catenin and RUNX2
(Figs. 6 and 7) were significantly increased in the
NS-treated ACLT + MMx rats compared with the NS-treated sham rats
at 2 and 6 weeks (P<0.001). Compared with NS-treated sham rats,
the expression of PTH1R was increased by 12 and 25% (P<0.001;
Fig. 3), and β-catenin was
increased by 76 and 81% (P<0.001; Fig. 7) at 2 and 6 weeks, respectively, in
the PTH 1–34-treated sham rats. Furthermore, the expression of
sclerostin was decreased by 16 and 27% (P<0.01; Fig. 5), DKK1 was decreased by 10 and 23%
(P<0.05; Fig. 5), and RUNX2 was
decreased by 10 and 42% (P<0.01; Fig. 7) at 2 and 6 weeks, respectively, in
PTH 1–34-treated sham rats compared with NS-treated sham rats. The
expression of PTH1R was increased by 27 and 67% (P<0.001;
Fig. 3), and β-catenin was
increased by 6 and 8% (P<0.05; Fig.
7), while the expression of sclerostin was decreased by 16 and
54% (P<0.001; Fig. 5), DKK1 was
decreased by 10 and 28% (P<0.001; Fig. 5), and RUNX2 was decreased by 9 and
19% (P<0.01; Fig. 7) at 2 and 6
weeks, respectively, in the PTH 1–34-treated ACLT + MMx rats
compared with the NS-treated ACLT + MMx rats.
RT-qPCR analysis
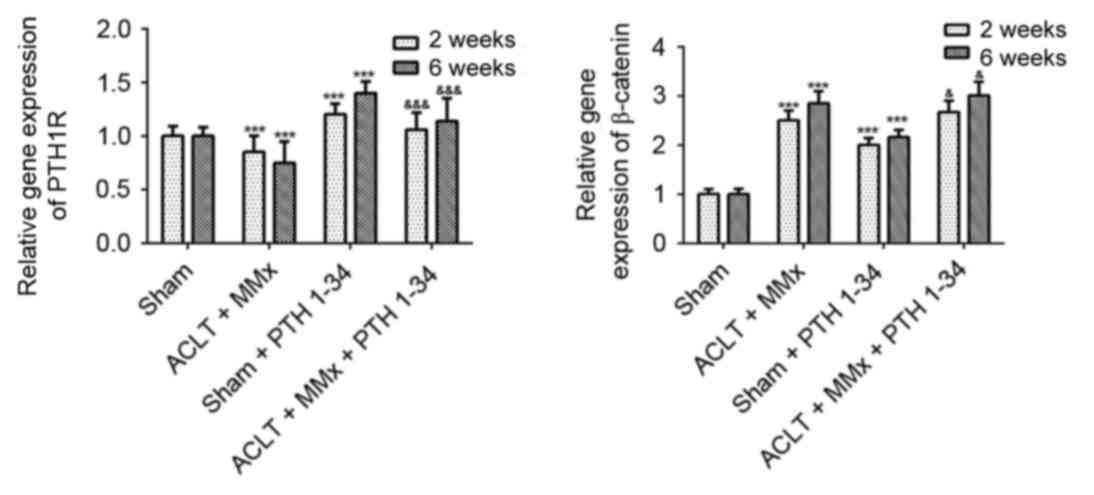
To further investigate the mechanism, RT-qPCR was
performed to detect the mRNA expression levels of PTH1R and
β-catenin in cartilage (Fig. 8).
At 2 and 6 weeks, the gene expression of PTH1R (Fig. 8) was decreased by 15 and 25%,
respectively, in NS-treated ACLT + MMx rats compared with
NS-treated sham rats (P<0.001). PTH1R expression was increased
by 20 and 40% in PTH 1–34-treated sham rats compared with
NS-treated sham rats (P<0.001), and was increased by 24 and 51%
in PTH 1–34-treated ACLT + MMx rats compared with NS-treated ACLT +
MMx rats (P<0.001). The present study also demonstrated that the
gene expression of β-catenin (Fig.
8) was increased by 150 and 185% in NS-treated ACLT + MMx rats
compared with NS-treated sham rats (P<0.001), was increased by
101 and 116% in PTH 1–34-treated sham rats compared with NS-treated
sham rats (P<0.001), and was increased by 8 and 9% in PTH
1–34-treated ACLT + MMx rats compared with NS-treated ACLT + MMx
rats (P<0.05).
