Introduction
Chronic heart failure (CHF) occurs as a result of a
number of cardiovascular diseases and is a complex
pathophysiological process; neurohormonal disorders and ventricular
remodeling are important pathophysiological changes for patients
with CHF (1). A large dose (>85
mg/kg) of isoproterenol (ISO) may cause diffuse myocardial necrosis
and fibrosis, which may gradually develop into dilated
cardiomyopathy heart failure (2).
ISO may cause myocardial necrosis similar to myocardial infarction,
but it maintains effective coronary circulation (3). This necrosis depends on dose and
time, and ranges between endocardial focal necrosis and transmural
necrosis. There are multiple factors underlying the induction of
myocardial necrosis by ISO, primarily associated with its cardiac
toxicity, and a previous study demonstrated that the following
factors are relevant: Relative hypoxia; microcirculation;
permeability alterations in myocardial cell membranes; overload of
Ca2+; toxic effects of ISO oxidation products; and
myocardial ischemia-reperfusion injury (3).
CHF is the end result of the majority of
cardiovascular diseases. Despite progress in treatment methods and
an improved prognosis for CHF during the past two decades, the
overall morbidity, mortality and readmission rates for heart
failure remain high (4). At
present, clinicians recognize that heart failure is an inflammatory
reaction, and increasing evidence has demonstrated that
inflammation serves an important role in the development of heart
failure (5). Previous reports have
indicated that pro-inflammatory cytokines are frequently
overexpressed in patients with CHF, including C-reactive protein,
tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6 and
monocyte chemoattractant protein-1, which induce myocardial
apoptosis and fibrosis, result in cardiac remodeling, and promote
the development of CHF; these effects are positively correlated
with the severity of heart failure, indicating a poor prognosis for
patients with CHF (6,7). Therefore, anti-inflammatory treatment
for heart failure may represent a novel approach.
A recent study demonstrated that for CHF, in
addition to enhanced sympathetic excitability and abnormal
secretion of various humoral factors, overexpression of
inflammatory cytokines and imbalances within the immune system are
important aspects of its complex pathophysiology (8). The abnormal inflammatory response
mediated by pro-inflammatory cytokines is associated with left
ventricular remodeling, left ventricular failure, endothelial
injury, myocardial apoptosis of endothelial cells and cachexia in
patients with CHF, and promotes the development of heart failure
(9). Immune system disorders are
associated with myocardial cell death, fibrosis, systolic
dysfunction and deterioration, and heart failure severity (10).
The phosphatidylinositol 3-kinase (PI3K)/RAC-α
serine/threonine protein kinase (Akt) signaling pathway is widely
present in mammalian cells and serves a complex role (11). When CHF occurs, PI3K/Akt may
achieve a cardioprotective effect by regulating downstream target
genes (12).
Mitogen-activated protein kinase (MAPK) belongs to
the family of serine/threonine protein kinases, which may be
activated by certain ligands, including receptors, growth factors,
G-protein-coupled receptors and certain stressors (13). p38 MAPK belongs to the same MAPK
system as extracellular signal-regulated kinases 1/2 and 5, and
c-Jun N-terminal kinase (JNK), and may be activated by stressors.
JNK, p38 MAPK and extracellular signal-regulated kinase 5 are
expressed in the human heart (14). The activities of JNK and p38 MAPK
are markedly increased in CHF with ischemic cardiomyopathy
(14). p38 MAPK are selectively
expressed in myocardial cells of mice with myocardial infarction
(15).
Ulinastatin is a type of glycoprotein, isolated and
purified from the fresh urine of healthy adult males, which acts as
a broad-spectrum enzyme inhibitor and may block the release of
inflammatory cytokines, prevent the initiation of the cytokine
cascade, suppress excessive activation of leukocytes, and block
cycle of feedback activation among cytokines, inflammatory
mediators and leukocytes (16).
Previous studies have demonstrated, regarding the
anti-inflammatory, immunomodulatory and visceral protective effects
of ulinastatin, that ulinastatin exhibits a cellular protective
function in ischemia-reperfusion injury in the liver, kidney, heart
and lung, and improves immune function (16–18).
The present study examined the hypothesis that the cardioprotective
effect of ulinastatin may prevent ISO-induced CHF, and aimed to
elucidate the possible mechanism.
Materials and methods
Subjects, grouping and the CHF
model
Male Sprague-Dawley rats (6–8 weeks old) weighing
200–220 g were purchased from Beijing Vital River Laboratory Animal
Technology Co., Ltd. (Beijing, China) and caged individually at
controlled temperature (22–23°C) and humidity (45–55%) with a
12-hour light/dark cycle and free access to food, and water. All
procedures were performed in accordance with the Guide for the Care
and Use of Laboratory Animals and were approved by the medical
Ethics Committee of the First Affiliated Hospital of PLA General
Hospital (Beijing, China). A total of 30 male Sprague Dawley rats
were randomly assigned to three groups: Control group (n=6),
ISO-induced CHF model (n=20), and ulinastatin group (n=20). In the
control group, rats were intraperitoneally injected with normal
saline. In the ISO-induced CHF model group, rats were
intraperitoneally injected with 5 mg/kg/day isoproterenol
hydrochloride (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 5
days, and subsequently with normal saline for 5 days. In the
ulinastatin group, ISO-induced CHF model rats were pretreated with
a one off dose of 2,500 IU/kg ulinastatin (Sigma-Aldrich; Merck
KGaA) for 1 week, followed by treatment with 5 mg/kg/day
isoproterenol hydrochloride (Sigma-Aldrich; Merck KGaA) for 5 days,
and subsequently with normal saline for 5 days.
