Introduction
Van der wood Syndrome [VWS; Online Mendelian
Inheritance in Man (www.omim.org)
119300] is an autosomal dominant inheritance of cleft lip and
palate deformities. The primary clinical features of VWS are lower
lip fistula, teeth agenesis or pits, simple cleft palate, simple
cleft lip or cleft palate and cleft palate merger, and several of
the features may appear at the same time. VWS is a syndrome in
cleft lip and palate, and accounts for 2% of all patients with
cleft lip and palate (1). Kondo
et al (2) succeeded in
cloning the interferon regulatory factor (IRF) 6 gene that was the
disease-causing gene of VWS, and VWS has been considered to be the
best genetic study model of non-syndromic cleft lip with or without
cleft palate. In 1986, the global incidence of VWS was 1~3/30,000,
and the incidence in newborns was 1/100,000-1/40,000 (3), but there was a big difference in
phenotypes (3,4). The clinical phenotype of VWS patients
differs with families and even among VWS patients within the same
family. Due to the different phenotypes, the clinical diagnosis of
VWS has often been confused with non-syndromic cleft lip with or
without cleft palate (NSCL/P), with the lower lip pits easily
mistaken for bite marks. Peyrard-Janvid et al (5) studied 17 families from Sweden,
Finland, Norway, Thailand and Singapore, and reported that 80% of
VWS patients exhibited an IRF6 gene mutation, thus demonstrating
that it was a reliable marker of VWS. IRF6 in VWS has not been so
well reported in China, although previous studies have confirmed
that IRF6 is associated with VWS in a clinical study of a VWS
family (6,7). By a joint analysis of clinical data
and genetic testing results of a VWS family, the present study
aimed to clarify the nosogenesis of these patients from the
perspective of genetics, and analyze the association between
genotype and phenotype. It also aimed to clarify the VWS pathogenic
mutations of Chinese VWS patients, and to identify the suspected
gene mutation areas of interest. The pathogenic gene detection of