Discussion
OA is a common degenerative disease that seriously
affects daily life and causes an economic burden. The pathogenesis
of OA is complex. Cartilage matrix degeneration and hypertrophic
chondrocytes have key roles in the progression of OA, therefore, it
is particularly important to inhibit the development of these
factors. The present study demonstrated that PTH 1–34 reduced the
Mankin scores and increased the mRNA expression and protein levels
of PTH1R and β-catenin, and decreased protein levels of sclerostin,
DKK1 and RUNX2, in ACLT + MMx rats with PTH 1–34-treatment compared
with ACLT + MMx rats with NS-treatment. Similar results for mRNA
expression and protein levels were observed between sham rats with
PTH 1–34-treatment and sham rats with NS-treatment. These results
have, to the best of our knowledge, demonstrated for the first time
that PTH 1–34 may upregulate the Wnt/β-catenin signaling pathway,
and that PTH 1–34 may downregulate RUNX2 through an alternative
pathway to the Wnt/β-catenin signaling pathway in a rat model of
OA.
In OA experimental animal models, surgically-induced
destabilization of joints is the most widely used induction method.
These models control the timing and follow predictable progression
of the disease. Of these OA models, the ACLT + MMx rat model is one
of the most common (19). Early
cartilage degradation, subchondral osteopenia followed by sclerosis
and late osteophyte formation emerge sequentially in the ACLT + MMx
rat model (15). In the present
study, the ACLT + MMx rat model exhibited cartilage degeneration at
2 weeks post-surgery, which gradually increased over time.
In guinea pigs with spontaneous OA, papain-induced
OA in rats, meniscal/ligamentous injury-induced OA in mice or in
osteochondral defects in rabbits, PTH1R expression gradually
decreased with progression of OA. Systemic application of PTH 1–34
significantly upregulated PTH1R expression in cartilage (8–10,20).
Similar to these previous results, the present study demonstrated
that, as OA progressed, the protein levels and mRNA expression of
PTH1R gradually declined and systemic application of PTH 1–34
significantly upregulated them in cartilage in the ACLT + MMx rat
model.
Sclerostin, a potent inhibitor of Wnt/β-catenin
signaling that binds to LDL receptor related protein (LRP)5/6, is
encoded by the SOST gene and is expressed by osteocytes and
articular chondrocytes. Sclerostin is increased in OA cartilage
compared with normal cartilage (8,21–23).
In bone, inhibiting sclerostin increases bone formation, bone mass
and bone strength (24). The role
of sclerostin in OA progression is not well established. Knockout
of the SOST gene promoted OA in mice via β-catenin-dependent and
-independent Wnt pathways, therefore, sclerostin has a role in the
maintenance of cartilage integrity in OA (25). However, increasing sclerostin using
a recombinant sclerostin may retard cartilage degradation (26). The present study demonstrated that
with the progression of OA, sclerostin expression gradually
increases. To the best of our knowledge, the present study is the
first to demonstrate that administration of PTH 1–34 reduced
sclerostin expression in cartilage in the ACLT + MMx rat model,
similar to guinea pigs with spontaneous OA (8).
DKK1 is a secreted protein and a member of a
multigene family. It acts as a direct inhibitor of Wnt/β-catenin
signaling by binding to LRP5/6 and forms a ternary complex with
Kremen and LRP6. DKK1 is a master regulator of joint remodeling
(27,28). Various studies have indicated that
DKK1 is associated with OA development, however, these results are
controversial. A previous study demonstrated that, as joint
severity was increased, DKK1 levels were decreased in plasma and
synovial fluid, thus, DKK1 may have a role in the progression of OA
(29). Previous results
demonstrated that DKK1 was increased in OA cartilage when compared
with normal cartilage (30). In a
previous study that used the destabilization of the medial meniscus
mouse model of OA, which was performed using intra-articular
injection of an adenovirus expression DKK1 to increase DKK1
expression in chondrocytes, DKK1 inhibited cartilage destruction
(30). Increased DKK1 expression
in ACLT and collagenase-induced OA rat models, via intra-peritoneal
injection of DKK1 antisense oligonucleotides, was previously
observed to downregulate DKK1 expression and subsequently
ameliorate chondrocyte apoptosis and cartilage destruction in OA
(31). The present study
demonstrated that DKK1 expression gradually increased with OA
progression, and that administration of PTH 1–34 downregulated DKK1
expression in cartilage in the ACLT + MMx rat model. A previous
report demonstrated that PTH 1–34 inhibited the expression of DKK1
in osteoblasts (32). Although the
results of the present study also demonstrated a similar inhibition
of PTH 1–34 on DKK1, the results of the present study were in OA
cartilage, and further investigation is required to determine the
specific mechanisms involved.