Echocardiographic assessment of heart
function
Rats were anesthetized with 30 mg/kg pentobarbital
sodium and an ultrasound probe was placed in the left sternal
border. Interventricular septal thickness (IVS) and left
ventricular posterior wall thickness (LVPW), the left ventricular
ejection fraction (LVEF), left ventricular fractional shortening
(LVFS) and peak E to peak A ratio were obtained using the long axis
of left ventricle and the maximum diameter, and calculated using
ECToolbox™ for Xeleris™ version 2 software (GE Healthcare, Chicago,
IL, USA).
Measurements
Rats were anesthetized with 30 mg/kg pentobarbital
sodium and sacrificed by decollation. Peripheral blood was
collected and serum was absorbed following centrifugation at 5,000
× g for 10 min at 4°C. Subsequently, serum was used to measure
NF-κB (cat no. H202; Nanjing Jiancheng Biology Engineering
Institute, Nanjing, China), TNF-α (cat no. PT51), IL-1β (cat no.
PI303), IL-6 (cat no. PI328), glutathione peroxidase (GSH-PX;
A005), glutathione (GSH; cat no. A005), superoxide dismutase (SOD;
cat no. A001-3), malondialdehyde (MDA; cat no. A003-1), caspase-3
(cat no. C1115) and caspase-9 (cat no. C1157) (all from Beyotime
Institute of Biotechnology, Haimen, China) using commercial kits.
The optical density (OD) of NF-κB, TNF-α, IL-1β, IL-6, GSH-PX, GSH,
SOD and MDA was measured using an ELX-800 microplate assay reader
(BioTek Instruments, Inc., Winooski, VT, USA) at 450 nm. The OD of
caspase-3 and caspase-9 was measured using the ELX-800 microplate
assay reader at 405 nm.
Western blotting
Rats were anesthetized with 30 mg/kg pentobarbital
sodium and sacrificed by decollation. Heart tissue samples were
collected and washed with PBS. The frozen myocardial tissues were
homogenized in tissue lysis buffer (radioimmunoprecipitation
buffer; Beyotime Institute of Biotechnology) on ice for 20 min. The
supernatant was collected following centrifugation at 5,000 × g for
10 min at 4°C and protein concentration was determined using a
bicinchoninic acid kit (Beyotime Institute of Biotechnology).
Protein samples (50–80 µg) were subjected to SDS-PAGE on 6–10% gels
and transferred to a nitrocellulose membrane (EMD Millipore,
Billerica, MA, USA). The membrane was blocked with 5% skim milk
powder in Tris-buffered saline with Tween-20 (TBST) for 1 h at 37°C
and probed with the following primary antibodies: Anti-p65 (cat no.
8242; 1:2,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-apoptosis regulator Bcl-2 (Bcl-2; cat no. sc-783; 1:500; Santa
Cruz Biotechnology, Inc., Dallas, TZ, USA), anti-apoptosis
regulator BAX (Bax; cat no. 2772; 1:5,000), anti-Akt (cat no. 4685;
1:2,000), anti-phosphorylated (p)-Akt (cat no. 4060; 1:2,000),
anti-p-p38 (cat no. 4511; 1:2,000) and anti-GAPDH (cat no. 5174)
(all from Cell Signaling Technology, Inc.), at 4°C overnight. The
membranes were washed with TBST and incubated with anti-rabbit
horseradish peroxidase-conjugated secondary antibodies (cat no.
sc-2030; 1:5,000; Santa Cruz Biotechnology, Inc.) for 1 h at 37°C.
Protein bands were visualized using the Enhanced Chemiluminescence
Plus western blotting detection system (PerkinElmer, Inc., Waltham,
MA, USA) and quantified using Image Studio version 1.1 software
(LI-COR Biosciences, Lincoln, NE, USA).
Statistical analysis
Quantitative data are expressed as the mean ±
standard error of the mean (n=3) using SPSS (version 17.0; SPSS,
Inc., Chicago, IL, USA). Data were analyzed using one-way analysis
of variance followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cardioprotective effect of ulinastatin
against alterations in cardiac structure
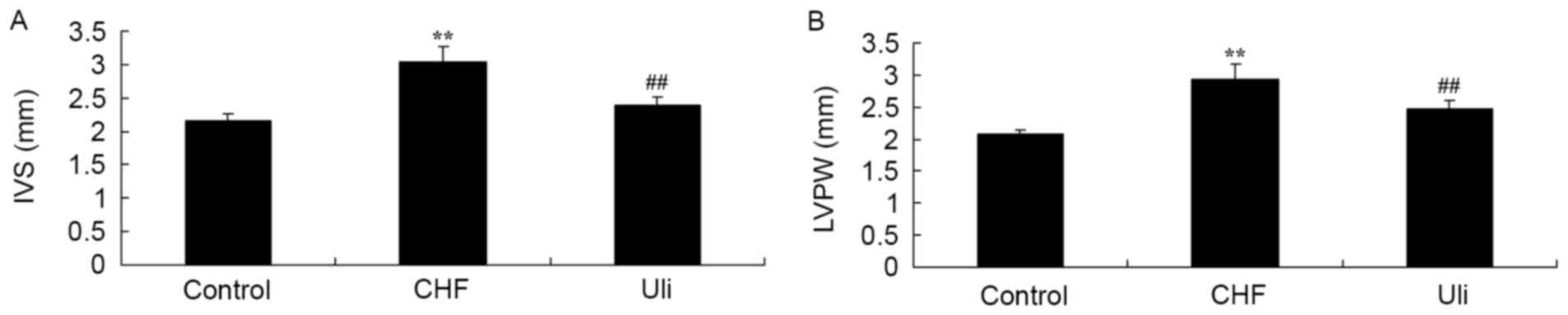
At week 5, the IVS and LVPW of the ISO-induced CHF
model animals were increased compared with those of the control
group (Fig. 1). Treatment with
Ulinastatin prevented IVS and LVPW in ISO-induced CHF rat, compared
with the ISO-induced CHF model rat group (Fig. 1).