VWS may be used for aiding clinical assessment in the future, and
for prenatal diagnosis.
Materials and methods
General
The probands from two families were inpatients in
the oral and maxillofacial surgery ward of Handan Central Hospital
(Handan, China). The two families had 24 members, including 6 with
VWS. At the same time, 200 peripheral blood samples from normal
individuals who did not possess a family history of cleft lip or
palate or other immune organic diseases were collected between
January and March 2014; these individuals were recruited from the
Department of Clinical Laboratory, Handan Central Hospital (Hebei,
China). The genetic and patient information of probands were
provided by their parents. The present study was conducted in
accordance with the declaration of Helsinki and with approval from
the Ethics Committee of Handan Central Hospital. Written informed
consent was obtained from all participants' guardians and the 200
normal individuals.
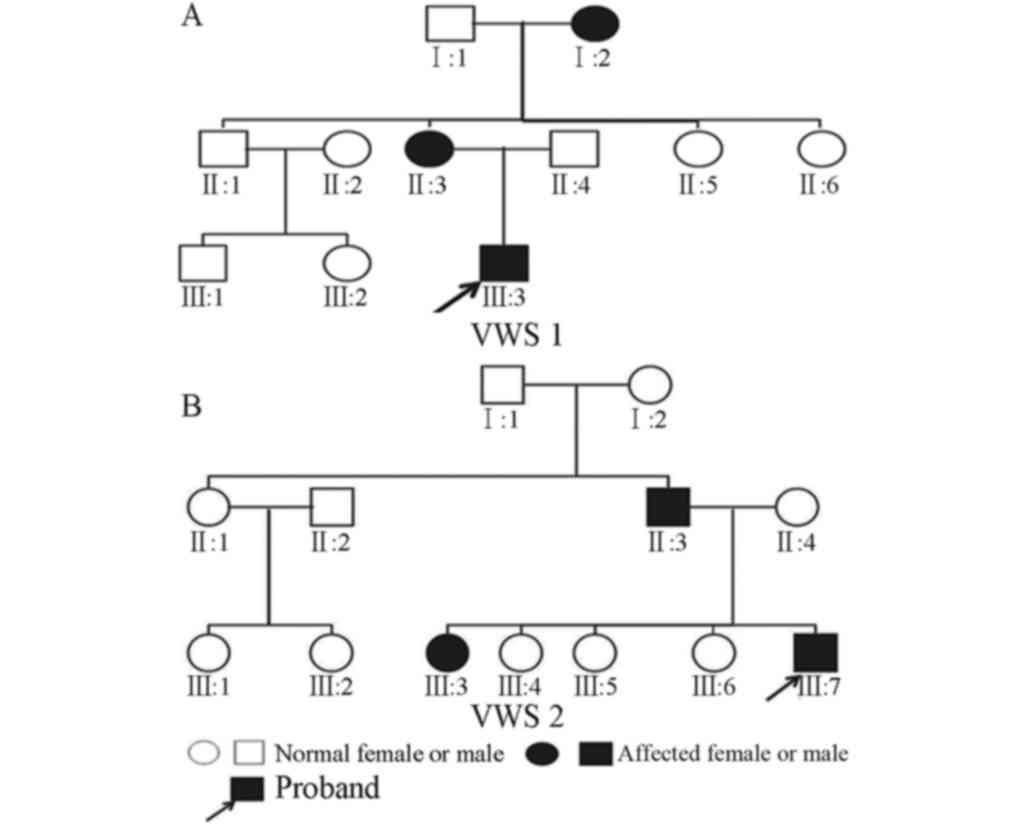
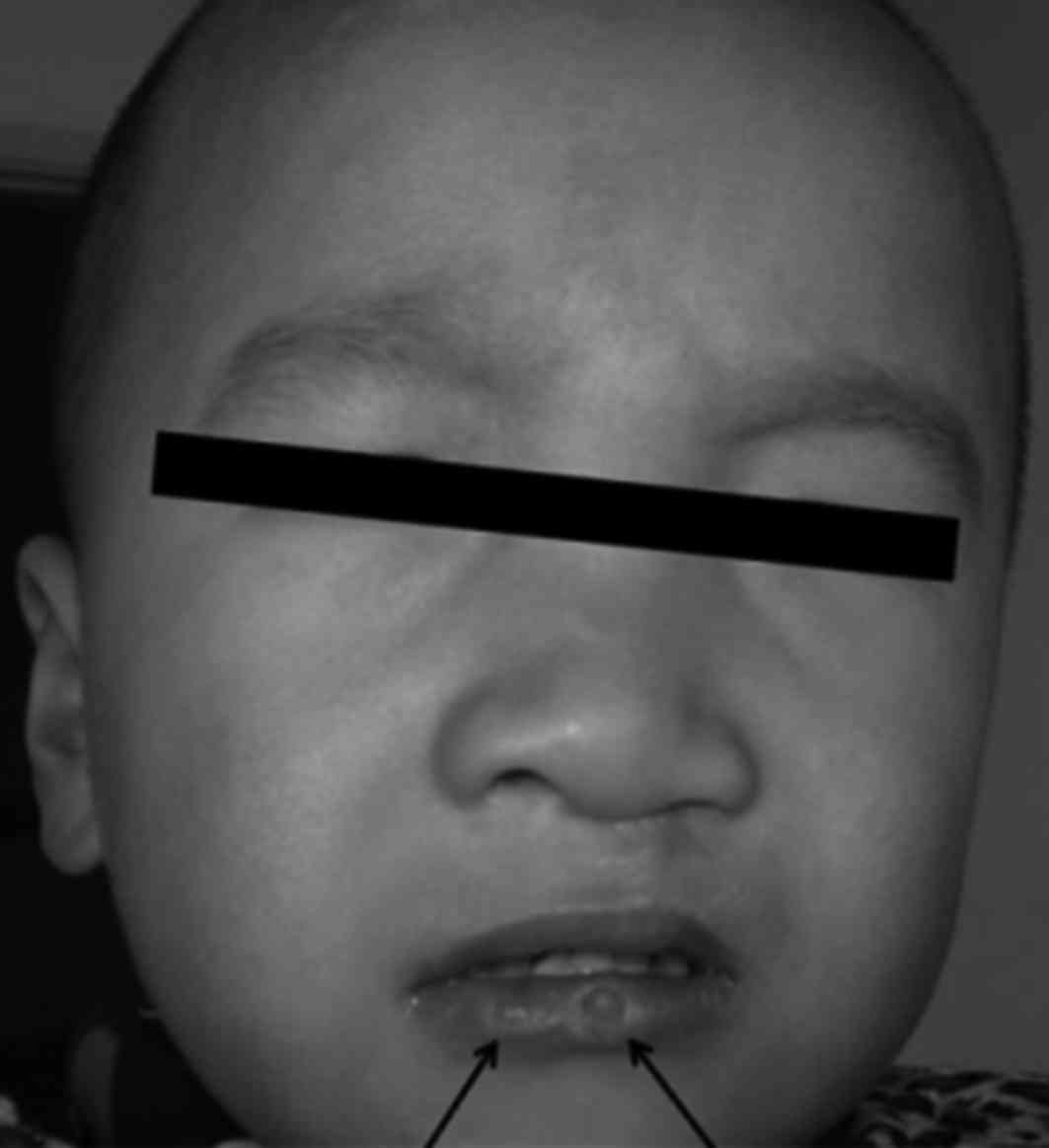
Family 1 (VWS1): Proband A (III:3; Figs. 1A and 2), male, 2 years old, had congenital
cleft palate and bilateral labium fistula. His mother did not
experience a cold or a fever or use drugs during pregnancy. There
were 3 generations and 11 members in this family. The VWS patients
in this family were 3 in total, and distributed in each generation.
Pedigree analysis was compatible with the law of autosomal dominant
inheritance. Through detailed examination by maxillofacial
surgeons, congenital malformations, and cardiovascular,
cerebrovascular and mental diseases were excluded for the patients
in this family. Although the patients in this family expressed
typical symptoms of VWS, the individual phenotype in each
generation was different. The symptoms of the patient in the first
generation (I:2) were characterized by simple red small pits on the
lower lip, not easy to distinguish from bite marks. The patients in
the second (II:3) and third (III:3) generations demonstrated
serious cleft lip and palate deformities and lower lip fistula.