Signaling pathways that control joint and articular
cartilage are particularly important for the treatment of OA. The
hypertrophy of chondrocytes and degradation of the extracellular
matrix of cartilage lead to a loss of articular cartilage that is
characteristic of OA (3). Wnt
signaling has major roles in the majority of aspects of skeletal
development and homeostasis, and abnormal signaling causes various
human skeletal diseases (11). The
Wnt pathway is a key regulator of joint remodeling and also has a
critical role in OA pathogenesis. Wnt/β-catenin signaling is
associated with a loss of differentiated phenotype and chondrocyte
matrix catabolic action, which may contribute to cartilage
destruction (33). β-catenin is a
key factor of the Wnt/β-catenin pathway that is encoded by the
CTNNB1 gene. The nuclear levels of β-catenin directly reflect the
activation level of this signaling pathway. β-catenin is stable in
the cytoplasm. Upon entering the nucleus, it interacts with a
transcription factor and subsequently forms a complex with TCF/LEF
transcription factors and regulates the expression of target genes
(34). Conditional activation of
the β-catenin gene in articular chondrocytes increases MMP-9,
MMP-13, alkaline phosphatase and collagen X in chondrocytes, which
leads to premature chondrocyte differentiation and the development
of an OA-like phenotype in mice (35). The results of the present study
demonstrated that the protein levels and mRNA expression of
β-catenin gradually increased with OA progression, and
administration of PTH 1–34 upregulated the protein levels and mRNA
expression of β-catenin in cartilage in the ACLT + MMx rat model.
According to a study in osteoblasts, PTH activated β-catenin via
recruitment of LRP6 to the PTH/PTH1R complex (36). PTH increased β-catenin expression
by protein kinase A (PKA) and PKC pathways, and SMAD3 in mouse
osteoblastic cells (37). A
previous study also demonstrated that PTH1R activated β-catenin via
direct recruitment of disheveled segment polarity proteins,
independent of Wnt or LRP5/6, which induced osteoclastogenesis
(38). These studies demonstrated
that PTH increases the expression of β-catenin in osteoblasts and
osteoclasts. Sclerostin and DKKs negatively regulate Wnt/β-catenin
signaling in bone formation, and PTH inhibits the expression of
sclerostin and DKK1. The current study demonstrated that PTH 1–34
upregulated β-catenin in cartilage in the ACLT + MMx rat model. PTH
may increase β-catenin expression by decreasing sclerostin and DKK1
expression. However, identification of the specific mechanism
requires further investigation.
RUNX2, a transcription factor that belongs to the
runt homology domain protein family, is strongly expressed in
osteoblasts, and also in pre-hypertrophic and hypertrophic
chondrocytes (39,40). RUNX2 is a major positive regulator
of chondrocyte differentiation (39). RUNX2 is constitutively expressed in
non-hypertrophic chondrocytes that induce hypertrophic chondrocyte
differentiation and partially rescued the chondrocyte phenotype in
RUNX2-deficient mice (41). In
mice, following induction of joint instability, RUNX2 may
successively induce type X collagen and MMP-13 expression, which
contribute to chondrocyte hypertrophy and matrix breakdown,
therefore causing the pathogenesis of OA (41). Inhibition of RUNX2 delayed
endochondral ossification or chondrocyte hypertrophy in a previous
study (42). The present study
demonstrated that RUNX2 expression gradually increased with OA
progression and administration of PTH 1–34 downregulated RUNX2
expression in cartilage in the ACLT + MMx rat model. The
association between increased expression of RUNX2 in articular
cartilage and articular cartilage degeneration was similar to a
previous report (41). A previous
study demonstrated that Wnt/β-catenin signaling induced chondrocyte
hypertrophy through RUNX2 (41).