Cardioprotective effect of ulinastatin
on cardiac function
At week 5, the data presented in Fig. 2 revealed significant inhibitions of
LVEF, LVFS and peak E/A ratio in the ISO-induced CHF model rat,
compared with the control group. Treatment with ulinastatin
significantly increased LVEF, LVFS and peak E/A ratio in the
ISO-induced CHF rat, compared with ISO-induced CHF model rat group
(Fig. 2).
Cardioprotective effect of ulinastatin
against inflammatory factors
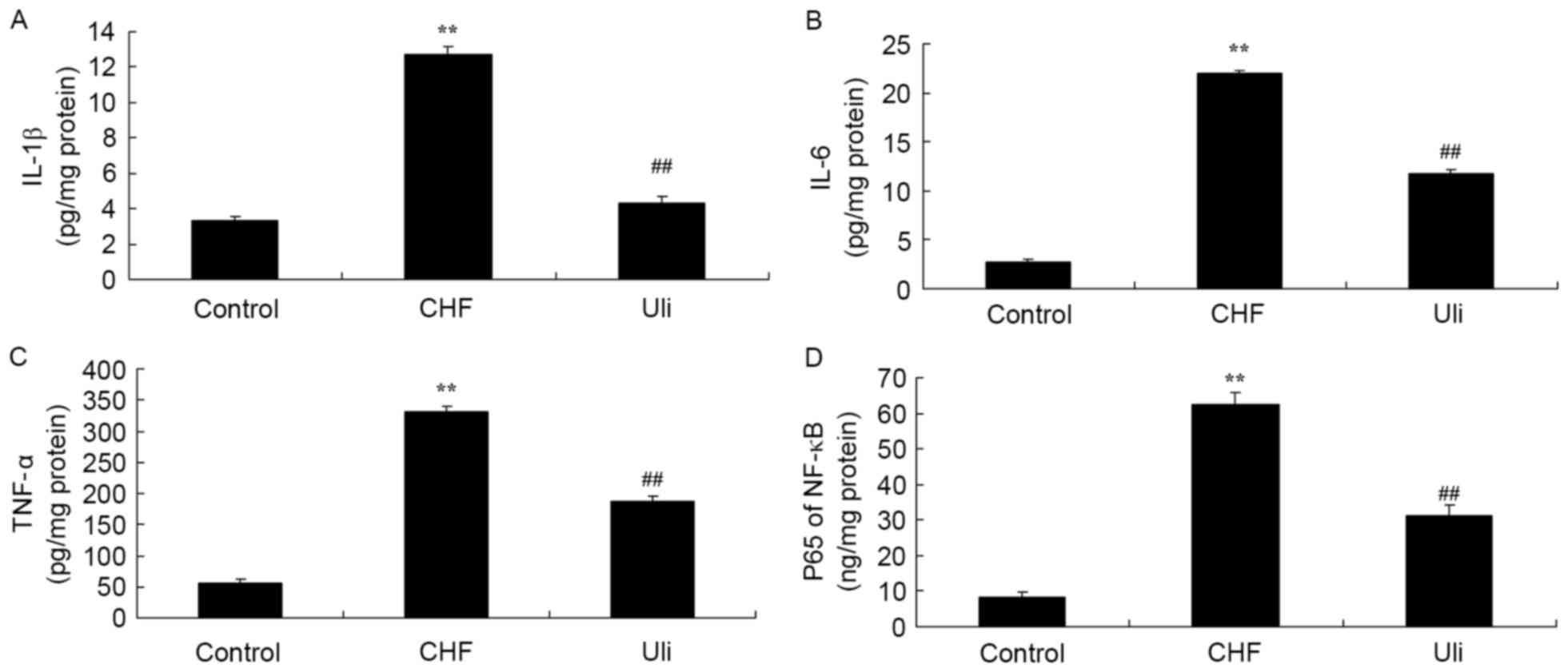
In order to examine the cardioprotective effect of
ulinastatin against inflammatory factors, NF-κB, TNF-α, IL-1β and
IL-6 expression levels in the ISO-induced CHF model rat were
analyzed. As presented in Fig. 3,
NF-κB, TNF-α, IL-1β and IL-6 levels in the ISO-induced CHF model
rat were significantly increased compared with the control group.
Ulinastatin significantly decreased NF-κB, TNF-α, IL-1β and IL-6
levels in the ISO-induced CHF model rat, compared with the
ISO-induced CHF model group (Fig.
3).