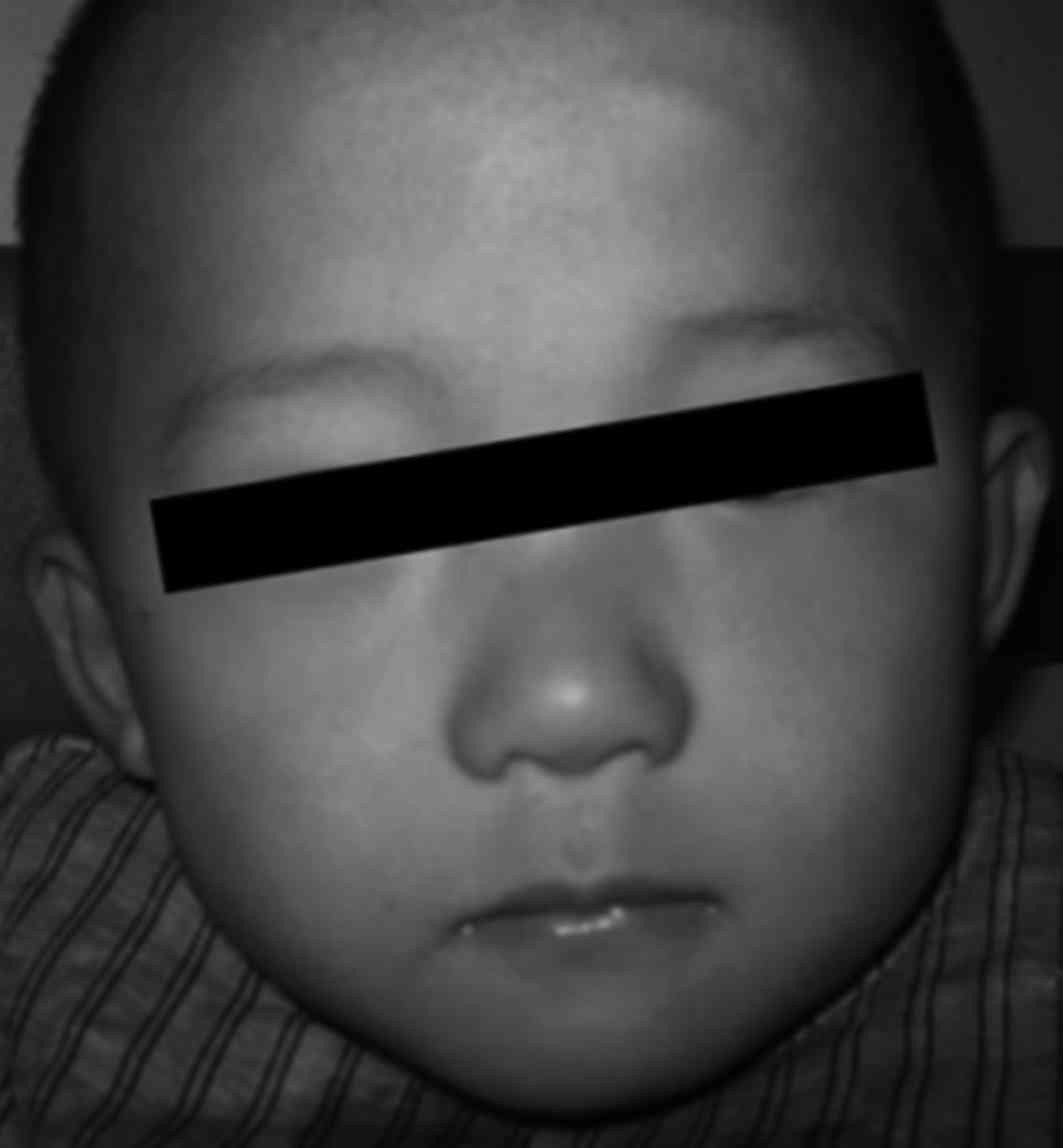
Family 2 (VWS2): Proband B (III:7; Figs. 1B and 3), male, 3 years old, had congenital
cleft soft palate. His mother did not experience a cold or a fever
or use drugs during pregnancy. There were 3 generations and 13
members in this family and congenital malformations, and
cardiovascular, cerebrovascular and mental diseases were excluded.
The VWS patients in this family were 3 in total, and appeared in
the second and third generation. Pedigree analysis was also
compatible with law of autosomal dominant inheritance. Through
detailed examination by maxillofacial surgeons, the symptoms of VWS
patients in the second generation were characterized by bite marks
in the red defects of the lower lip and occult cleft palate. The
VWS patients in the third generation were siblings. The elder
sister (III:3) demonstrated cleft palate and lower lip fistula. The
young brother was the proband and demonstrated cleft soft
palate.