The current study demonstrated that the expression of β-catenin and
RUNX2 was increased in the cartilage of ACLT + MMx rats with
NS-treatment compared with NS-treated sham rats. Furthermore, in
the PTH 1–34-treated ACLT + MMx rats, the expression of β-catenin
was increased, and the expression of RUNX2 decreased. Increasing
β-catenin did not upregulate RUNX2, which contradicts the results
of a previous study (14).
Parathyroid hormone like hormone (PTHrP) and PTH have similar
N-terminal regions, activate the same G-protein-coupled receptor,
PTH1R, and also have similar potential effects (7). A previous study reported that PTHrP
downregulated RUNX2 expression, which was accompanied by
suppression of type X collagen through the PKA signaling pathway
(43). Therefore, PTH 1–34 may
decrease RUNX2 levels through pathways other than the Wnt/β-catenin
signaling pathway.
In conclusion, the results of the present study
indicate that intermittent application of the anabolic bone agent,
PTH 1–34, reduced the Mankin scores in the OA model, ACLT + MMx,
compared with NS-treated ACLT + MMx rats, with no significant
effect observed in sham rats. This may be associated with PTH 1–34
increasing the expression of PTH1R and β-catenin, and reducing the
expression of sclerostin, DKK1 and RUNX2 in cartilage. Although the
experimental models used in the present study allowed us to control
the time of disease onset and follow predictable progression of the
disease, rats have a faster growth rate and higher disease severity
than humans and they do not completely imitate OA development in
humans. The present study investigated the association between
PTH1R and the factors associated with the Wnt/β-catenin pathway,
and did not detect downstream factors, including type X collagen
and MMP-13. Therefore, further investigation is needed to determine
the association between PTH 1–34 and downstream factors in
articular cartilage. Additionally, integration of information from
different signaling pathways is required, which may provide useful
information for the investigation of the pathogenesis of OA.
References
|
1
|
Allen KD and Golightly YM: State of the
evidence. Curr Opin Rheumatol. 27:276–283. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pulsatelli L, Addimanda O, Brusi V,
Pavloska B and Meliconi R: New findings in osteoarthritis
pathogenesis: Therapeutic implications. Ther Adv Chronic Dis.
4:23–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhong L, Huang X, Karperien M and Post JN:
The regulatory role of signaling crosstalk in hypertrophy of MSCs
and human articular chondrocytes. Int J Mol Sci. 16:19225–19247.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuasa T, Otani T, Koike T, Iwamoto M and
Enomoto-Iwamoto M: Wnt/beta-catenin signaling stimulates matrix
catabolic genes and activity in articular chondrocytes: Its
possible role in joint degeneration. Lab Invest. 88:264–274. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harrington EK, Roddy GW, West R and
Svoboda KK: Parathyroid hormone/parathyroid hormone-related peptide
modulates growth of avian sternal cartilage via chondrocytic
proliferation. Anat Rec (Hoboken). 290:155–167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neer RM, Arnaud CD, Zanchetta JR, Prince
R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S,
Genant HK, et al: Effect of parathyroid hormone (1–34) on fractures
and bone mineral density in postmenopausal women with osteoporosis.
N Engl J Med. 344:1434–1441. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abou-Samra AB, Jüppner H, Force T, Freeman
MW, Kong XF, Schipani E, Urena P, Richards J, Bonventre JV, Potts
JT Jr, et al: Expression cloning of a common receptor for
parathyroid hormone and parathyroid hormone-related peptide from
rat osteoblast-like cells: A single receptor stimulates
intracellular accumulation of both cAMP and inositol trisphosphates
and increases intracellular free calcium. Proc Natl Acad Sci USA.
89:pp. 2732–2736. 1992; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan JY, Tian FM, Wang WY, Cheng Y, Song
HP, Zhang YZ and Zhang L: Parathyroid hormone (1–34) prevents
cartilage degradation and preserves subchondral bone
micro-architecture in guinea pigs with spontaneous osteoarthritis.