Cardioprotective effect of ulinastatin
against NF-κB signaling pathway activation and an increased
Bax/Bcl-2 ratio
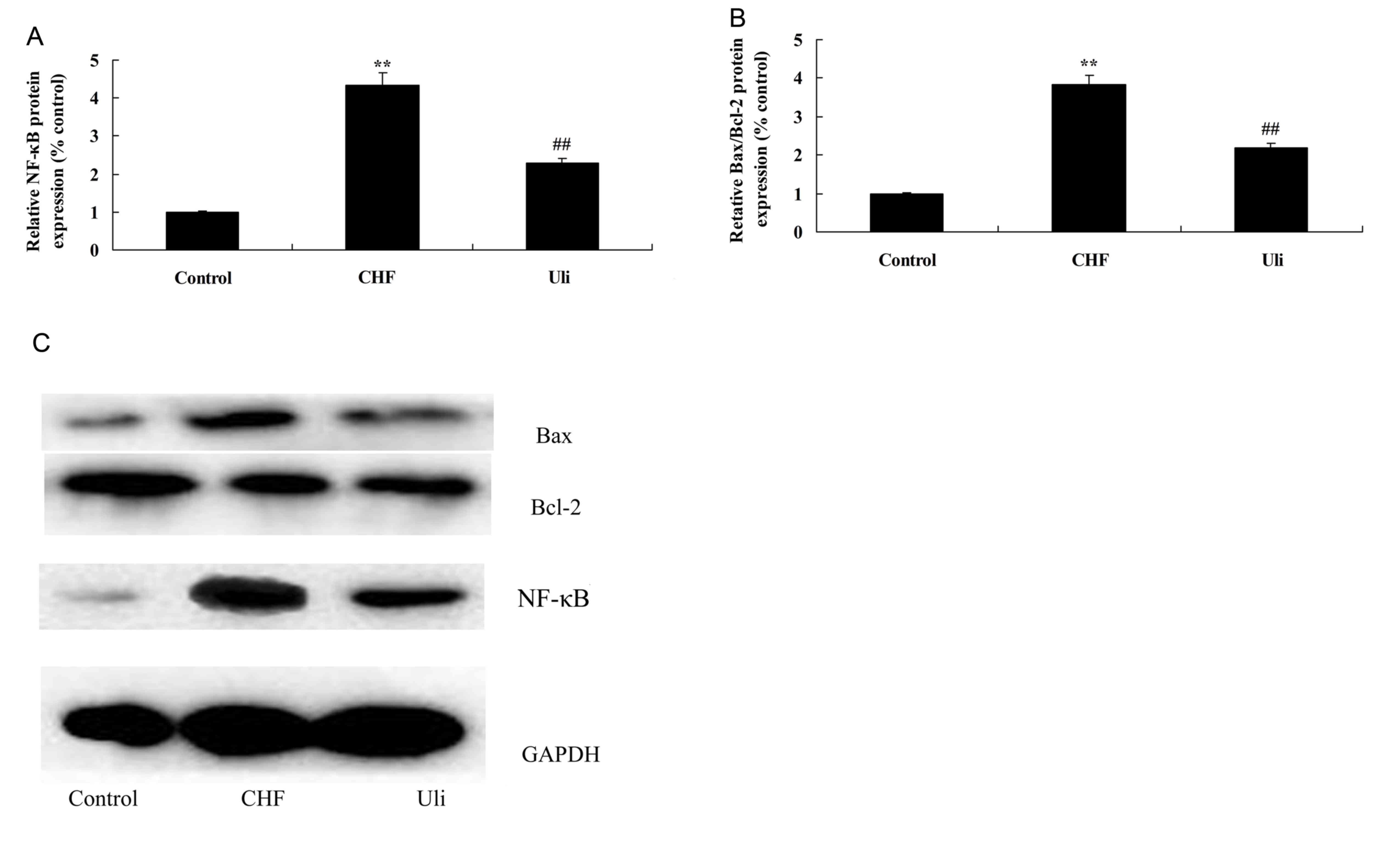
In order to further examine the mechanism underlying
the anti-inflammatory effect of ulinastatin, the NF-κB pathway and
the expression of Bax/Bcl-2 were analyzed in the present study. The
data presented in Fig. 4
demonstrated that NF-κB protein expression and the Bax/Bcl-2 ratio
were significantly upregulated in ISO-induced CHF model rats,
compared with the control group. Treatment with ulinastatin
significantly suppressed NF-κB protein expression and the Bax/Bcl-2
ratio in ISO-induced CHF rats, compared with the ISO-induced CHF
model group (Fig. 4).
Cardioprotective effect of ulinastatin
against oxidative stress
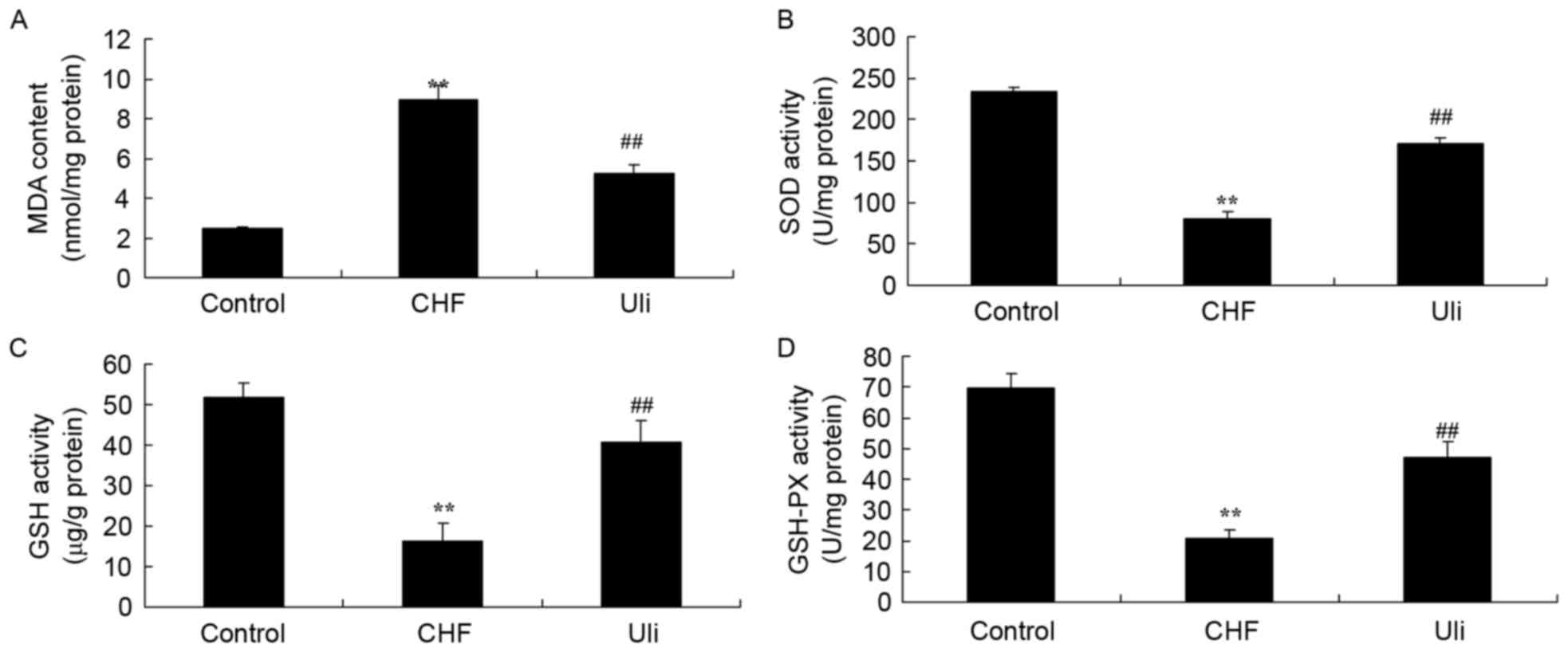
The effects of ulinastatin on the expression levels
of CHF, GSH-PX, GSH, SOD and MDA in ISO-induced CHF rats were
measured in the present study. Fig.