Medical history collection
The genetic and patient information of probands were
filled in registration forms by their parents. The information in
the hereditary information registration form included general
information concerning the patients, pregnancy risk factors,
pregnancy situation, family history, the VWS patient situation and
laboratory tests. The information in the patient information forms
included the patients' general information, general situation,
history of mother's pregnancy (including history of infection, drug
administration and childbirth), physical examination (including
systemic physical examination and professional oral medical
examination) and various clinical data. All family members from
whom peripheral blood samples were collected provided written
informed consent. The family members who agreed to donate blood
were provided with a medical examination and 5 ml of peripheral
venous blood was extracted for subsequent testing.
Genomic DNA extraction
The genomic DNA was extracted by AxyPrep blood
genomic DNA extraction kit (Axygen Scientific, Inc., Union City,
CA, USA) in strict accordance with the manufacturer's protocol. DNA
concentration and purity were detected by NanoDrop 1000
determination (Thermo Fisher Scientific, Inc., Wilmington, DE,
USA). When the extraction process was complete, all DNA samples
were diluted to 10 ng/µl in 96 well plates and stored at −20°C.
Gene sequencing
The amplification primer sequences of IRF6 were
designed by online software Primer 3 version 3.0.0 (www.biotools.umassmed.edu/bioapps/primer3_www.cgi).
The amplified fragment was the exons between 3 and 9, since exons 1
and 2 were encoding fragments. Polymerase chain reaction (PCR)
primer sequences and amplified fragment length are listed in
Table I. Primers were synthesized
by Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
The PCR reaction conditions were: Pre-denaturation at 94°C for 5
min, denaturation at 94°C for 30 sec, annealing at 6°C for 30 sec,
extension at 72°C for 35 sec, 35 cycles in total; extension at 72°C
for 5 min. The PCR product was subjected to electrophoresis on 1.5%
agarose gel and the results were analyzed by a gel imaging and
analysis system (Gel Doc™ EZ; Bio-Rad Laboratories,
Inc., Hercules, CA, USA). PCR products were sequenced by an ABI
3730 (Applied Biosystems; Thermo Fisher Scientific, Inc.), and the
mutation positive samples were sequence reversed for verification.
The sequencing results were compared with the standard IRF6 genetic
sequence on National Center for Biotechnology Information
(https://www.ncbi.nlm.nih.gov; NM_006147)
by Mutation Surveyor software version 4.0 (SoftGenetics, LLC.,
State College, PA, USA).
 | Table I.Primer sequences and fragment lengths
for IRF6 gene in polymerase chain reaction. |
Table I.
Primer sequences and fragment lengths
for IRF6 gene in polymerase chain reaction.
| Primer name | Primer sequences
(5′-3′) | Fragment length
(bp) |
|---|
| IRF6-3F |
CCACCTGGCACAGCTTATTC | 492 |
| IRF6-3R |
AGACATGCCCCCAAAAGAG |
|
| IRF6-4F |
TCTCTGTAAATCGGGGTTGG | 527 |
| IRF6-4R |
TTTGTTTTCTGGGTCCTTCC |
|
| IRF6-5F |
GGAGGTCCTTCCATGAGAGA | 496 |
| IRF6-5R |
GGGATAACTGAGCCAACAGG |
|
| IRF6-6F |
GGAGCAGGGGAACCTTATTT | 500 |
| IRF6-6R |
CCTGGTGACTCATGGGCTA |
|
| IRF6-7F |
CTATGGCAGTGGCCTTCCT | 700 |
| IRF6-7R |
GTTGCCTAGGTCACCTCCAA |
|
| IRF6-8F |
TGACTAATGTGACCCAGGAACT | 430 |
| IRF6-8R |
AGCATGAGTCCTACTGCACAAA |
|
| IRF6-9F |
TCATTTGAACCAACTTCCACAG | 600 |
| IRF6-9R |
AGTCTGAAGGGTGATTTTTCCA |
|
Results
Clinical examination results
There were 6 VWS patients in the two families that
comprised 24 individuals in total. Of the 6 patients, 5 (83.3%)
presented cleft lip and palate, 5 (83.3%) presented labial fistula
and 1 (III:7) in the second family was without labial fistula.
These VWS patients presented no teeth dysplasia or other organ
deformity, although the phenotypic lip fistula with cleft lip was
common. Although the patients belonged to the same family, their
clinical phenotype differed: 3 patients in the first family all had
lower lip fistula or labium fistula with cleft lip and palate,
patient I:2 was the only one exhibiting bilateral labium fistula,
patient II:3 demonstrated degree II right cleft palate and patient
III:3 demonstrated degree III left cleft palate. Two of the total 3
patients in the second family had lower lip bite marks. Patient
II:3 presented lower lip bite marks on the sides and cracked
palate, patient III:3 was characterized by degree II left cleft
palate for and lower lip bite marks, and patient III:7 demonstrated
soft cleft palate alone.