Osteoarthritis Cartilage. 22:1869–1877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sampson ER, Hilton MJ, Tian Y, Chen D,
Schwarz EM, Mooney RA, Bukata SV, O'Keefe RJ, Awad H, Puzas JE, et
al: Teriparatide as a chondroregenerative therapy for
injury-induced osteoarthritis. Sci Transl Med. 3:101ra932011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang JK, Chang LH, Hung SH, Wu SC, Lee
HY, Lin YS, Chen CH, Fu YC, Wang GJ and Ho ML: Parathyroid hormone
1–34 inhibits terminal differentiation of human articular
chondrocytes and osteoarthritis progression in rats. Arthritis
Rheum. 60:3049–3060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Regard JB, Zhong Z, Williams BO and Yang
Y: Wnt signaling in bone development and disease: Making stronger
bone with Wnts. Cold Spring Harb Perspect Biol. 4:pii: a0079972012.
View Article : Google Scholar
|
|
12
|
Yasuhara R, Ohta Y, Yuasa T, Kondo N,
Hoang T, Addya S, Fortina P, Pacifici M, Iwamoto M and
Enomoto-Iwamoto M: Roles of β-catenin signaling in phenotypic
expression and proliferation of articular cartilage superficial
zone cells. Lab Invest. 91:1739–1752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loeser RF: Osteoarthritis Year in Review
2013: Biology. Osteoarthritis Cartilage. 21:1436–1442. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong YF, do Y Soung, Schwarz EM, O'Keefe
RJ and Drissi H: Wnt induction of chondrocyte hypertrophy through
the Runx2 transcription factor. J Cell Physiol. 208:77–86. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pickarski M, Hayami T, Zhuo Y and Duong
LT: Molecular changes in articular cartilage and subchondral bone
in the rat anterior cruciate ligament transection and
meniscectomized models of osteoarthritis. BMC Musculoskelet Disord.
12:1972011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rossi R, Dettoni F, Bruzzone M, Cottino U,
D'Elicio DG and Bonasia DE: Clinical examination of the knee: Know
your tools for diagnosis of knee injuries. Sports Med Arthrosc
Rehabil Ther Technol. 3:252011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mankin HJ, Dorfman H, Lippiello L and
Zarins A: Biochemical and metabolic abnormalities in articular
cartilage from osteo-arthritic human hips. II. Correlation of
morphology with biochemical and metabolic data. J Bone Joint Surg
Am. 53:523–537. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Longo UG, Loppini M, Fumo C, Rizzello G,
Khan WS, Maffulli N and Denaro V: Osteoarthritis: New insights in
animal models. Open Orthop J. 6:558–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orth P, Cucchiarini M, Zurakowski D,
Menger MD, Kohn DM and Madry H: Parathyroid hormone [1-34] improves
articular cartilage surface architecture and integration and
subchondral bone reconstitution in osteochondral defects in vivo.
Osteoarthritis Cartilage. 21:614–624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roudier M, Li X, Niu QT, Pacheco E,
Pretorius JK, Graham K, Yoon BR, Gong J, Warmington K, Ke HZ, et
al: Sclerostin is expressed in articular cartilage but loss or
inhibition does not affect cartilage remodeling during aging or
following mechanical injury. Arthritis Rheum. 65:721–731. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Bezooijen RL, Roelen BA, Visser A, Van
der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE,
ten Dijke P and Löwik CW: Sclerostin is an osteocyte-expressed
negative regulator of bone formation, but not a classical BMP
antagonist. J Exp Med. 199:805–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang
J, Harris SE and Wu D: Sclerostin binds to LRP5/6 and antagonizes
canonical Wnt signaling. J Biol Chem. 280:19883–19887. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Ominsky MS, Warmington KS, Morony S,
Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, et al:
Sclerostin antibody treatment increases bone formation, bone mass,
and bone strength in a rat model of postmenopausal osteoporosis. J
Bone Miner Res. 24:578–588. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bouaziz W, Funck-Brentano T, Lin H, Marty
C, Ea HK, Hay E and Cohen-Solal M: Loss of sclerostin promotes
osteoarthritis in mice via beta-catenin-dependent and -independent
Wnt pathways. Arthritis Res Ther. 17:242015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan BY, Fuller ES, Russell AK, Smith SM,
Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN and
Little CB: Increased chondrocyte sclerostin may protect against
cartilage degradation in osteoarthritis. Osteoarthritis Cartilage.