5 illustrates that the levels of GSH-PX, GSH and SOD were
significantly decreased and the MDA level was significantly
increased in the ISO-induced CHF rat, compared with the control
group. In ISO-induced CHF rats treated with ulinastatin, a
significant increase in GSH-PX, GSH and SOD levels, and an
inhibition of MDA, were observed compared with the ISO-induced CHF
model group (Fig. 5).
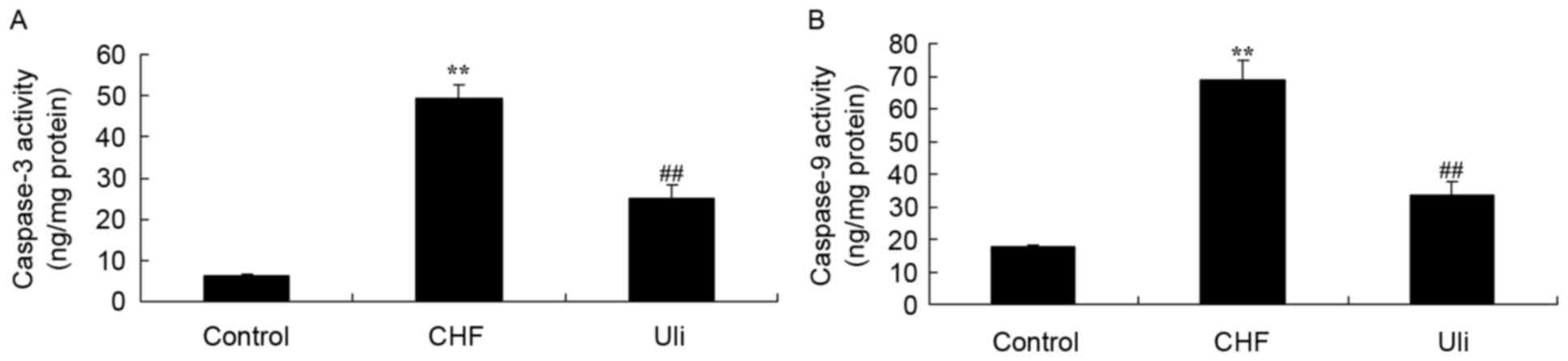
Cardioprotective effect of ulinastatin
against caspase-3 and caspase-9 activity
The expression of caspase-3 and caspase-9 was
analyzed using commercial kits, in order to further examine the
effect of ulinastatin on apoptosis in CHF. As presented in Fig. 6, significant increases in caspase-3
and caspase-9 expression were observed in the ISO-induced CHF rat
model group, compared with the control group. Following treatment
with isoproterenol hydrochloride for 5 days, ulinastatin
significantly inhibited the expression of caspase-3 and caspase-9
in ISO-induced CHF rats, compared with the ISO-induced CHF model
group (Fig. 6).
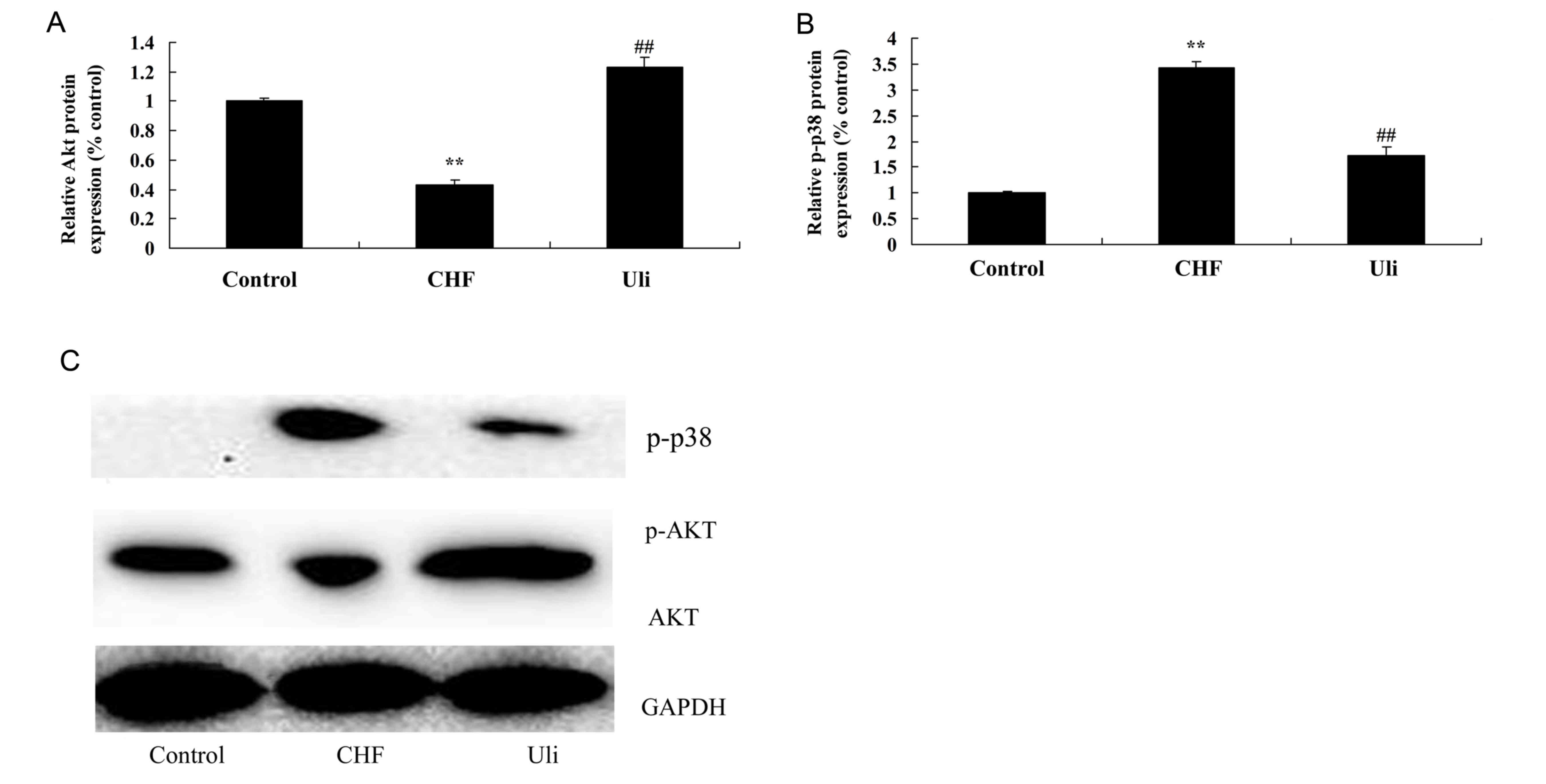
Cardioprotective effect of ulinastatin
against an increased p-Akt/Akt ratio and p-38 expression
In order to assay the alteration in p-Akt/Akt
protein expression in heart tissue samples, western blotting was
performed on cardiac tissues from each group. As presented in
Fig. 7, the p-Akt/Akt ratio was
decreased and p-38 protein expression of the ISO-induced CHF model
was increased compared with the control group. Pretreatment with
ulinastatin significantly inhibited the alterations in the
p-Akt/Akt ratio and p-38 protein expression in ISO-induced CHF
rats, compared with the ISO-induced CHF model group (Fig. 7).
Discussion
CHF is the most severe form of cardiovascular
disease (19). Due to improvements
in medical technology, the incidences of other cardiovascular
diseases are decreasing; however the incidence of CHF exhibits the
opposite trend, and the World Health Organization has established
the pathogenesis and treatment of heart failure as the focus of the
prevention and treatment of cardiovascular diseases (20,21).
A previous study has demonstrated that, in the pathogenesis of CHF,
the NF-κB signal transduction pathway is associated with apoptosis
and myocardial remodeling, which serve important roles (22). The results of the present study
demonstrated that ulinastatin prevented the increase in IVS and
LVPW, and increased LVEF and LVFS in ISO-induced CHF rats.
As a transcription factor, NF-κB is able to regulate
the immune response, including effects on innate immunity and
acquired immunity, which regulates the expressions of a series of
inflammatory cytokines and serves an important role in the
pathogenesis of CHF (23). During
CHF compensation to decompensation, the amount of cytokines
released has been demonstrated to be increased compared with sham
controls, and is positively-associated with the activation of