Genetic testing results
The gene sequencing of IFR6 between exons 3 and 9
was conducted on 14 members of the 2 families. The results
demonstrated that the 6 VWS patients all had genetic mutations. The
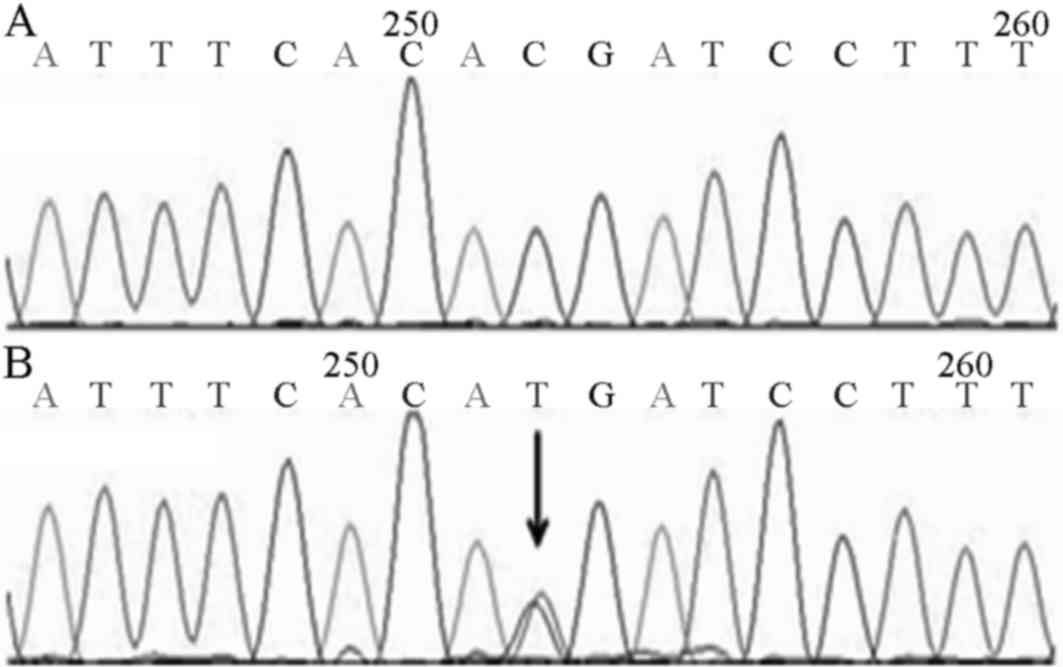
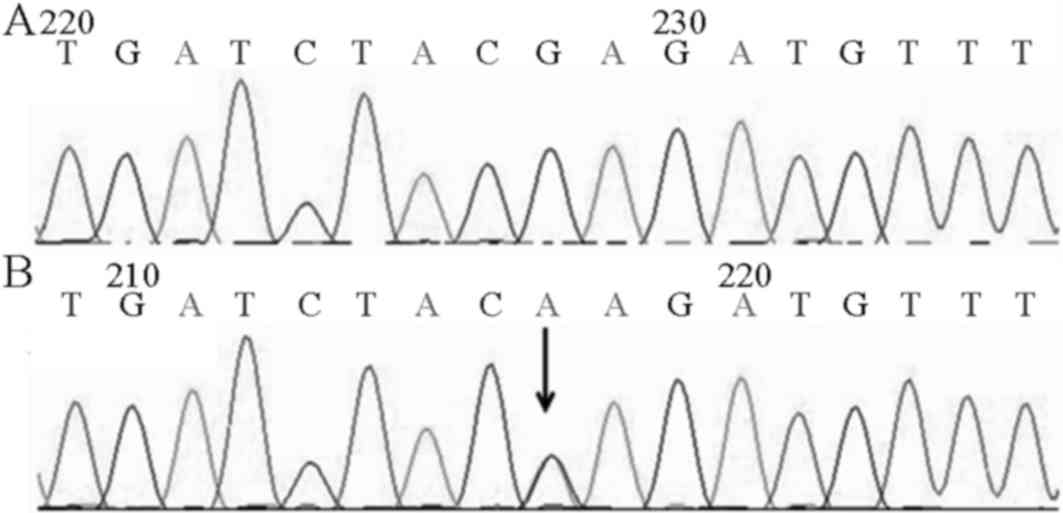
3 patients in the first family all carried c.1234C>T (p.R412X)
heterozygous mutation on exon 9 of the IRF6 gene (Fig. 4). In family 2, 3 patients all
carried c.1210G>A (p.E404K) heterozygous mutation on exon 9 of
the IRF6 gene (Fig. 5). In
addition, II:3 and III:3 in family 2 carried the rs2235373 single
nucleotide polymorphism (SNP) on exon 7 of the IRF6 gene. Other
family members who were not VWS patients did not carry IRR6 gene
mutations. Genetic testing results demonstrated that the mutation
and disease exhibited co-segregation.
Normal test results
In order to rule out that the two mutation sites
detected in the two families were not SNPs in local people, the
exon 9 gene sequencing of IFR6 was conducted on 200 local normal
individuals and the c.1234C>T and c.1210G>A mutations were
not observed.