19:874–885. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao B, Wu W, Davidson G, Marhold J, Li M,
Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, et al: Kremen
proteins are Dickkopf receptors that regulate Wnt/beta-catenin
signalling. Nature. 417:664–667. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niehrs C: Function and biological roles of
the Dickkopf family of Wnt modulators. Oncogene. 25:7469–7481.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Honsawek S, Tanavalee A, Yuktanandana P,
Ngarmukos S, Saetan N and Tantavisut S: Dickkopf-1 (Dkk-1) in
plasma and synovial fluid is inversely correlated with radiographic
severity of knee osteoarthritis patients. BMC Musculoskelet Disord.
11:2572010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh H, Chun CH and Chun JS: Dkk-1
expression in chondrocytes inhibits experimental osteoarthritic
cartilage destruction in mice. Arthritis Rheum. 64:2568–2578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weng LH, Wang CJ, Ko JY, Sun YC and Wang
FS: Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage
destruction, and subchondral bone deterioration in osteoarthritic
knees. Arthritis Rheum. 62:1393–1402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo J, Liu M, Yang D, Bouxsein ML, Saito
H, Galvin RJ, Kuhstoss SA, Thomas CC, Schipani E, Baron R, et al:
Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated
stromal cell response and new bone formation. Cell Metab.
11:161–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hwang SG, Ryu JH, Kim IC, Jho EH, Jung HC,
Kim K, Kim SJ and Chun JS: Wnt-7a causes loss of differentiated
phenotype and inhibits apoptosis of articular chondrocytes via
different mechanisms. J Biol Chem. 279:26597–26604. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Voronkov A and Krauss S: Wnt/beta-catenin
signaling and small molecule inhibitors. Curr Pharm Des.
19:634–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang M, Xie R, Hou W, Wang B, Shen R,
Wang X, Wang Q, Zhu T, Jonason JH and Chen D: PTHrP prevents
chondrocyte premature hypertrophy by inducing cyclin-D1-dependent
Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal
degradation. J Cell Sci. 122:1382–1389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H,
He X, Nie S, Chang C and Cao X: Parathyroid hormone signaling
through low-density lipoprotein-related protein 6. Genes Dev.
22:2968–2979. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tobimatsu T, Kaji H, Sowa H, Naito J,
Canaff L, Hendy GN, Sugimoto T and Chihara K: Parathyroid hormone
increases beta-catenin levels through Smad3 in mouse osteoblastic
cells. Endocrinology. 147:2583–2590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Romero G, Sneddon WB, Yang Y, Wheeler D,
Blair HC and Friedman PA: Parathyroid hormone receptor directly
interacts with dishevelled to regulate beta-Catenin signaling and
osteoclastogenesis. J Biol Chem. 285:14756–14763. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim IS, Otto F, Zabel B and Mundlos S:
Regulation of chondrocyte differentiation by Cbfa1. Mech Dev.
80:159–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Takeda S, Bonnamy JP, Owen MJ, Ducy P and
Karsenty G: Continuous expression of Cbfa1 in nonhypertrophic
chondrocytes uncovers its ability to induce hypertrophic
chondrocyte differentiation and partially rescues Cbfa1-deficient
mice. Genes Dev. 15:467–481. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kamekura S, Kawasaki Y, Hoshi K, Shimoaka
T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G,
et al: Contribution of runt-related transcription factor 2 to the
pathogenesis of osteoarthritis in mice after induction of knee
joint instability. Arthritis Rheum. 54:2462–2470. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ueta C, Iwamoto M, Kanatani N, Yoshida C,
Liu Y, Enomoto-Iwamoto M, Ohmori T, Enomoto H, Nakata K, Takada K,
et al: Skeletal malformations caused by overexpression of Cbfa1 or
its dominant negative form in chondrocytes. J Cell Biol.
153:87–100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li TF, Dong Y, Ionescu AM, Rosier RN,
Zuscik MJ, Schwarz EM, O'Keefe RJ and Drissi H: Parathyroid
hormone-related peptide (PTHrP) inhibits Runx2 expression through
the PKA signaling pathway. Exp Cell Res. 299:128–136. 2004.
View Article : Google Scholar : PubMed/NCBI
|