NF-κB, suggesting that the generation and activation of cytokines
may serve a regulatory role through the activation of NF-κB
(24,25). The results of the present study
demonstrated that ulinastatin significantly inhibited NF-κB, TNF-α,
IL-1β and IL-6 levels in ISO-induced CHF rats through suppression
of NF-κB. Hou et al (16)
posited that ulinastatin may inhibit inflammation via regulation of
the 5′-AMP-activated protein kinase/NF-κB pathway in
lipopolysaccharide-induced acute lung injury in mice.
CHF is the final result of a number of
cardiovascular diseases; the pathogenesis of heart failure is very
complex, and involves apoptosis, inflammation, myocardial
remodeling and mitochondrial injury of nerve-humoral factors
regulating system are interacted with one another. In addition, the
associated cell signal transduction mechanisms are complex
(26). The PI3K/Akt signaling
pathway serves an important role the regulation of a series of
myocardial protective functions, including myocardial cell
survival, apoptosis, myocardial remodeling and inflammation, in the
pathogenesis of heart failure (27). A previous study has confirmed that
there is an important response relationship between the
phosphorylation of Akt and myocardial protection (27). Pathological cardiac hypertrophy is
an important indicator of the development of heart failure
(28). Excessive cardiac
hypertrophy results in heart compliance, decreased contractility,
myocardial fibrosis and other irreversible alterations (29). Therefore, the inhibition of
pathological cardiac hypertrophy and the delay in ventricular
remodeling is an important potential strategy for the prevention
and treatment of CHF (29).
Protein kinase mTOR and endothelial nitric oxide synthase (eNOS)
are considered to be the important regulators of pathological
myocardial hypertrophy (27). In
the present study, it was observed that ulinastatin significantly
decreased the p-Akt/Akt ratio in ISO-induced CHF rats.
Myocardial apoptosis is an important mechanism in
the pathogenesis of heart failure, and is considered to be the
threshold at which heart failure develops into decompensation. The
anti-apoptotic effect mediated by the PI3K/Akt signaling pathway is
of importance in the regulation of myocardial cellular apoptosis
(15). The PI3K/Akt signal
transduction pathway exerts its anti-apoptotic effect through the
regulation of its downstream target proteins, including glycogen
synthase kinase-3β, caspase family proteins, apoptosis regulator
Bcl-2 family proteins and eNOS (30). The results of the present study
demonstrated that ulinastatin significantly suppressed the
Bax/Bcl-2 ratio in ISO-induced CHF rats via the PI3K/Akt signaling
pathway. Kim et al (31)
reported that ulinastatin exerted a protective effect against
regional myocardial I/R injury through activation of PI3K-Akt
signal transduction and inhibition of p38 MAPK.