Discussion
VWS, an autosomal dominant inherited clefting
syndrome first described by Demarquay in 1845, is a rare congenital
syndrome (8). The characteristics
of the phenotype are labium fistula with cleft lip and palate. In
VWS, the formation of congenital lip pits is associated with the
occurrence of labium fistula with cleft lip and palate; this is a
common clinical problem that occurs in 80% of patients with VWS
(9). Therefore, certain patients
with VWS were characterized only by simple cleft lip or cleft
palate, and this made VWS and NSCL/P indistinguishable from the
outward appearance of patients. However, for the congenital anomaly
of epidemiology and etiology, accurate phenotyping is important. If
the heterogeneous group is regarded as a single group, the effect
of detection may be weakened (10). Consequently, for the diagnosis of
VWS, a detailed survey of the patient's family and clinical
phenotyping is required.
With the development of molecular biology and the
completion of the human genome map, research has made progress
regarding the disease genes associated with VWS. In 1954, Van der
Woude first described the clinical manifestation of VWS and
discussed its hereditary pattern (11). Then, in 1987, Bocian and Walker
(12) first reported that these
patients were missing chromosome lq32-q41. In 2002, Kondo et
al (2) analyzed a large VWS
family: No matter what the phenotype of VWS family members, there
was an 18 bp fragment missing in the chromosome lq32-q41 areas of
the IRF6 genes of the affected individual. Kondo et al
(2) successfully cloned the VWS
IRF6 disease-causing genes and similar results have also been
identified in other VWS families. Therefore, the study demonstrated
that VWS had significant phenotypic differences. Certain reports
have confirmed that the IRF6 gene is one of the pathogenic genes
for VWS (13,14). The present study demonstrated that
IRF6 is important in jaw development and fusion. IRF6 gene
expression analysis demonstrated that the gene is not only widely
expressed in embryonic and adult mice, it is also expressed in
adult and fetal tissue. There was a wide range of expression in the
secondary cleft palate in 14.5–15 day mouse embryos and adult skin
tissue (2).
The IRF6 gene possesses a total length of 2,171 bp,
comprising 10 exons. Exon 1, 2 and 10 are non-coding regions. The
coding regions of IRF6 gene, a total of 1,404 bp, encodes 467 amino
acids (2). It is a member of the
interferon regulatory factor family and this gene family is
characterized by a highly-conserved helix-turn-helix DNA binding
domain and a protein binding domain termed Smad-interferon
regulatory domain (SMIR) (15).
The DNA binding domain mediates the adhesion between IRF6 and the
target sequence, while the SMIR domain is necessary to form
homologous dimers or heterologous dimers, to translocate dimers to
the nucleus, along with other transcription factors in target genes
(16). The SMIR domain mediates
the morphological development of the mouth-face region through
transforming growth factor-β signal transduction pathways. At
present, the majority of the mutations are in these two domains.
According to The Human Gene Mutation Database statistics
(www.hgmd.cf.ac.uk/ac/index.php), as of September 2012
there are a total of 219 IRF6 gene mutations, including 191 IRF6
gene mutations identified in patients with VWS. These mutations
include missense/nonsense, control area, frameshift and various
other different forms of mutations, however the majority appear to
be missense/nonsense mutations.
The VWS families in the present study exhibited two
mutations, which were located on exon 9 in the coding region of the
IRF6 gene. In the first VWS family, c.1234C>T resulted in a
truncation mutation, and the 412th amino acid appeared as an early
termination codon (p.R412X). The first generation of VWS family
members with this mutation were generally characterized by concave
lower lip or fistula; the second and third generations demonstrated
serious cleft palate. In 2009, this mutation was reported in a
European VWS family (17). The
emergence of these nonsense mutations resulted in partial IRF6 gene
encoding protein and thus loss of normal function: IRF6 decreased,
leading to developmental disorders in the maxillofacial development
process that affected the lips and/or jaw. In the present study,
the second VWS family carried the c.1210G>A mutation, an
alteration in the 404th amino acid which replaced glutamic acid
with lysine (p.E404K). The first-generation members of this VWS
family did not carry the IRF6 mutations and were not VWS cases;
II:3 may therefore be an instance of a novel mutation.
In 2009, de Lima (18) reported this genetic mutation in
VWS. However, the pathogenic mechanism of the mutation remains to
be elucidated. In the present study, the three websites consulted
(www.genetics.bwh.harvard.edu,
www.sift.jcvi.org and www.pantherdb.org) all suggested that the mutations
may result in damaging effects on the IRF6 protein. It was
hypothesized that as the mutation was only 10 amino acids from the
downstream SMIR function structure domain, the mutation may lead to
an alteration in the nature of the amino acids, thus affecting the
function of the protein. Two mutations in line with the White
family (17) were detected in the
mutation. In order to confirm if these two mutations are VWS hot
spot mutations, further samples and analyses are required.
The two mutations identified are VWS heterozygous
mutations, resulting in an insufficient haploid dose of the IRF6
gene, affecting the maxillofacial growth of the patients. However,
it was also identified that even the same mutations present in
patients from the same family exhibited different clinical
phenotypes. The propositus of the second VWS Family (III:7) was
only characterized by simple cleft palate, whereas the other two
patients in the family were characterized by lip and cleft palate
with fistula phenotype. In the same family, syndrome and
non-syndrome occurred in patients with cleft lip and palate cleft
lip, which is relatively rare. Therefore, the IRF6 gene sequencing
results for the 3 patients of the second VWS family were analyzed.
The results demonstrated that II:3 and III:3 exhibited lip fistula
and carried SNP loci (rs2235373), and the propositus simply carried
p.E404k heterozygous mutations. However, the rs2235373 polymorphic
loci was located in the IRF6 gene downstream of the exon 7 of 135
bp, so its effect on IRF6 encoding protein requires further
studies. Currently, there are no reports that these mutations
affect IRF6 protein production, therefore further studies are
required. Salahshourifar (19)
summarized the gene mutation and IRF6 introns and exons of mutation
loci in different VWS phenotypes. VWS patients with cleft palate II
phenotype carried rs2235373 and exon 7 p.V274I loci. These
phenomena illustrated that rs2235373 may be associated with the VWS
phenotype loci. The present study also identified a phenotypic
generational aggravating phenomenon in the first VWS family, a
phenomenon that has occurred in other similar studies in the
literature (20–22). It has additionally been suggested
that there may be other modifiers in virulent VWS genes (23). Due to the diversity of clinical
phenotypes in VWS patients, even individuals with a family genetic
disposition, may be misdiagnosed by antenatal ultrasound
examination; however, genetic testing may help to remedy these
shortcomings.
Recently there have been a large number of studies
on the VWS IRF6 gene in patients with novel mutations (17,18,23,24).
From these studies, it may be suggested that the majority of
mutations associated with VWS are point mutations, although splice
site mutations resulted in a more intense clinical phenotype
(17,19,25).
The majority of these reports were from Asian patients and
consistent with the previously identified higher percentage
incidence of cleft palate among them. Tan et al (25) identified that the 396C-T mutation
in the IRF6 gene fragment may be a IRF6 pathogenic factor. In
addition, the study suggests that the IRF6 gene mutation
distribution in VWS patients is not random and the majority of
mutations are located on exons 3, 4, 7 and 9, with ~1/5 of the
mutations present on exon 9 (18–20,23,24,26).
The pathogenic mutation research for VWS has been performed with a
relatively small Chinese cohort. The present study collected data
from two VWS families and identified that the pathogenic mutations
were all located on exon 9. Therefore, the exon 9 may be a hot spot
mutation in VWS occurring in Chinese patients. It will be necessary
to expand the sample size in the future in order to conduct
in-depth research.
In conclusion, the accumulation of these clinical
data and the study of pathogenic mutations may prove useful for the
differential diagnosis of VWS and NSCL/P. In addition, the results
of the present study may identify potential therapeutic targets.
They also provide the basis and support for genetic counseling and
prenatal diagnosis, and the prevention of cleft lip and palate with
gene therapy. With the advent of the genome, improved pathogenic
mutation research into the syndrome of cleft lip and palate may be
achieved, and the association between VWS and NSCL/P analyzed and
clarified in the Chinese population. It is hypothesized that
genetic research regarding NSCL/P and study of the underlying
molecular mechanism of the occurrence of cleft lip and palate, will
make greater progress in the future. The authors believe that the
study of mutations in this syndrome will aid therapeutic research
and improve the quality of life in patients with VWS.
Acknowledgements
The present study was funded by the Science and
Technology Project of Hebei Province (grant no. 1427781D) and the
Science and Technology Research and Development Program of Handan
(grant no. 1223108085).
References
|
1
|
Schutte BC, Bjork BC, Coppage KB, Malik
MI, Gregory SG, Scott DJ, Brentzell LM, Watanabe Y, Dixon MJ and
Murray JC: A preliminary gene map for the Van der Woude syndrome
critical region derived from 900 kb of genomic sequence at
1q32-q41. Genome Res. 10:81–94. 2000.PubMed/NCBI
|
|
2
|
Kondo S, Schutte BC, Richardson RJ, Bjork
BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S,
Sander A, et al: Mutations in IRF6 cause Van der Woude and
popliteal pterygium syndromes. Nat Genet. 32:285–289. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burdick AB: Genetic epidemiology and
control of genetic expression in van der Woude syndrome. J
Craniofac Genet Dev Biol Suppl. 2:99–105. 1986.PubMed/NCBI
|
|
4
|
Pingul MM and Quintos JB: Van der woude
syndrome (lip pit-cleft lip syndrome). J Pediatr. 164:12352014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peyrard-Janvid M, Pegelow M, Koillinen H,
Larsson C, Fransson I, Rautio J, Hukki J, Larson O, Karsten AL and
Kere J: Novel and de novo mutations of the IRF6 gene detected in
patients with Van der Woude or popliteal pterygium syndrome. Eur J
Hum Genet. 13:1261–1267. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mijiti A, Ling W, Guli and Moming A:
Association of single-nucleotide polymorphisms in the IRF6 gene
with non-syndromic cleft lip with or without cleft palatein the
Xinjiang Uyghur population. Br J Oral Maxillofac Surg. 53:268–274.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
XQ Ye, Zhou B and Jin H: Clinical and
genetic features in five chinese kindreds with Van der Woude
syndrome. Med J Wuhan University. 26:609–611. 2005.