Cardiac remodeling is an underlying process in heart
failure, which includes remodeling associated with organizational
structural alterations in myocardial cells, the extracellular
matrix and the collagen fiber network, for example, during the
processes of heart chamber expansion and ventricular hypertrophy,
in addition to electrical remodeling associated with a variety of
signal pathway alterations (32).
Studies have demonstrated that p38 MAPK may be activated in the
process of heart failure development, involved in the process of
ventricular remodeling, and that the inhibition of p38 MAPK is
beneficial for the improvement of ventricular remodeling (14,33).
During treatment of cardiac remodeling, p38 MAPK activity has been
demonstrated to be inhibited, suggesting that p38 MAPK activation
serves a positive role in the induction of cardiac remodeling
fibrosis (33). The results of the
present study indicated that ulinastatin significantly suppressed
p-p-38 protein expression in ISO-induced CHF rats. Liu et al
(34) reported that ulinastatin
protected against lung injury via the p38 signaling pathway.
In the present study, it was observed that the
cardioprotective effect of ulinastatin protected against
ISO-induced CHF, inflammation, oxidative stress and apoptosis via
the PI3K-Akt, p38 MAPK and NF-κB pathways. Inhibiting inflammation,
oxidative stress and apoptosis in ISO-induced CHF with ulinastatin,
and illustrating its effect in decelerating the progression of
cardiac remodeling requires further investigation.
References
|
1
|
Kitzman DW, Brubaker P, Morgan T,
Haykowsky M, Hundley G, Kraus WE, Eggebeen J and Nicklas BJ: Effect
of caloric restriction or aerobic exercise training on peak oxygen
consumption and quality of life in obese older patients with heart
failure with preserved ejection fraction: A randomized clinical
trial. JAMA. 315:36–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhong M, Zhou H, Long C, Zhang Y, Cui W,
Zhang H and Wang H: Natakalim ameliorates isoproterenol-induced
chronic heart failure by protecting against endothelial
dysfunction. Pharmacology. 98:99–110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang GX, Ohmori K, Nagai Y, Fujisawa Y,
Nishiyama A, Abe Y and Kimura S: Role of AT1 receptor in
isoproterenol-induced cardiac hypertrophy and oxidative stress in
mice. J Mol Cell Cardiol. 42:804–811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sente T, Van Berendoncks AM, Jonckheere
AI, Rodenburg RJ, Lauwers P, Van Hoof V, Wouters A, Lardon F,
Hoymans VY and Vrints CJ: Primary skeletal muscle myoblasts from
chronic heart failure patients exhibit loss of anti-inflammatory
and proliferative activity. BMC Cardiovasc Disord. 16:1072016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cabassi A, Binno SM, Tedeschi S, Graiani
G, Galizia C, Bianconcini M, Coghi P, Fellini F, Ruffini L, Govoni
P, et al: Myeloperoxidase-related chlorination activity is
positively associated with circulating ceruloplasmin in chronic
heart failure patients: Relationship with neurohormonal,
inflammatory and nutritional parameters. Biomed Res Int.
2015:6916932015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brouwers C, Kupper N, Pelle AJ, Szabó BM,
Westerhuis BL and Denollet J: Depressive symptoms in outpatients
with heart failure: Importance of inflammatory biomarkers, disease
severity and personality. Psychol Health. 29:564–582. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brouwers C, Mommersteeg PM, Nyklíček I,
Pelle AJ, Westerhuis BL, Szabó BM and Denollet J: Positive affect
dimensions and their association with inflammatory biomarkers in
patients with chronic heart failure. Biol Psychol. 92:220–226.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ranganathan P, Jayakumar C, Tang Y, Park
KM, Teoh JP, Su H, Li J, Kim IM and Ramesh G: MicroRNA-150 deletion
in mice protects kidney from myocardial infarction-induced acute
kidney injury. Am J Physiol Renal Physiol. 309:F551–F558. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bodén S, Wennberg M, Van Guelpen B,
Johansson I, Lindahl B, Andersson J, Shivappa N, Hebert JR and
Nilsson LM: Dietary inflammatory index and risk of first myocardial
infarction; a prospective population-based study. Nutr J.
16:212017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
D'Amario D, Cabral-Da-Silva MC, Zheng H,
Fiorini C, Goichberg P, Steadman E, Ferreira-Martins J, Sanada F,
Piccoli M, Cappetta D, et al: Insulin-like growth factor-1 receptor
identifies a pool of human cardiac stem cells with superior
therapeutic potential for myocardial regeneration. Circ Res.
108:1467–1481. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu LJ, Ren WY, Shen QJ, Ji HY and Zhu L:
Inflammation in lung after acute myocardial infarction is induced
by dendritic cell-mediated immune response. J Biol Regul Homeost
Agents. 31:29–40. 2017.PubMed/NCBI
|
|
12
|
Rocha JA, Ribeiro SP, França CM, Coelho O,
Alves G, Lacchini S, Kallás EG, Irigoyen MC and Consolim-Colombo
FM: Increase in cholinergic modulation with pyridostigmine induces
anti-inflammatory cell recruitment soon after acute myocardial
infarction in rats. Am J Physiol Regul Integr Comp Physiol.
310:R697–R706. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu L, Mei L, Chong L, Huang Y, Li Y, Chu M
and Yang X: Olmesartan ameliorates pressure overload-induced
cardiac remodeling through inhibition of TAK1/p38 signaling in
mice. Life Sci. 145:121–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li CY, Zhou Q, Yang LC, Chen YH, Hou JW,
Guo K, Wang YP and Li YG: Dual-specificity phosphatase 14 protects
the heart from aortic banding-induced cardiac hypertrophy and
dysfunction through inactivation of TAK1-P38MAPK/-JNK1/2 signaling
pathway. Basic Res Cardiol. 111:192016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao Y, Ruan Y, Shen T, Huang X, Li M, Yu
W, Zhu Y, Man Y, Wang S and Li J: Astragalus polysaccharide
suppresses doxorubicin-induced cardiotoxicity by regulating the
PI3k/Akt and p38MAPK pathways. Oxid Med Cell Longev.