|
|
8
|
Burdick AB, Bixler D and Puckett CL:
Genetic analysis in families with van der Woude syndrome. J
Craniofac Genet Dev Biol. 5:181–208. 1985.PubMed/NCBI
|
|
9
|
Hersh JH and Verdi GD: Natal teeth in
monozygotic twins with Van der Woude syndrome. Cleft Palate
Craniofac J. 29:279–281. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dixon MJ, Marazita ML, Beaty TH and Murray
JC: Cleft lip and palate: Understanding genetic and environmental
influences. Nat Rev Genet. 12:167–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Der Woude A: Fistula labii inferioris
congenita and its association with cleft lip and palate. Am J Hum
Genet. 6:244–256. 1954.PubMed/NCBI
|
|
12
|
Bocian M and Walker AP: Lip pits and
deletion 1q32-41. Am J Med Genet. 26:437–443. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou Q, Li M, Zhu W, Guo J, Wang Y, Li Y
and Li S: Association between interferon regulatory factor 6 gene
polymorphisms and nonsyndromic cleft lip with or without cleft
palate in a Chinese population. Cleft Palate Craniofac J.
50:570–576. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leslie EJ, Standley J, Compton J, Bale S,
Schutte BC and Murray JC: Comparative analysis of IRF6 variants in
families with Van der Woude syndrome and popliteal pterygium
syndrome using public whole-exome databases. Genet Med. 15:338–344.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eroshkin A and Mushegian A: Conserved
transactivation domain shared by interferon regulatory factors and
Smad morphogens. J Mol Med (Berl). 77:403–405. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mamane Y, Heylbroeck C, Génin P, Algarté
M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R and Hiscott J:
Interferon regulatory factors: The next generation. Gene. 237:1–14.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Houweling AC, Gille JJ, Baart JA, van
Hagen JM and Lachmeijer AM: Variable phenotypic manifestation of
IRF6 mutations in the Van der Woude syndrome and popliteal
pterygium syndrome: Implications for genetic counseling. Clin
Dysmorphol. 18:225–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Lima RL, Hoper SA, Ghassibe M, Cooper
ME, Rorick NK, Kondo S, Katz L, Marazita ML, Compton J, Bale S, et
al: Prevalence and nonrandom distribution of exonic mutations in
interferon regulatory factor 6 in 307 families with Van der Woude
syndrome and 37 families with popliteal pterygium syndrome. Genet
Med. 11:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Salahshourifar I, Sulaiman Wan WA, Halim
AS and Zilfalil BA: Mutation screening of IRF6 among families with
non-syndromic oral clefts and identification of two novel variants:
Review of the literature. Eur J Med Genet. 55:389–393. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Soekarman D, Cobben JM, Vogels A, Spauwen
PH and Fryns JP: Variable expression of the popliteal pterygium
syndrome in two 3-generation families. Clin Genet. 47:169–174.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marazita ML, Lidral AC, Murray JC, Field
LL, Maher BS, Goldstein McHenry T, Cooper ME, Govil M, Daack-Hirsch
S, Riley B, et al: Genome scan, fine-maping, and candidate gene
analysis of non-syndomic cleft lip with or without cleft palate
reveals phenotype-sppecific diferences in linkage and association
results. Hum Hered. 68:151–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jugessur A, Shi M, Gjessing HK, Lie RT,
Wilcox AJ, Weinberg CR, Christensen K, Boyles AL, Daack-Hirsch S,
Nguyen TT, et al: Fetal genetic risk of isolated cleft lip only
(CLO) versus isolated cleft lip and palate (CLP): A sub-phenotype
analysis usingtwo population-based studies of orofacial clefts in
Scandinavia. Birth Defects Res A Clin Mol Teratol. 91:85–92. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de la Garza G, Schleiffarth JR, Dunnwald
M, Mankad A, Weirather JL, Bonde G, Butcher S, Mansour TA, Kousa
YA, Fukazawa CF, et al: Interferon regulatory factor6 promotes
differentiation of the peridermby activating expression of
Grainyhead-like 3. J Invest Dermatol. 133:68–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peyrard-Janvid M, Leslie EJ, Kousa YA,
Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson
I, Koillinen HK, et al: Dominant mutations in GRHL3 cause Van der
Woude Syndrome and disrupt oral periderm development. Am J Hum
Genet. 94:23–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan EC, Lim EC, Yap SH, Lee ST, Cheng J,
Por YC and Yeow V: Identification of IRF6 gene variants in three
families with Van der Woude syndrome. Int J Mol Med. 21:747–751.
2008.PubMed/NCBI
|
|
26
|
Morton NE: Sequential tests for the
detection of linkage. Am J Hum Genet. 7:277–318. 1955.PubMed/NCBI
|