2014:6742192014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou S, Fang M, Zhu Q, Liu Y, Liu L and Li
X: MicroRNA-939 governs vascular integrity and angiogenesis through
targeting γ-catenin in endothelial cells. Biochem Biophys Res
Commun. 484:27–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Häyry P, Aavik E and Myllärniemi M:
Blockade of growth factor synthesis and growth factor action: Two
possible sites of interference in allograft vessel disease and
coronary bypass or balloon injury. Metabolism. 45 8 Suppl
1:S101–S103. 1996. View Article : Google Scholar
|
|
18
|
Delafontaine P, Song YH and Li Y:
Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1
binding proteins in blood vessels. Arterioscler Thromb Vasc Biol.
24:435–444. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang H, Zhang FF, Peng XH, Zhao DH and
Peng J: Efficacy of medication directed by home-monitoring cardiac
resynchronization therapy in chronic heart failure patients. Chin
Med Sci J. 29:61–62. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vergaro G, Prud'homme M, Fazal L, Merval
R, Passino C, Emdin M, Samuel JL, Solal Cohen A and Delcayre C:
Inhibition of galectin-3 pathway prevents isoproterenol-induced
left ventricular dysfunction and fibrosis in mice. Hypertension.
67:606–612. 2016.PubMed/NCBI
|
|
21
|
Pitt B, Anker SD, Böhm M, Gheorghiade M,
Køber L, Krum H, Maggioni AP, Ponikowski P, Voors AA, Zannad F, et
al: Rationale and design of MinerAlocorticoid Receptor antagonist
Tolerability Study-Heart Failure (ARTS-HF): A randomized study of
finerenone vs. Eplerenone in patients who have worsening chronic
heart failure with diabetes and/or chronic kidney disease. Eur J
Heart Fail. 17:224–232. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Javan H, Szucsik AM, Li L, Schaaf CL,
Salama ME and Selzman CH: Cardiomyocyte p65 nuclear factor-κB is
necessary for compensatory adaptation to pressure overload. Circ
Heart Fail. 8:109–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao W, Chen J, Chen Y, Chen S, Chen X,
Huang H and Liu P: Advanced glycation end products induced immune
maturation of dendritic cells controls heart failure through NF-κB
signaling pathway. Arch Biochem Biophys. 580:112–120. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Killeen MJ, Linder M, Pontoniere P and
Crea R: NF-κβ signaling and chronic inflammatory diseases:
Exploring the potential of natural products to drive new
therapeutic opportunities. Drug Discov Today. 19:373–378. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maier HJ, Schips TG, Wietelmann A, Krüger
M, Brunner C, Sauter M, Klingel K, Böttger T, Braun T and Wirth T:
Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces
reversible inflammatory cardiomyopathy and heart failure. Proc Natl
Acad Sci USA. 109:11794–11799. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siman FD, Silveira EA, Fernandes AA,
Stefanon I, Vassallo DV and Padilha AS: Ouabain induces nitric
oxide release by a PI3K/Akt-dependent pathway in isolated aortic
rings from rats with heart failure. J Cardiovasc Pharmacol.
65:28–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cui J, Zhang F, Wang Y, Liu J, Ming X, Hou
J, Lv B, Fang S and Yu B: Macrophage migration inhibitory factor
promotes cardiac stem cell proliferation and endothelial
differentiation through the activation of the PI3K/Akt/mTOR and
AMPK pathways. Int J Mol Med. 37:1299–1309. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sagara S, Osanai T, Itoh T, Izumiyama K,
Shibutani S, Hanada K, Yokoyama H, Yamamoto Y, Yokota T, Tomita H,
et al: Overexpression of coupling factor 6 attenuates
exercise-induced physiological cardiac hypertrophy by inhibiting
PI3K/Akt signaling in mice. J Hypertens. 30:778–786. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun GW, Qiu ZD, Wang WN, Sui X and Sui DJ:
Flavonoids extraction from propolis attenuates pathological cardiac
hypertrophy through PI3K/AKT signaling pathway. Evid Based
Complement Alternat Med. 2016:62813762016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He SF, Jin SY, Wu H, Wang B, Wu YX, Zhang
SJ, Irwin MG, Wong TM and Zhang Y: Morphine preconditioning confers
cardioprotection in doxorubicin-induced failing rat hearts via
ERK/GSK-3β pathway independent of PI3K/Akt. Toxicol Appl Pharmacol.
288:349–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim SJ, Yoo KY, Jeong CW, Kim WM, Lee HK,
Bae HB, Kwak SH, Li M and Lee J: Urinary trypsin inhibitors afford
cardioprotective effects through activation of PI3K-Akt and ERK
signal transduction and inhibition of p38 MAPK and JNK. Cardiology.
114:264–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gaur M, Ritner C, Sievers R, Pedersen A,
Prasad M, Bernstein HS and Yeghiazarians Y: Timed inhibition of
p38MAPK directs accelerated differentiation of human embryonic stem
cells into cardiomyocytes. Cytotherapy. 12:807–817. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sucharov CC: Role of p38MAPK in
beta(2)AR-induced cardiomyopathy: At the heart of the matter?
Future Cardiol. 3:387–389. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Xiao X, Shen Y, Chen L, Xu C, Zhao
H, Wu Y, Zhang Q, Zhong J, Tang Z, et al: MicroRNA-32 promotes
calcification in vascular smooth muscle cells: Implications as a
novel marker for coronary artery calcification. PLoS One.
12:e01741382017. View Article : Google Scholar : PubMed/NCBI
|